

The Multifaceted Role of Connexins in Tumor Microenvironment Initiation and Maintenance



{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
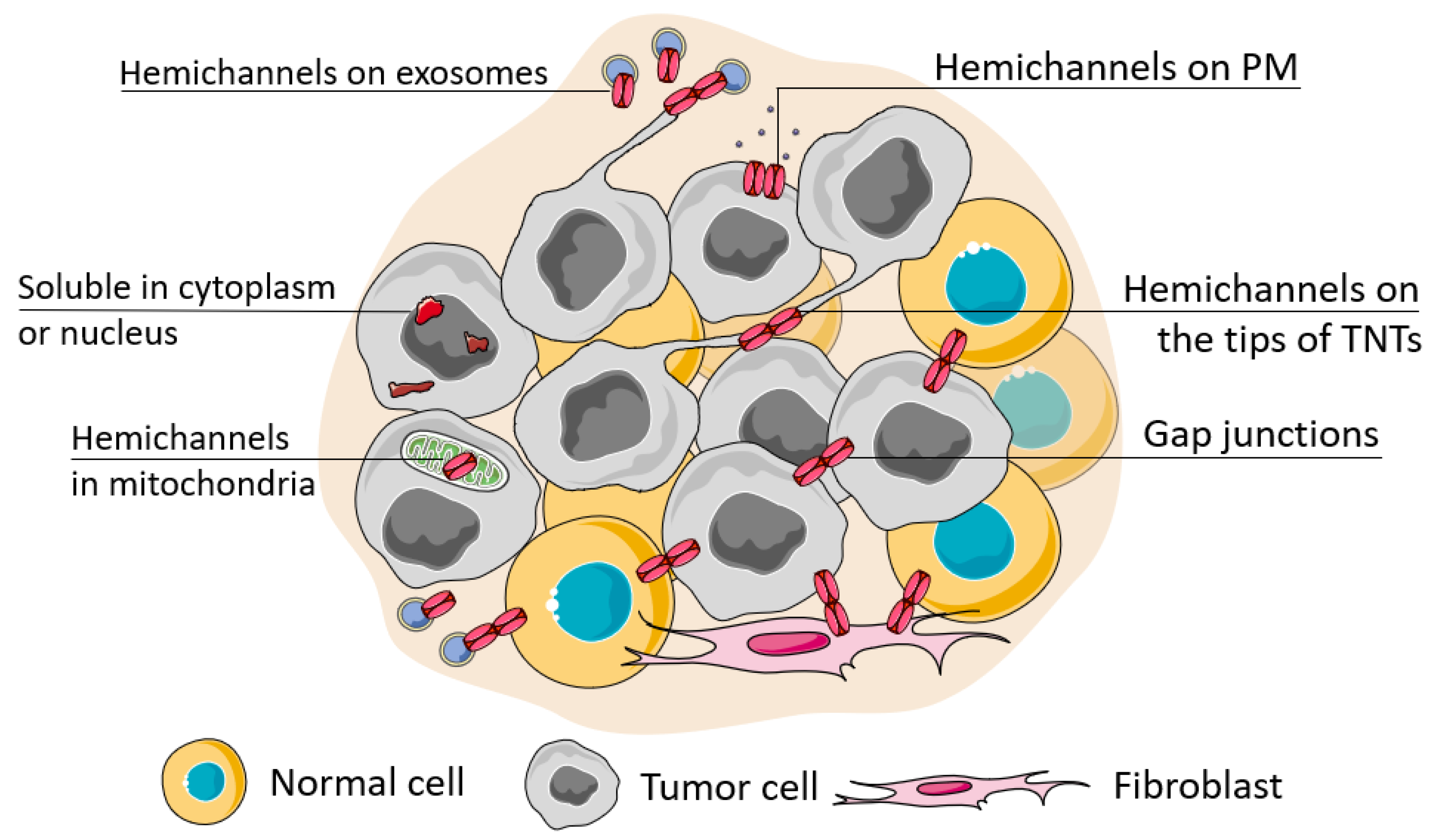
2. Connexin Localization and Its Role in Cancer
2.1. Connexin Crosstalk with Other Adhesion Proteins
2.2. Connexin on the Tips of Tunneling Nanotubes
2.3. Connexin Localization in Exosomes
2.4. Connexin Hemichannel Functioning
2.5. Intracellular Localization of Connexins
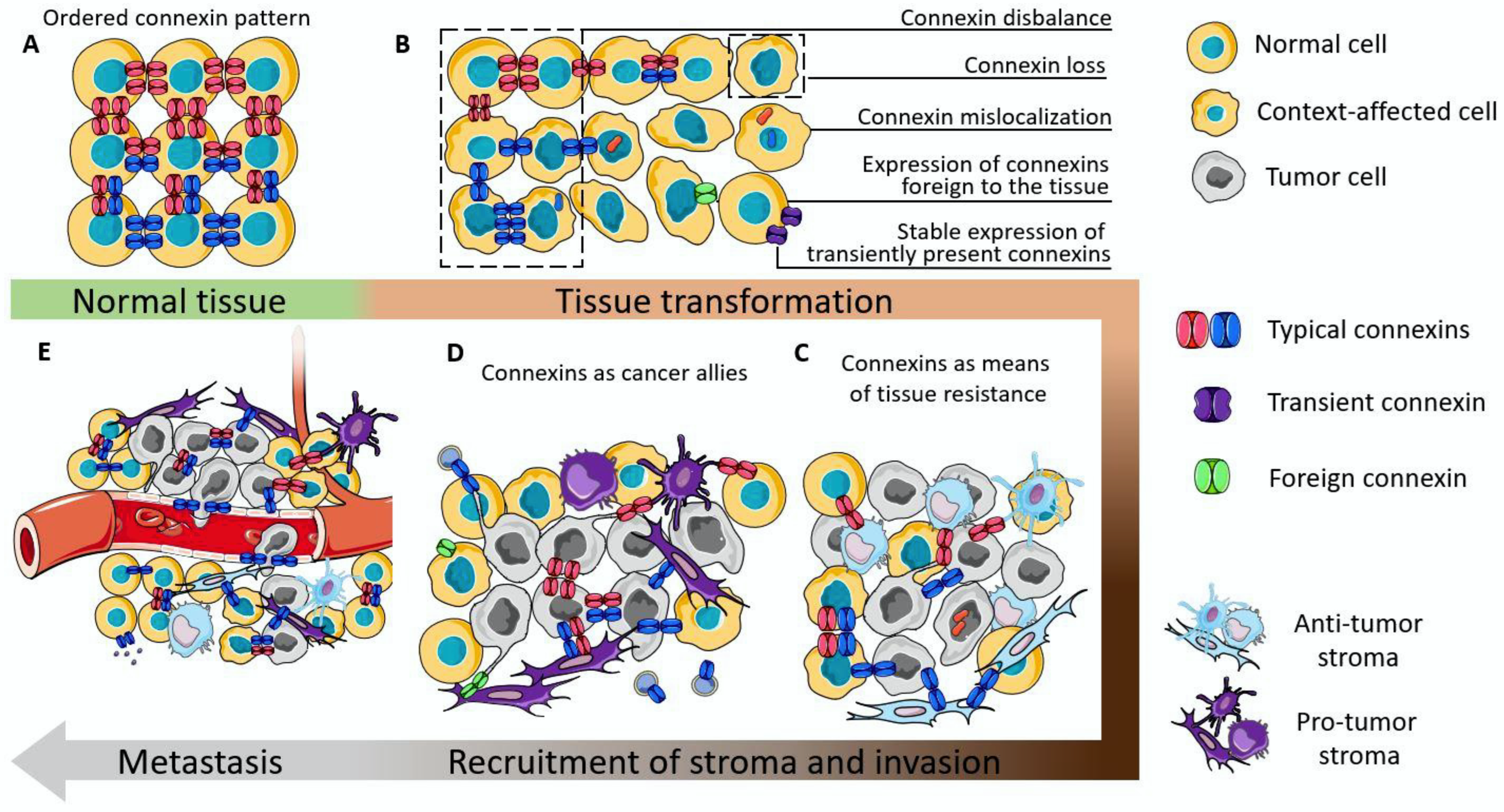
3. Connexin Participation in the Structural and Functional Integration of Malignant, Stromal, and Immune Cells within the Tumor
3.1. Connexins Integrate Tumor Cells
3.2. Connexins Integrate into the Bone Metastaic Niche
3.3. Connexins in the Brain Niche
3.4. Connexins Mediate Interactions of Cancer Cells and Cells of the Immune System
3.5. Connexins in the Interactions between Tumor Cells and Other Types of Stromal Cells
4. Involvement of Connexins in Cancer Initiation
5. Role of Connexins in Forming a Hypoxia-Resistant Cancer Cell Phenotype
6. The Multifaceted Role of Connexins in Both Tumor Progression and Suppression Due to the Intracellular Transfer of miRNA
7. Connexins and Cancer Stemness
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Brücher, B.L.; Jamall, I.S. Somatic Mutation Theory—Why it’s Wrong for Most Cancers. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. BioEssays News Rev. Mol. Cell. Dev. Biol. 2011, 33, 332–340. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Soto, A.M. Over a century of cancer research: Inconvenient truths and promising leads. PLoS Biol. 2020, 18, e3000670. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Zefferino, R.; Piccoli, C.; Di Gioia, S.; Capitanio, N.; Conese, M. How Cells Communicate with Each Other in the Tumor Microenvironment: Suggestions to Design Novel Therapeutic Strategies in Cancer Disease. Int. J. Mol. Sci. 2021, 22, 2550. [Google Scholar] [CrossRef]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Et Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Tepass, U. Adherens junctions: From molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.H. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Beyer, E.; Berthoud, V. The Family of Connexin Genes; Humana Press: Berlin/Heidelberg, Germany, 2009; pp. 3–26. [Google Scholar]
- Willecke, K.; Eiberger, J.; Degen, J.; Eckardt, D.; Romualdi, A.; Güldenagel, M.; Deutsch, U.; Söhl, G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002, 383, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Et Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Jayalakshmi, J.; Vanisree, A.J.; Ravisankar, S.; K, R. Site specific hypermethylation of CpGs in Connexin genes 30, 26 and 43 in different grades of glioma and attenuated levels of their mRNAs. Int. J. Neurosci. 2019, 129, 273–282. [Google Scholar] [CrossRef]
- Sirnes, S.; Honne, H.; Ahmed, D.; Danielsen, S.A.; Rognum, T.O.; Meling, G.I.; Leithe, E.; Rivedal, E.; Lothe, R.A.; Lind, G.E. DNA methylation analyses of the connexin gene family reveal silencing of GJC1 (Connexin45) by promoter hypermethylation in colorectal cancer. Epigenetics 2011, 6, 602–609. [Google Scholar] [CrossRef]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Kleber, A.G.; Hong, T.; et al. Auxiliary trafficking subunit GJA1-20k protects connexin-43 from degradation and limits ventricular arrhythmias. J. Clin. Investig. 2020, 130, 4858–4870. [Google Scholar] [CrossRef]
- Basheer, W.A.; Xiao, S.; Epifantseva, I.; Fu, Y.; Kleber, A.G.; Hong, T.; Shaw, R.M. GJA1-20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circ. Res. 2017, 121, 1069–1080. [Google Scholar] [CrossRef]
- Dubina, M.V.; Iatckii, N.A.; Popov, D.E.; Vasil’ev, S.V.; Krutovskikh, V.A. Connexin 43, but not connexin 32, is mutated at advanced stages of human sporadic colon cancer. Oncogene 2002, 21, 4992–4996. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef]
- Sakabe, J.; Yoshiki, R.; Sugita, K.; Haruyama, S.; Sawada, Y.; Kabashima, R.; Bito, T.; Nakamura, M.; Tokura, Y. Connexin 26 (GJB2) mutations in keratitis-ichthyosis-deafness syndrome presenting with squamous cell carcinoma. J. Derm. 2012, 39, 814–815. [Google Scholar] [CrossRef]
- Tang, T.; Tan, X.; Wang, Z.; Wang, S.; Wang, Y.; Xu, J.; Wei, X.; Zhang, D.; Liu, Q.; Jiang, J. Germline Mutations in Patients With Early-Onset Prostate Cancer. Front. Oncol. 2022, 12. [Google Scholar] [CrossRef]
- Son, H.J.; An, C.H.; Yoo, N.J.; Lee, S.H. Tight Junction-Related CLDN5 and CLDN6 Genes, and Gap Junction-Related GJB6 and GJB7 Genes Are Somatically Mutated in Gastric and Colorectal Cancers. Pathol. Oncol. Res. 2020, 26, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.W.; Minogue, P.J.; Thomas, B.C.; Domowicz, D.A.; Berthoud, V.M.; Hanck, D.A.; Beyer, E.C. An intact connexin N-terminus is required for function but not gap junction formation. J. Cell Sci. 2008, 121, 2744–2750. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.A.; Chang, H.C.; Spaeth, K.E.; Nitsche, J.M.; Nicholson, B.J. The permeability of gap junction channels to probes of different size is dependent on connexin composition and permeant-pore affinities. Biophys. J. 2004, 87, 958–973. [Google Scholar] [CrossRef] [PubMed]
- Fleishman, S.J.; Unger, V.M.; Yeager, M.; Ben-Tal, N. A Calpha model for the transmembrane alpha helices of gap junction intercellular channels. Mol. Cell 2004, 15, 879–888. [Google Scholar] [CrossRef]
- Skerrett, I.M.; Williams, J.B. A structural and functional comparison of gap junction channels composed of connexins and innexins. Dev. Neurobiol. 2017, 77, 522–547. [Google Scholar] [CrossRef]
- Zhou, X.W.; Pfahnl, A.; Werner, R.; Hudder, A.; Llanes, A.; Luebke, A.; Dahl, G. Identification of a pore lining segment in gap junction hemichannels. Biophys. J. 1997, 72, 1946–1953. [Google Scholar] [CrossRef]
- Kronengold, J.; Srinivas, M.; Verselis, V.K. The N-terminal half of the connexin protein contains the core elements of the pore and voltage gates. J. Membr. Biol. 2012, 245, 453–463. [Google Scholar] [CrossRef]
- Flores, J.A.; Haddad, B.G.; Dolan, K.A.; Myers, J.B.; Yoshioka, C.C.; Copperman, J.; Zuckerman, D.M.; Reichow, S.L. Connexin-46/50 in a dynamic lipid environment resolved by CryoEM at 1.9 Å. Nat. Commun. 2020, 11, 4331. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.K.; Jagielnicki, M.; McIntire, W.E.; Purdy, M.D.; Dharmarajan, V.; Griffin, P.R.; Yeager, M. A Steric “Ball-and-Chain” Mechanism for pH-Mediated Regulation of Gap Junction Channels. Cell Rep. 2020, 31, 107482. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeong, H.; Hyun, J.; Ryu, B.; Park, K.; Lim, H.H.; Yoo, J.; Woo, J.S. Cryo-EM structure of human Cx31.3/GJC3 connexin hemichannel. Sci. Adv. 2020, 6, eaba4996. [Google Scholar] [CrossRef]
