
Molecular Mechanisms and Clinical Phenotypes of GJB2 Missense Variants
 ,
,
Simple Summary
Abstract
1. Introduction