- Khan, A.K.; Jagielnicki, M.; Bennett, B.C.; Purdy, M.D.; Yeager, M. Cryo-EM structure of an open conformation of a gap junction hemichannel in lipid bilayer nanodiscs. Structure 2021, 29, 1040–1047.e1043. [Google Scholar] [CrossRef]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Et Biophys. Acta. Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef]
- Retamal, M.A.; García, I.E.; Pinto, B.I.; Pupo, A.; Báez, D.; Stehberg, J.; Del Rio, R.; González, C. Extracellular Cysteine in Connexins: Role as Redox Sensors. Front. Physiol. 2016, 7, 1. [Google Scholar] [CrossRef]
- Olbina, G.; Eckhart, W. Mutations in the second extracellular region of connexin 43 prevent localization to the plasma membrane, but do not affect its ability to suppress cell growth. Mol. Cancer Res. MCR 2003, 1, 690–700. [Google Scholar]
- Fernández-Olivares, A.; Durán-Jara, E.; Verdugo, D.A.; Fiori, M.C.; Altenberg, G.A.; Stehberg, J.; Alfaro, I.; Calderón, J.F.; Retamal, M.A. Extracellular Cysteines Are Critical to Form Functional Cx46 Hemichannels. Int. J. Mol. Sci. 2022, 23, 7252. [Google Scholar] [CrossRef]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Et Biophys. Acta. Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Koval, M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006, 16, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Lagree, V.; Brunschwig, K.; Lopez, P.; Gilula, N.B.; Richard, G.; Falk, M.M. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J. Cell Sci. 2003, 116, 3189–3201. [Google Scholar] [CrossRef]
- Smith, T.D.; Mohankumar, A.; Minogue, P.J.; Beyer, E.C.; Berthoud, V.M.; Koval, M. Cytoplasmic amino acids within the membrane interface region influence connexin oligomerization. J. Membr. Biol. 2012, 245, 221–230. [Google Scholar] [CrossRef]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef]
- Tong, X.; Lopez, W.; Ramachandran, J.; Ayad, W.A.; Liu, Y.; Lopez-Rodriguez, A.; Harris, A.L.; Contreras, J.E. Glutathione release through connexin hemichannels: Implications for chemical modification of pores permeable to large molecules. J. Gen. Physiol. 2015, 146, 245–254. [Google Scholar] [CrossRef]
- Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J.D. Connexins in Cancer: Jekyll or Hyde? Biomolecules 2020, 10, 1654. [Google Scholar] [CrossRef]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef]
- Loewenstein, W.R.; Kanno, Y. Intercellular communication and the control of tissue growth: Lack of communication between cancer cells. Nature 1966, 209, 1248–1249. [Google Scholar] [CrossRef]
- Zhang, K.; Guan, Q.W.; Zhou, X.Y.; Xia, Q.X.; Yin, X.X.; Zhou, H.H.; Mao, X.Y. The mutual interplay of redox signaling and connexins. J. Mol. Med. 2021, 99, 933–941. [Google Scholar] [CrossRef]
- Cascio, M. Connexins and their environment: Effects of lipids composition on ion channels. Biochim. Et Biophys. Acta 2005, 1711, 142–153. [Google Scholar] [CrossRef]
- Govindarajan, R.; Chakraborty, S.; Johnson, K.E.; Falk, M.M.; Wheelock, M.J.; Johnson, K.R.; Mehta, P.P. Assembly of connexin43 into gap junctions is regulated differentially by E-cadherin and N-cadherin in rat liver epithelial cells. Mol. Biol. Cell 2010, 21, 4089–4107. [Google Scholar] [CrossRef]
- Chakraborty, S.; Mitra, S.; Falk, M.M.; Caplan, S.H.; Wheelock, M.J.; Johnson, K.R.; Mehta, P.P. E-cadherin differentially regulates the assembly of Connexin43 and Connexin32 into gap junctions in human squamous carcinoma cells. J. Biol. Chem. 2010, 285, 10761–10776. [Google Scholar] [CrossRef]
- Kanczuga-Koda, L.; Wincewicz, A.; Fudala, A.; Abrycki, T.; Famulski, W.; Baltaziak, M.; Sulkowski, S.; Koda, M. E-cadherin and β-catenin adhesion proteins correlate positively with connexins in colorectal cancer. Oncol. Lett. 2014, 7, 1863–1870. [Google Scholar] [CrossRef]
- Sinha, G.; Ferrer, A.I.; Ayer, S.; El-Far, M.H.; Pamarthi, S.H.; Naaldijk, Y.; Barak, P.; Sandiford, O.A.; Bibber, B.M.; Yehia, G.; et al. Specific N-cadherin-dependent pathways drive human breast cancer dormancy in bone marrow. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Xu, X.; Li, W.E.; Huang, G.Y.; Meyer, R.; Chen, T.; Luo, Y.; Thomas, M.P.; Radice, G.L.; Lo, C.W. Modulation of mouse neural crest cell motility by N-cadherin and connexin 43 gap junctions. J. Cell Biol. 2001, 154, 217–230. [Google Scholar] [CrossRef]
- Perrot, C.Y.; Herrera, J.L.; Fournier-Goss, A.E.; Komatsu, M. Prostaglandin E2 breaks down pericyte-endothelial cell interaction via EP1 and EP4-dependent downregulation of pericyte N-cadherin, connexin-43, and R-Ras. Sci. Rep. 2020, 10, 11186. [Google Scholar] [CrossRef]
- Cohen-Barak, E.; Godsel, L.M.; Koetsier, J.L.; Hegazy, M.; Kushnir-Grinbaum, D.; Hammad, H.; Danial-Farran, N.; Harmon, R.; Khayat, M.; Bochner, R.; et al. The Role of Desmoglein 1 in Gap Junction Turnover Revealed through the Study of SAM Syndrome. J. Investig. Dermatol. 2020, 140, 556–567.e559. [Google Scholar] [CrossRef] [PubMed]
- Leech, A.O.; Cruz, R.G.B.; Hill, A.D.K.; Hopkins, A.M. Paradigms lost—An emerging role for over-expression of tight junction adhesion proteins in cancer pathogenesis. Ann. Transl. Med. 2015, 3, 13. [Google Scholar]
- Liu, Y.Q.; Zou, H.Y.; Xie, J.J.; Fang, W.K. Paradoxical Roles of Desmosomal Components in Head and Neck Cancer. Biomolecules 2021, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Venhuizen, J.H.; Jacobs, F.J.C.; Span, P.N.; Zegers, M.M. P120 and E-cadherin: Double-edged swords in tumor metastasis. Semin. Cancer Biol. 2020, 60, 107–120. [Google Scholar] [CrossRef]
- Caillou, B.; Talbot, M.; Weyemi, U.; Pioche-Durieu, C.; Al Ghuzlan, A.; Bidart, J.M.; Chouaib, S.; Schlumberger, M.; Dupuy, C. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PloS One 2011, 6, e22567. [Google Scholar] [CrossRef] [PubMed]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. United States Am. 2010, 107, 17194–17199. [Google Scholar] [CrossRef]
- Desir, S.; O’Hare, P.; Vogel, R.I.; Sperduto, W.; Sarkari, A.; Dickson, E.L.; Wong, P.; Nelson, A.C.; Fong, Y.; Steer, C.J.; et al. Chemotherapy-Induced Tunneling Nanotubes Mediate Intercellular Drug Efflux in Pancreatic Cancer. Sci. Rep. 2018, 8, 9484. [Google Scholar] [CrossRef] [PubMed]
- Tishchenko, A.; Azorín, D.D.; Vidal-Brime, L.; Muñoz, M.J.; Arenas, P.J.; Pearce, C.; Girao, H.; Ramón, Y.C.S.; Aasen, T. Cx43 and Associated Cell Signaling Pathways Regulate Tunneling Nanotubes in Breast Cancer Cells. Cancers 2020, 12, 2798. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Kameritsch, P.; Wallner, S.; Pohl, U.; Pogoda, K. The carboxyl tail of Cx43 augments p38 mediated cell migration in a gap junction-independent manner. Eur. J. Cell Biol. 2010, 89, 828–838. [Google Scholar] [CrossRef]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. Biochim. Et Biophys. Acta 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [PubMed]
- Pulze, L.; Congiu, T.; Brevini, T.A.L.; Grimaldi, A.; Tettamanti, G.; D’Antona, P.; Baranzini, N.; Acquati, F.; Ferraro, F.; de Eguileor, M. MCF7 Spheroid Development: New Insight about Spatio/Temporal Arrangements of TNTs, Amyloid Fibrils, Cell Connections, and Cellular Bridges. Int. J. Mol. Sci. 2020, 21, 5400. [Google Scholar] [CrossRef]
- Bao, B.; Jiang, J.; Yanase, T.; Nishi, Y.; Morgan, J.R. Connexon-mediated cell adhesion drives microtissue self-assembly. FASEB J. 2011, 25, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Martinez, L.; Villafranca-Baughman, D.; Quintero, H.; Kacerovsky, J.B.; Dotigny, F.; Murai, K.K.; Prat, A.; Drapeau, P.; Di Polo, A. Interpericyte tunnelling nanotubes regulate neurovascular coupling. Nature 2020, 585, 91–95. [Google Scholar] [CrossRef]
- Errede, M.; Mangieri, D.; Longo, G.; Girolamo, F.; de Trizio, I.; Vimercati, A.; Serio, G.; Frei, K.; Perris, R.; Virgintino, D. Tunneling nanotubes evoke pericyte/endothelial communication during normal and tumoral angiogenesis. Fluids Barriers CNS 2018, 15, 28. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Thayanithy, V.; Babatunde, V.; Dickson, E.L.; Wong, P.; Oh, S.; Ke, X.; Barlas, A.; Fujisawa, S.; Romin, Y.; Moreira, A.L.; et al. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 2014, 323, 178–188. [Google Scholar] [CrossRef]
- Acuña, R.A.; Varas-Godoy, M.; Berthoud, V.M.; Alfaro, I.E.; Retamal, M.A. Connexin-46 Contained in Extracellular Vesicles Enhance Malignancy Features in Breast Cancer Cells. Biomolecules 2020, 10, 676. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Martins-Marques, T.; Ribeiro-Rodrigues, T.; Ferreira, J.V.; Catarino, S.; Pinho, M.J.; Zuzarte, M.; Isabel Anjo, S.; Manadas, B.; Joost, P.G.S.; et al. Gap junctional protein Cx43 is involved in the communication between extracellular vesicles and mammalian cells. Sci. Rep. 2015, 5, 13243. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Zhang, L.L.; Bi, Q.C.; Gan, L.J.; Wei, M.J.; Hong, T.; Tan, R.J.; Lan, X.M.; Liu, L.H.; Han, X.J.; et al. Exosomal connexin 43 regulates the resistance of glioma cells to temozolomide. Oncol. Rep. 2021, 45. [Google Scholar] [CrossRef] [PubMed]
- Varela-Eirin, M.; Varela-Vazquez, A.; Rodríguez-Candela Mateos, M.; Vila-Sanjurjo, A.; Fonseca, E.; Mascareñas, J.L.; Eugenio Vázquez, M.; Mayan, M.D. Recruitment of RNA molecules by connexin RNA-binding motifs: Implication in RNA and DNA transport through microvesicles and exosomes. Biochim. Et Biophys. Acta. Mol. Cell Res. 2017, 1864, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Martins-Marques, T.; Costa, M.C.; Catarino, S.; Simoes, I.; Aasen, T.; Enguita, F.J.; Girao, H. Cx43-mediated sorting of miRNAs into extracellular vesicles. EMBO Rep. 2022, 23, e54312. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Kawamoto, E.; Gaowa, A.; Okamoto, T.; Park, E.J. Connexins and Integrins in Exosomes. Cancers 2019, 11, 106. [Google Scholar] [CrossRef]
- Decrock, E.; Vinken, M.; De Vuyst, E.; Krysko, D.V.; D’Herde, K.; Vanhaecke, T.; Vandenabeele, P.; Rogiers, V.; Leybaert, L. Connexin-related signaling in cell death: To live or let die? Cell Death Differ. 2009, 16, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.A.; Ilina, O.; Vasaturo, A.; Venhuizen, J.H.; Vullings, M.; Venhuizen, V.; Bilos, A.; Figdor, C.G.; Span, P.N.; Friedl, P. Collective invasion induced by an autocrine purinergic loop through connexin-43 hemichannels. J. Cell Biol. 2020, 219, e201911120. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Riquelme, M.A.; Tu, C.; Quan, Y.; Liu, X.; Sun, L.-Z.; Jiang, J.X. Osteocytic Connexin Hemichannels Modulate Oxidative Bone Microenvironment and Breast Cancer Growth. Cancers 2021, 13, 6343. [Google Scholar] [CrossRef] [PubMed]
- Turovsky, E.A.; Varlamova, E.G. Mechanism of Ca(2+)-Dependent Pro-Apoptotic Action of Selenium Nanoparticles, Mediated by Activation of Cx43 Hemichannels. Biology 2021, 10, 743. [Google Scholar] [CrossRef]
- Nardin, C.; Peres, C.; Putti, S.; Orsini, T.; Colussi, C.; Mazzarda, F.; Raspa, M.; Scavizzi, F.; Salvatore, A.M.; Chiani, F.; et al. Connexin Hemichannel Activation by S-Nitrosoglutathione Synergizes Strongly with Photodynamic Therapy Potentiating Anti-Tumor Bystander Killing. Cancers 2021, 13, 5062. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Riquelme, M.A.; Gu, S.; Kar, R.; Gao, X.; Sun, L.; Jiang, J.X. Osteocytic connexin hemichannels suppress breast cancer growth and bone metastasis. Oncogene 2016, 35, 5597–5607. [Google Scholar] [CrossRef] [PubMed]
- Kozoriz, M.G.; Church, J.; Ozog, M.A.; Naus, C.C.; Krebs, C. Temporary sequestration of potassium by mitochondria in astrocytes. J. Biol. Chem. 2010, 285, 31107–31119. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Smith, K.; Yu, Q.; Miller, C.; Singh, K.; Sen, C.K. Mitochondrial connexin 43 in sex-dependent myocardial responses and estrogen-mediated cardiac protection following acute ischemia/reperfusion injury. Basic Res. Cardiol. 2019, 115, 1. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Kim, I.S.; Ganesan, P.; Choi, D.K. Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2016, 17, 1819. [Google Scholar] [CrossRef] [PubMed]
- Uzu, M.; Sato, H.; Shimizu, A.; Shibata, Y.; Ueno, K.; Hisaka, A. Connexin 43 enhances Bax activation via JNK activation in sunitinib-induced apoptosis in mesothelioma cells. J. Pharmacol. Sci. 2017, 134, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Artesi, M.; Kroonen, J.; Bredel, M.; Nguyen-Khac, M.; Deprez, M.; Schoysman, L.; Poulet, C.; Chakravarti, A.; Kim, H.; Scholtens, D.; et al. Connexin 30 expression inhibits growth of human malignant gliomas but protects them against radiation therapy. Neuro-Oncol. 2015, 17, 392–406. [Google Scholar] [CrossRef]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological Functions of Connexin43 Beyond Intercellular Communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef]
- Peracchia, C. Connexin/Innexin Channels in Cytoplasmic Organelles. Are There Intracellular Gap Junctions? A Hypothesis! Int. J. Mol. Sci. 2020, 21, 2163. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Mitochondria and Preconditioning. Circ. Res. 2006, 99, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Haider, H.; Porollo, A.; Ashraf, M. Mitochondria-specific transgenic overexpression of connexin-43 simulates preconditioning-induced cytoprotection of stem cells. Cardiovasc. Res. 2010, 88, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Ruiz-Meana, M.; Gent, S.; Ungefug, E.; Soetkamp, D.; Miro-Casas, E.; Cabestrero, A.; Fernandez-Sanz, C.; Semenzato, M.; Di Lisa, F.; et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J. Cell. Mol. Med. 2012, 16, 1649–1655. [Google Scholar] [CrossRef]
- Wörsdörfer, P.; Wagner, N.; Ergün, S. The role of connexins during early embryonic development: Pluripotent stem cells, gene editing, and artificial embryonic tissues as tools to close the knowledge gap. Histochem. Cell Biol. 2018, 150, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Iikawa, N.; Yamamoto, Y.; Kawasaki, Y.; Nishijima-Matsunobu, A.; Suzuki, M.; Yamada, T.; Omori, Y. Intrinsic Oncogenic Function of Intracellular Connexin26 Protein in Head and Neck Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2018, 19, 2134. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.G.; Lin, X.; Liu, J.C.; Zhou, J. Hypoxia-induced internalization of connexin 26 and connexin 43 in pulmonary epithelial cells is involved in the occurrence of non-small cell lung cancer via the P53/MDM2 signaling pathway. Int. J. Oncol. 2019, 55, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, X.; Ge, H.; Zheng, N.; Peng, F.; Fu, Y.; Tao, L.; Wang, Q. Inhibition of ubiquitin-specific protease 14 promotes connexin 32 internalization and counteracts cisplatin cytotoxicity in human ovarian cancer cells. Oncol. Rep. 2019, 42, 1237–1247. [Google Scholar] [CrossRef]
- Leroy, K.; Silva Costa, C.J.; Pieters, A.; Dos Santos Rodrigues, B.; Van Campenhout, R.; Cooreman, A.; Tabernilla, A.; Cogliati, B.; Vinken, M. Expression and Functionality of Connexin-Based Channels in Human Liver Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 12187. [Google Scholar] [CrossRef]
- Sozen, B.; Can, A.; Demir, N. Cell fate regulation during preimplantation development: A view of adhesion-linked molecular interactions. Dev. Biol. 2014, 395, 73–83. [Google Scholar] [CrossRef]
- Li, J.; Cheng, L.; Wang, L.J.; Liu, H.C.; Li, L.; Wang, X.L.; Geng, M.Y. Cell surface sialic acid inhibits Cx43 gap junction functions in constructed Hela cancer cells involving in sialylated N-cadherin. Mol. Cell. Biochem. 2010, 344, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Conduction in cardiac tissue. Historical reflections. Physiol. Rep. 2019, 7, e13860. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, E.; Han, Y.L.; Guo, M.; Shenoy, V.B. Gap junctions amplify spatial variations in cell volume in proliferating tumor spheroids. Nat. Commun. 2020, 11, 6148. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Hong, Q.; Wu, H.; Wang, F.; Zhong, L.; Shen, L.; Xu, P.; Zhang, W.; Cao, H.; Zhan, Y.Y.; et al. Gap junctions mediate glucose transfer to promote colon cancer growth in three-dimensional spheroid culture. Cancer Lett. 2022, 531, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Balandre, A.C.; Debant, M.; Vix, J.; Harnois, T.; Bourmeyster, N.; Péraudeau, E.; Chépied, A.; Clarhaut, J.; Debiais, F.; et al. Cx43 Present at the Leading Edge Membrane Governs Promigratory Effects of Osteoblast-Conditioned Medium on Human Prostate Cancer Cells in the Context of Bone Metastasis. Cancers 2020, 12, 3013. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, L.; Liu, J.; Goldstein, A.; Bado, I.; Zhang, W.; Arenkiel, B.R.; Li, Z.; Yang, M.; Du, S.; et al. The Osteogenic Niche Is a Calcium Reservoir of Bone Micrometastases and Confers Unexpected Therapeutic Vulnerability. Cancer Cell 2018, 34, 823–839.e827. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Guise, T.A.; Mohammad, K.S. A “Connexin” Responsible for the Fatal Attraction of Cancer to Bone. Cell Metab. 2019, 29, 6–8. [Google Scholar] [CrossRef]
- Suzhi, Z.; Liang, T.; Yuexia, P.; Lucy, L.; Xiaoting, H.; Yuan, Z.; Qin, W. Gap Junctions Enhance the Antiproliferative Effect of MicroRNA-124-3p in Glioblastoma Cells. J. Cell. Physiol. 2015, 230, 2476–2488. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- McCutcheon, S.; Spray, D.C. Abstract 2885: Connexin 43-dependent miRNA transfer drives perivascular glioma invasion through dysregulation of astrocytes. Cancer Res. 2021, 81, 2885. [Google Scholar] [CrossRef]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, S.; Spray, D.C. Glioblastoma–Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol. Cancer Res. 2021, 20, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Talaverón, R.; Matarredona, E.R.; Herrera, A.; Medina, J.M.; Tabernero, A. Connexin43 Region 266-283, via Src Inhibition, Reduces Neural Progenitor Cell Proliferation Promoted by EGF and FGF-2 and Increases Astrocytic Differentiation. Int. J. Mol. Sci. 2020, 21, 8852. [Google Scholar] [CrossRef] [PubMed]
- Figueira, I.; Galego, S.; Custódio-Santos, T.; Vicente, R.; Molnár, K.; Haskó, J.; Malhó, R.; Videira, M.; Wilhelm, I.; Krizbai, I.; et al. Picturing Breast Cancer Brain Metastasis Development to Unravel Molecular Players and Cellular Crosstalk. Cancers 2021, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Cha, J.; Kim, S.K.; Park, J.H.; Song, K.H.; Kim, P.; Kim, M.Y. c-MYC Drives Breast Cancer Metastasis to the Brain, but Promotes Synthetic Lethality with TRAIL. Mol. Cancer Res. MCR 2019, 17, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A.; Navarrete, M.; Gleisner, M.A.; Gebicke-Haerter, P.; Salazar-Onfray, F. Connexin-Mediated Signaling at the Immunological Synapse. Int. J. Mol. Sci. 2020, 21, 3736. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A. Connexin channels modulation in pathophysiology and treatment of immune and inflammatory disorders. Biochim. Et Biophys. Acta. Mol. Basis Dis. 2021, 1867, 166258. [Google Scholar] [CrossRef]
- Mendoza-Naranjo, A.; Bouma, G.; Pereda, C.; Ramírez, M.; Webb, K.F.; Tittarelli, A.; López, M.N.; Kalergis, A.M.; Thrasher, A.J.; Becker, D.L.; et al. Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J. Immunol. 2011, 187, 3121–3132. [Google Scholar] [CrossRef]
- Tittarelli, A.; Mendoza-Naranjo, A.; Farías, M.; Guerrero, I.; Ihara, F.; Wennerberg, E.; Riquelme, S.; Gleisner, A.; Kalergis, A.; Lundqvist, A.; et al. Gap junction intercellular communications regulate NK cell activation and modulate NK cytotoxic capacity. J. Immunol. 2014, 192, 1313–1319. [Google Scholar] [CrossRef]
- Benlalam, H.; Carré, T.; Jalil, A.; Noman, Z.; Caillou, B.; Vielh, P.; Tittarelli, A.; Robert, C.; Chouaib, S. Regulation of gap junctions in melanoma and their impact on Melan-A/MART-1-specific CD8+ T lymphocyte emergence. J. Mol. Med. (Berl. Ger.) 2013, 91, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Navarrete, M.; Álvarez, J.; Guerrero, I.; Gleisner, M.A.; Tittarelli, A.; Salazar-Onfray, F. Cx43-Gap Junctions Accumulate at the Cytotoxic Immunological Synapse Enabling Cytotoxic T Lymphocyte Melanoma Cell Killing. Int. J. Mol. Sci. 2019, 20, 4509. [Google Scholar] [CrossRef] [PubMed]
- Benlalam, H.; Jalil, A.; Hasmim, M.; Pang, B.; Tamouza, R.; Mitterrand, M.; Godet, Y.; Lamerant, N.; Robert, C.; Avril, M.F.; et al. Gap junction communication between autologous endothelial and tumor cells induce cross-recognition and elimination by specific CTL. J. Immunol. 2009, 182, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Ji, L.; Wang, H.; Wang, W.; Zheng, H.; Zou, J.; Liu, L.; Qi, X.; Liu, Z.; Du, B.; et al. Connexin 43 upregulation by dioscin inhibits melanoma progression via suppressing malignancy and inducing M1 polarization. Int. J. Cancer 2017, 141, 1690–1703. [Google Scholar] [CrossRef]
- Tittarelli, A.; Navarrete, M.; Lizana, M.; Hofmann-Vega, F.; Salazar-Onfray, F. Hypoxic Melanoma Cells Deliver microRNAs to Dendritic Cells and Cytotoxic T Lymphocytes through Connexin-43 Channels. Int. J. Mol. Sci. 2020, 21, 7567. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Zhou, Y.; Zheng, J.; Jin, G.; Tao, L.; Yao, H.; Zhang, J.; Sun, Y.; Liu, Y.; Hu, L.P. GJB3 promotes pancreatic cancer liver metastasis by enhancing the polarization and survival of neutrophil. Front. Immunol. 2022, 13, 983116. [Google Scholar] [CrossRef]
- Balla, P.; Maros, M.E.; Barna, G.; Antal, I.; Papp, G.; Sapi, Z.; Athanasou, N.A.; Benassi, M.S.; Picci, P.; Krenacs, T. Prognostic impact of reduced connexin43 expression and gap junction coupling of neoplastic stromal cells in giant cell tumor of bone. PLoS ONE 2015, 10, e0125316. [Google Scholar] [CrossRef]
- Dilley, T.K.; Bowden, G.T.; Chen, Q.M. Novel mechanisms of sublethal oxidant toxicity: Induction of premature senescence in human fibroblasts confers tumor promoter activity. Exp. Cell Res. 2003, 290, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kiszner, G.; Balla, P.; Wichmann, B.; Barna, G.; Baghy, K.; Nemeth, I.B.; Varga, E.; Furi, I.; Toth, B.; Krenacs, T. Exploring Differential Connexin Expression across Melanocytic Tumor Progression Involving the Tumor Microenvironment. Cancers 2019, 11, 165. [Google Scholar] [CrossRef]
- Haass, N.K.; Ripperger, D.; Wladykowski, E.; Dawson, P.; Gimotty, P.A.; Blome, C.; Fischer, F.; Schmage, P.; Moll, I.; Brandner, J.M. Melanoma progression exhibits a significant impact on connexin expression patterns in the epidermal tumor microenvironment. Histochem. Cell Biol. 2010, 133, 113–124. [Google Scholar] [CrossRef]
- Au, A.; Shao, Q.; White, K.K.; Lucaciu, S.A.; Esseltine, J.L.; Barr, K.; Laird, D.W. Comparative Analysis of Cx31 and Cx43 in Differentiation-Competent Rodent Keratinocytes. Biomolecules 2020, 10, 1443. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmann, D.; Ale-Agha, N.; Reinehr, R.; Steinbrenner, H.; Ramos, M.C.; Sies, H.; Brenneisen, P. Modulation of homologous gap junctional intercellular communication of human dermal fibroblasts via a paracrine factor(s) generated by squamous tumor cells. Carcinogenesis 2003, 24, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Essa, A.A.; Yamazaki, M.; Maruyama, S.; Abé, T.; Babkair, H.; Raghib, A.M.; Megahed, E.M.; Cheng, J.; Saku, T. Tumour-associated macrophages are recruited and differentiated in the neoplastic stroma of oral squamous cell carcinoma. Pathology 2016, 48, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Zoellner, H.; Paknejad, N.; Cornwell, J.A.; Chami, B.; Romin, Y.; Boyko, V.; Fujisawa, S.; Kelly, E.; Lynch, G.W.; Rogers, G.; et al. Potential Hydrodynamic Cytoplasmic Transfer between Mammalian Cells: Cell-Projection Pumping. Biophys. J. 2020, 118, 1248–1260. [Google Scholar] [CrossRef] [PubMed]
- Khawar, I.A.; Park, J.K.; Jung, E.S.; Lee, M.A.; Chang, S.; Kuh, H.-J. Three Dimensional Mixed-Cell Spheroids Mimic Stroma-Mediated Chemoresistance and Invasive Migration in hepatocellular carcinoma. Neoplasia 2018, 20, 800–812. [Google Scholar] [CrossRef]
- Masamune, A.; Suzuki, N.; Kikuta, K.; Ariga, H.; Hayashi, S.; Takikawa, T.; Kume, K.; Hamada, S.; Hirota, M.; Kanno, A.; et al. Connexins regulate cell functions in pancreatic stellate cells. Pancreas 2013, 42, 308–316. [Google Scholar] [CrossRef]
- Avanzo, J.L.; Mesnil, M.; Hernandez-Blazquez, F.J.; Mackowiak, I.I.; Mori, C.M.; da Silva, T.C.; Oloris, S.C.; Gárate, A.P.; Massironi, S.M.; Yamasaki, H.; et al. Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis 2004, 25, 1973–1982. [Google Scholar] [CrossRef]
- De Oliveira, K.D.; Tedardi, M.V.; Cogliati, B.; Dagli, M.L.Z. Higher incidence of lung adenocarcinomas induced by DMBA in connexin 43 heterozygous knockout mice. Biomed. Res. Int. 2013, 2013, 618475. [Google Scholar] [CrossRef]
- Evert, M.; Ott, T.; Temme, A.; Willecke, K.; Dombrowski, F. Morphology and morphometric investigation of hepatocellular preneoplastic lesions and neoplasms in connexin32-deficient mice. Carcinogenesis 2002, 23, 697–703. [Google Scholar] [CrossRef]
- King, T.J.; Lampe, P.D. Mice deficient for the gap junction protein Connexin32 exhibit increased radiation-induced tumorigenesis associated with elevated mitogen-activated protein kinase (p44/Erk1, p42/Erk2) activation. Carcinogenesis 2004, 25, 669–680. [Google Scholar] [CrossRef]
- Naiki-Ito, A.; Kato, H.; Naiki, T.; Yeewa, R.; Aoyama, Y.; Nagayasu, Y.; Suzuki, S.; Inaguma, S.; Takahashi, S. A novel model of non-alcoholic steatohepatitis with fibrosis and carcinogenesis in connexin 32 dominant-negative transgenic rats. Arch. Toxicol. 2020, 94, 4085–4097. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, D.; Hu, S.; Huang, L.; Sun, H.; Yang, S.; Wu, B.; Ji, F.; Zhou, D. Aging-dependent decrease in the numbers of enteric neurons, interstitial cells of Cajal and expression of connexin43 in various regions of gastrointestinal tract. Aging 2018, 10, 3851–3865. [Google Scholar] [CrossRef] [PubMed]
- Yassine, F.; Fostok, S.F.; Al Deen, N.N.; Talhouk, R.S. Endotoxin Triggers Tumor Initiation Events in Nontumorigenic Breast Epithelial Cells and Enhances Invasion-Related Phenotype in Pretumorigenic and Tumorigenic Breast Epithelial Cells. Int. J. Inflam 2021, 2021, 4666380. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deguchi, Y.; Tian, R.; Wei, D.; Wu, L.; Chen, W.; Xu, W.; Xu, M.; Liu, F.; Gao, S.; et al. Pleiotropic Effects of PPARD Accelerate Colorectal Tumorigenesis, Progression, and Invasion. Cancer Res. 2019, 79, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Y.; Cao, D.; Liu, X.; Zhang, L.; Cao, K.; Hu, T.; Qi, Y.; Xu, C. Effects of Helicobacter pylori on the expression levels of GATA-3 and connexin 32 and the GJIC function in gastric epithelial cells and their association by promoter analysis. Oncol. Lett. 2018, 16, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, C.X.; Gong, R.J.; Chi, J.S.; Liu, P.; Liu, X.M. How does Helicobacter pylori cause gastric cancer through connexins: An opinion review. World J. Gastroenterol. 2019, 25, 5220–5232. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Dong, L.; MacDonald, A.I.; Akbari, S.; Edward, M.; Hodgins, M.B.; Johnstone, S.R.; Graham, S.V. HPV16 E6 Controls the Gap Junction Protein Cx43 in Cervical Tumour Cells. Viruses 2015, 7, 5243–5256. [Google Scholar] [CrossRef] [PubMed]
- Polusani, S.R.; Huang, Y.W.; Huang, G.; Chen, C.W.; Wang, C.M.; Lin, L.L.; Osmulski, P.; Lucio, N.D.; Liu, L.; Hsu, Y.T.; et al. Adipokines Deregulate Cellular Communication via Epigenetic Repression of Gap Junction Loci in Obese Endometrial Cancer. Cancer Res. 2019, 79, 196–208. [Google Scholar] [CrossRef]
- Kim, E.M.; Bae, Y.M.; Choi, M.H.; Hong, S.T. Connexin 43 plays an important role in the transformation of cholangiocytes with Clonochis sinensis excretory-secretory protein and N-nitrosodimethylamine. PLoS Negl. Trop. Dis. 2019, 13, e0006843. [Google Scholar] [CrossRef]
- Ruch, R.J.; Trosko, J.E. The role of oval cells and gap junctional intercellular communication in hepatocarcinogenesis. Anticancer Res. 1999, 19, 4831–4838. [Google Scholar]
- Qin, J.; Chang, M.; Wang, S.; Liu, Z.; Zhu, W.; Wang, Y.; Yan, F.; Li, J.; Zhang, B.; Dou, G.; et al. Connexin 32-mediated cell-cell communication is essential for hepatic differentiation from human embryonic stem cells. Sci. Rep. 2016, 6, 37388. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Zhai, C.; Li, H.; Yan, F.; Qin, J.; Yuan, H.; Zhang, R.; Wang, S.; Zhang, W.; Chang, M.; et al. Connexin 32 and connexin 43 are involved in lineage restriction of hepatic progenitor cells to hepatocytes. Stem Cell Res. Ther. 2017, 8, 252. [Google Scholar] [CrossRef]
- Dianati, E.; Poiraud, J.; Weber-Ouellette, A.; Plante, I. Connexins, E-cadherin, Claudin-7 and β-catenin transiently form junctional nexuses during the post-natal mammary gland development. Dev. Biol. 2016, 416, 52–68. [Google Scholar] [CrossRef]
- Adak, A.; Unal, Y.C.; Yucel, S.; Vural, Z.; Turan, F.B.; Yalcin-Ozuysal, O.; Ozcivici, E.; Mese, G. Connexin 32 induces pro-tumorigenic features in MCF10A normal breast cells and MDA-MB-231 metastatic breast cancer cells. Biochim. Et Biophys. Acta. Mol. Cell Res. 2020, 1867, 118851. [Google Scholar] [CrossRef]
- Banerjee, D.; Gakhar, G.; Madgwick, D.; Hurt, A.; Takemoto, D.; Nguyen, T.A. A novel role of gap junction connexin46 protein to protect breast tumors from hypoxia. Int. J. Cancer 2010, 127, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Fostok, S.; El-Sibai, M.; Bazzoun, D.; Lelièvre, S.; Talhouk, R. Connexin 43 Loss Triggers Cell Cycle Entry and Invasion in Non-Neoplastic Breast Epithelium: A Role for Noncanonical Wnt Signaling. Cancers 2019, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Ravisankar, S.; Vanisree, A.J. Implication of connexin30 on the stemness of glioma: Connexin30 reverses the malignant phenotype of glioma by modulating IGF-1R, CD133 and cMyc. J. Neuro-Oncol. 2017, 135, 473–485. [Google Scholar] [CrossRef]
- Li, H.; Wang, B.; Qi, B.; Jiang, G.; Qin, M.; Yu, M. Connexin32 regulates expansion of liver cancer stem cells via the PI3K/Akt signaling pathway. Oncol. Rep. 2022, 48, 166. [Google Scholar] [CrossRef]
- Zhao, Y.; Lai, Y.; Ge, H.; Guo, Y.; Feng, X.; Song, J.; Wang, Q.; Fan, L.; Peng, Y.; Cao, M.; et al. Non-junctional Cx32 mediates anti-apoptotic and pro-tumor effects via epidermal growth factor receptor in human cervical cancer cells. Cell Death Dis. 2017, 8, e2773. [Google Scholar] [CrossRef]
- Liu, G.; Pang, Y.; Zhang, Y.; Fu, H.; Xiong, W.; Zhang, Y. GJB4 promotes gastric cancer cell proliferation and migration via Wnt/CTNNB1 pathway. OncoTargets Ther. 2019, 12, 6745–6755. [Google Scholar] [CrossRef]
- Bache, M.; Kappler, M.; Said, H.M.; Staab, A.; Vordermark, D. Detection and specific targeting of hypoxic regions within solid tumors: Current preclinical and clinical strategies. Curr. Med. Chem. 2008, 15, 322–338. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.C.; Lebedev, A.; Aten, E.; Madsen, K.; Marciano, L.; Kolb, H.C. The clinical importance of assessing tumor hypoxia: Relationship of tumor hypoxia to prognosis and therapeutic opportunities. Antioxid. Redox Signal. 2014, 21, 1516–1554. [Google Scholar] [CrossRef] [PubMed]
- Orlova, A.G.; Kirillin, M.Y.; Volovetsky, A.B.; Shilyagina, N.Y.; Sergeeva, E.A.; Golubiatnikov, G.Y.; Turchin, I.V. Diffuse optical spectroscopy monitoring of oxygen state and hemoglobin concentration during SKBR-3 tumor model growth. Laser Phys. Lett. 2016, 14, 015601. [Google Scholar] [CrossRef]
- Zibara, K.; Awada, Z.; Dib, L.; El-Saghir, J.; Al-Ghadban, S.; Ibrik, A.; El-Zein, N.; El-Sabban, M. Anti-angiogenesis therapy and gap junction inhibition reduce MDA-MB-231 breast cancer cell invasion and metastasis in vitro and in vivo. Sci. Rep. 2015, 5, 12598. [Google Scholar] [CrossRef]
- Elzarrad, K.; Haroon, A.; Reed, D.; Al-Mehdi, A.B. Early incorporated endothelial cells as origin of metastatic tumor vasculogenesis. Clin. Exp. Metastasis 2009, 26, 589–598. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Uruski, P.; Szubert, S.; Maksin, K.; Moszyński, R.; Szpurek, D.; Woźniak, A.; Sajdak, S.; Tykarski, A.; Książek, K. The Proangiogenic Capabilities of Malignant Ascites Generated by Aggressive Ovarian Tumors. Biomed. Res. Int. 2017, 2017, 2592496. [Google Scholar] [CrossRef]
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef]
- Wang, W.K.; Chen, M.C.; Leong, H.F.; Kuo, Y.L.; Kuo, C.Y.; Lee, C.H. Connexin 43 suppresses tumor angiogenesis by down-regulation of vascular endothelial growth factor via hypoxic-induced factor-1α. Int. J. Mol. Sci. 2014, 16, 439–451. [Google Scholar] [CrossRef]
- Dovmark, T.H.; Hulikova, A.; Niederer, S.A.; Vaughan-Jones, R.D.; Swietach, P. Normoxic cells remotely regulate the acid-base balance of cells at the hypoxic core of connexin-coupled tumor growths. FASEB J. 2018, 32, 83–96. [Google Scholar] [CrossRef]
- Dovmark, T.H.; Saccomano, M.; Hulikova, A.; Alves, F.; Swietach, P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene 2017, 36, 4538–4550. [Google Scholar] [CrossRef]
- Hulikova, A.; Black, N.; Hsia, L.T.; Wilding, J.; Bodmer, W.F.; Swietach, P. Stromal uptake and transmission of acid is a pathway for venting cancer cell-generated acid. Proc. Natl. Acad. Sci. USA 2016, 113, E5344–E5353. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Kucheryavykh, L.Y.; Benedikt, J.; Cubano, L.A.; Skatchkov, S.N.; Bukauskas, F.F.; Kucheryavykh, Y.V. Polyamines preserve connexin 43-mediated gap junctional communication during intracellular hypercalcemia and acidosis. Neuroreport 2017, 28, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Jego, G.; Berthenet, K.; Hammann, A.; Solary, E.; Garrido, C. Gap junction-mediated transfer of miR-145-5p from microvascular endothelial cells to colon cancer cells inhibits angiogenesis. Oncotarget 2016, 7, 28160–28168. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Akiyama, M.; Niki, Y.; Kawatsura, R.; Harada, H.; Nakahama, K.I. Inhibitory effects of miRNAs in astrocytes on C6 glioma progression via connexin 43. Mol. Cell. Biochem. 2021, 476, 2623–2632. [Google Scholar] [CrossRef]
- Aucher, A.; Rudnicka, D.; Davis, D.M. MicroRNAs transfer from human macrophages to hepato-carcinoma cells and inhibit proliferation. J. Immunol. 2013, 191, 6250–6260. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Boucher, J.; Jego, G.; Pernet, N.; Cronier, L.; Hammann, A.; Solary, E.; Garrido, C. Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget 2016, 7, 73925–73934. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef]
- Zong, L.; Zhu, Y.; Liang, R.; Zhao, H.B. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci. Rep. 2016, 6, 19884. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, X.; Guo, Y.; Peng, F.; Zheng, N.; He, B.; Ge, H.; Tao, L.; Wang, Q. Pattern of cell-to-cell transfer of microRNA by gap junction and its effect on the proliferation of glioma cells. Cancer Sci. 2019, 110, 1947–1958. [Google Scholar] [CrossRef]
- Prasad, S.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Srivastava, S.K. Cancer cells stemness: A doorstep to targeted therapy. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2020, 1866, 165424. [Google Scholar] [CrossRef] [PubMed]
- Ruch, R.J. Connexin43 Suppresses Lung Cancer Stem Cells. Cancers 2019, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, Y.; Ma, X.; Wan, Q.; Jiang, Z.; Liu, Y.; Zhang, D.; Liu, X.; Wu, W. Connexin 43 SUMOylation improves gap junction functions between liver cancer stem cells and enhances their sensitivity to HSVtk/GCV. Int. J. Oncol. 2018, 52, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, P.S.; Sinyuk, M.; Turaga, S.M.; Mulkearns-Hubert, E.E.; Hale, J.S.; Rao, V.; Demelash, A.; Saygin, C.; China, A.; Alban, T.J.; et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat. Commun. 2018, 9, 578. [Google Scholar] [CrossRef]
- Mulkearns-Hubert, E.E.; Torre-Healy, L.A.; Silver, D.J.; Eurich, J.T.; Bayik, D.; Serbinowski, E.; Hitomi, M.; Zhou, J.; Przychodzen, B.; Zhang, R.; et al. Development of a Cx46 Targeting Strategy for Cancer Stem Cells. Cell Rep. 2019, 27, 1062–1072.e1065. [Google Scholar] [CrossRef]
- Todorova, M.G.; Soria, B.; Quesada, I. Gap junctional intercellular communication is required to maintain embryonic stem cells in a non-differentiated and proliferative state. J. Cell. Physiol. 2008, 214, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Dave, M.A.; Bliss, S.A.; Giec-Ujda, A.B.; Bryan, M.; Pliner, L.F.; Rameshwar, P. T(reg)/Th17 polarization by distinct subsets of breast cancer cells is dictated by the interaction with mesenchymal stem cells. J. Cancer Stem Cell Res. 2014, 2014, e1003. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. AS602801, an Anti-Cancer Stem Cell Drug Candidate, Suppresses Gap-junction Communication Between Lung Cancer Stem Cells and Astrocytes. Anticancer Res. 2018, 38, 5093–5099. [Google Scholar] [CrossRef]
- Trosko, J.E. Cancer Prevention and Therapy of Two Types of Gap Junctional Intercellular Communication-Deficient “Cancer Stem Cell”. Cancers 2019, 11, 87. [Google Scholar] [CrossRef]
- Trosko, J.E. On the potential origin and characteristics of cancer stem cells. Carcinogenesis 2021, 42, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E. In Search of a Unifying Concept in Human Diseases. Diseases 2021, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.K.; Simek, J.; Laird, D.W. Insights into the role of connexins in mammary gland morphogenesis and function. Reprod 2015, 149, R279–R290. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhang, C.; Li, L.; Dong, S.; Zhang, N.; Tong, X. Cx43 reverses the resistance of A549 lung adenocarcinoma cells to cisplatin by inhibiting EMT. Oncol. Rep. 2014, 31, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.P.; Zhou, Y.; Hou, L.X.; Zhu, X.X.; Yi, W.; Yang, S.M.; Lin, T.Y.; Huang, J.L.; Zhang, B.; Yin, X.X. Cx43 deficiency confers EMT-mediated tamoxifen resistance to breast cancer via c-Src/PI3K/Akt pathway. Int. J. Biol. Sci. 2021, 17, 2380–2398. [Google Scholar] [CrossRef]
- Wu, L.; Wang, Z.; He, X.; Jiang, Y.; Pan, R.; Chen, S.; Chen, Y.; Han, Y.; Yu, H.; Zhang, T. GJA1 reverses arsenic-induced EMT via modulating MAPK/ERK signaling pathway. Toxicol. Appl. Pharmacol. 2022, 450, 116138. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, J.H.; Du, Q.Y.; Zhou, Y.C.; Yao, T.J.; Wu, Q.; Liu, J.; Ou, Y.R. Connexin 32 downregulation is critical for chemoresistance in oxaliplatin-resistant HCC cells associated with EMT. Cancer Manag. Res. 2019, 11, 5133–5146. [Google Scholar] [CrossRef]
- Ugur, D.; Gungul, T.B.; Yucel, S.; Ozcivici, E.; Yalcin-Ozuysal, O.; Mese, G. Connexin 32 overexpression increases proliferation, reduces gap junctional intercellular communication, motility and epithelial-to-mesenchymal transition in Hs578T breast cancer cells. J. Cell Commun. Signal. 2022, 16, 361–376. [Google Scholar] [CrossRef]
- Acuña, R.A.; Varas-Godoy, M.; Herrera-Sepulveda, D.; Retamal, M.A. Connexin46 Expression Enhances Cancer Stem Cell and Epithelial-to-Mesenchymal Transition Characteristics of Human Breast Cancer MCF-7 Cells. Int. J. Mol. Sci. 2021, 22, 12604. [Google Scholar] [CrossRef]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef]
- Kutova, O.M.; Sencha, L.M.; Pospelov, A.D.; Dobrynina, O.E.; Brilkina, A.A.; Cherkasova, E.I.; Balalaeva, I.V. Comparative Analysis of Cell-Cell Contact Abundance in Ovarian Carcinoma Cells Cultured in Two- and Three-Dimensional In Vitro Models. Biology 2020, 9, 446. [Google Scholar] [CrossRef]
- Bai, C.; Yang, M.; Fan, Z.; Li, S.; Gao, T.; Fang, Z. Associations of chemo- and radio-resistant phenotypes with the gap junction, adhesion and extracellular matrix in a three-dimensional culture model of soft sarcoma. J. Exp. Clin. Cancer Res. CR 2015, 34, 58. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Boicea, A.; Barrett, K.L.; James, C.O.; Bagchi, I.C.; Bagchi, M.K.; Nezhat, C.; Sidell, N.; Taylor, R.N. Reduced connexin 43 in eutopic endometrium and cultured endometrial stromal cells from subjects with endometriosis. Mol. Hum. Reprod. 2014, 20, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.; Zaccagnino, A.; Bernt, A.; Szczepanowski, M.; Klapper, W.; Schwab, A.; Kalthoff, H.; Trauzold, A. The A818-6 system as an in-vitro model for studying the role of the transportome in pancreatic cancer. BMC Cancer 2020, 20, 264. [Google Scholar] [CrossRef]
- Neveu, M.J.; Sattler, C.A.; Sattler, G.L.; Hully, J.R.; Hertzberg, E.L.; Paul, D.L.; Nicholson, B.J.; Pitot, H.C. Differences in the expression of connexin genes in rat hepatomas in vivo and in vitro. Mol. Carcinog. 1994, 11, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, E.A.; Vodeneev, V.A.; Deyev, S.M.; Balalaeva, I.V. 3D in vitro models of tumors expressing EGFR family receptors: A potent tool for studying receptor biology and targeted drug development. Drug Discov. Today 2019, 24, 99–111. [Google Scholar] [CrossRef]
- Pospelov, A.D.; Timofeeva, L.B.; Cherkasova, E.I.; Balalaeva, I.V. Comparative Analysis of Two Protocols of Mouse Tissues Decellularization for Application in Experimental Oncology. Opera Med. Et Physiol. 2020, 7, 13–21. [Google Scholar] [CrossRef]
- Dornhof, J.; Kieninger, J.; Muralidharan, H.; Maurer, J.; Urban, G.A.; Weltin, A. Microfluidic organ-on-chip system for multi-analyte monitoring of metabolites in 3D cell cultures. Lab Chip 2021, 22, 225–239. [Google Scholar] [CrossRef]
- Mendoza-Martinez, A.K.; Loessner, D.; Mata, A.; Azevedo, H.S. Modeling the Tumor Microenvironment of Ovarian Cancer: The Application of Self-Assembling Biomaterials. Cancers 2021, 13, 5745. [Google Scholar] [CrossRef]
- Sankarasubramanian, S.; Pfohl, U.; Regenbrecht, C.R.A.; Reinhard, C.; Wedeken, L. Context Matters-Why We Need to Change From a One Size Fits all Approach to Made-to-Measure Therapies for Individual Patients With Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 760705. [Google Scholar] [CrossRef]
- Orlando-Mathur, C.E.; Bechberger, J.F.; Goldberg, G.S.; Naus, C.C.; Kidder, G.M.; Kennedy, T.G. Rat endometrial stromal cells express the gap junction genes connexins 26 and 43 and form functional gap junctions during in vitro decidualization. Biol. Reprod. 1996, 54, 905–913. [Google Scholar] [CrossRef]
- Walsh, J.J.; Parent, M.; Akif, A.; Adam, L.C.; Maritim, S.; Mishra, S.K.; Khan, M.H.; Coman, D.; Hyder, F. Imaging Hallmarks of the Tumor Microenvironment in Glioblastoma Progression. Front. Oncol. 2021, 11, 692650. [Google Scholar] [CrossRef] [PubMed]
- Nabavizadeh, S.A.; Ware, J.B.; Wolf, R.L. Emerging Techniques in Imaging of Glioma Microenvironment. Top Magn Reson Imaging 2020, 29, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhu, Q.; Zeng, Y.; Zeng, Q.; Chen, X.; Zhan, Y. Manganese Oxide Nanoparticles As MRI Contrast Agents In Tumor Multimodal Imaging And Therapy. Int J. Nanomed. 2019, 14, 8321–8344. [Google Scholar] [CrossRef]
- Matusiak, N.; van Waarde, A.; Bischoff, R.; Oltenfreiter, R.; van de Wiele, C.; Dierckx, R.A.; Elsinga, P.H. Probes for non-invasive matrix metalloproteinase-targeted imaging with PET and SPECT. Curr. Pharm. Des. 2013, 19, 4647–4672. [Google Scholar] [CrossRef]
- Löser, R.; Bergmann, R.; Frizler, M.; Mosch, B.; Dombrowski, L.; Kuchar, M.; Steinbach, J.; Gütschow, M.; Pietzsch, J. Synthesis and radiopharmacological characterisation of a fluorine-18-labelled azadipeptide nitrile as a potential PET tracer for in vivo imaging of cysteine cathepsins. ChemMedChem 2013, 8, 1330–1344. [Google Scholar] [CrossRef]
- Haris, M.; Singh, A.; Mohammed, I.; Ittyerah, R.; Nath, K.; Nanga, R.P.; Debrosse, C.; Kogan, F.; Cai, K.; Poptani, H.; et al. In vivo magnetic resonance imaging of tumor protease activity. Sci. Rep. 2014, 4, 6081. [Google Scholar] [CrossRef]
- Abadjian, M.Z.; Edwards, W.B.; Anderson, C.J. Imaging the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 229–257. [Google Scholar] [CrossRef]
- Moody, A.S.; Dayton, P.A.; Zamboni, W.C. Imaging methods to evaluate tumor microenvironment factors affecting nanoparticle drug delivery and antitumor response. Cancer Drug Resist. 2021, 4, 382–413. [Google Scholar] [CrossRef]
- Zhou, Z.; Lu, Z.-R. Molecular imaging of the tumor microenvironment. Adv. Drug Deliv. Rev. 2017, 113, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Ni, C.; Lou, X.; Yao, X.; Wang, L.; Wan, J.; Duan, X.; Liang, J.; Zhang, K.; Yang, Y.; Zhang, L.; et al. ZIP1(+) fibroblasts protect lung cancer against chemotherapy via connexin-43 mediated intercellular Zn(2+) transfer. Nat. Commun. 2022, 13, 5919. [Google Scholar] [CrossRef]
- Xia, Z.A.; Zhou, Y.; Li, J.; He, J. Integrated Analysis of Single-Cell and Bulk RNA-Sequencing Reveals a Tissue-Resident Macrophage-Related Signature for Predicting Immunotherapy Response in Breast Cancer Patients. Cancers 2022, 14, 5506. [Google Scholar] [CrossRef]
- Shen, Y.; Stanislauskas, M.; Li, G.; Zheng, D.; Liu, L. Epigenetic and genetic dissections of UV-induced global gene dysregulation in skin cells through multi-omics analyses. Sci. Rep. 2017, 7, 42646. [Google Scholar] [CrossRef]
- Dong, K.; Gu, D.; Shi, J.; Bao, Y.; Fu, Z.; Fang, Y.; Qu, L.; Zhu, W.; Jiang, A.; Wang, L. Identification and Verification of m7G Modification Patterns and Characterization of Tumor Microenvironment Infiltration via Multi-Omics Analysis in Clear Cell Renal Cell Carcinoma. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Nalewajska, M.; Marchelek-Myśliwiec, M.; Opara-Bajerowicz, M.; Dziedziejko, V.; Pawlik, A. Connexins-Therapeutic Targets in Cancers. Int. J. Mol. Sci. 2020, 21, 9119. [Google Scholar] [CrossRef]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef]
- Chasampalioti, M.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; Jackson, A.M.; Spendlove, I.; Ramage, J.M. Connexin 43 is an independent predictor of patient outcome in breast cancer patients. Breast Cancer Res. Treat. 2019, 174, 93–102. [Google Scholar] [CrossRef]
- Lin, Y.P.; Wu, J.I.; Tseng, C.W.; Chen, H.J.; Wang, L.H. Gjb4 serves as a novel biomarker for lung cancer and promotes metastasis and chemoresistance via Src activation. Oncogene 2019, 38, 822–837. [Google Scholar] [CrossRef]
- Busby, M.; Hallett, M.T.; Plante, I. The Complex Subtype-Dependent Role of Connexin 43 (GJA1) in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 693. [Google Scholar] [CrossRef]
- Segura, I.G.; Secchi, D.G.; Galíndez, M.F.; Carrica, A.; Bologna-Molina, R.; Brunotto, M.; Centeno, V.A. Connexin 43, Bcl-2, Bax, Ki67, and E-cadherin patterns in oral squamous cell carcinoma and its relationship with GJA1 rs12197797 C/G. Med. Oral Patol. Oral Y Cir. Bucal 2022, 27, e366–e374. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutova, O.M.; Pospelov, A.D.; Balalaeva, I.V. The Multifaceted Role of Connexins in Tumor Microenvironment Initiation and Maintenance. Biology 2023, 12, 204. https://doi.org/10.3390/biology12020204
Kutova OM, Pospelov AD, Balalaeva IV. The Multifaceted Role of Connexins in Tumor Microenvironment Initiation and Maintenance. Biology. 2023; 12(2):204. https://doi.org/10.3390/biology12020204
Chicago/Turabian StyleKutova, Olga M., Anton D. Pospelov, and Irina V. Balalaeva. 2023. "The Multifaceted Role of Connexins in Tumor Microenvironment Initiation and Maintenance" Biology 12, no. 2: 204. https://doi.org/10.3390/biology12020204
APA StyleKutova, O. M., Pospelov, A. D., & Balalaeva, I. V. (2023). The Multifaceted Role of Connexins in Tumor Microenvironment Initiation and Maintenance. Biology, 12(2), 204. https://doi.org/10.3390/biology12020204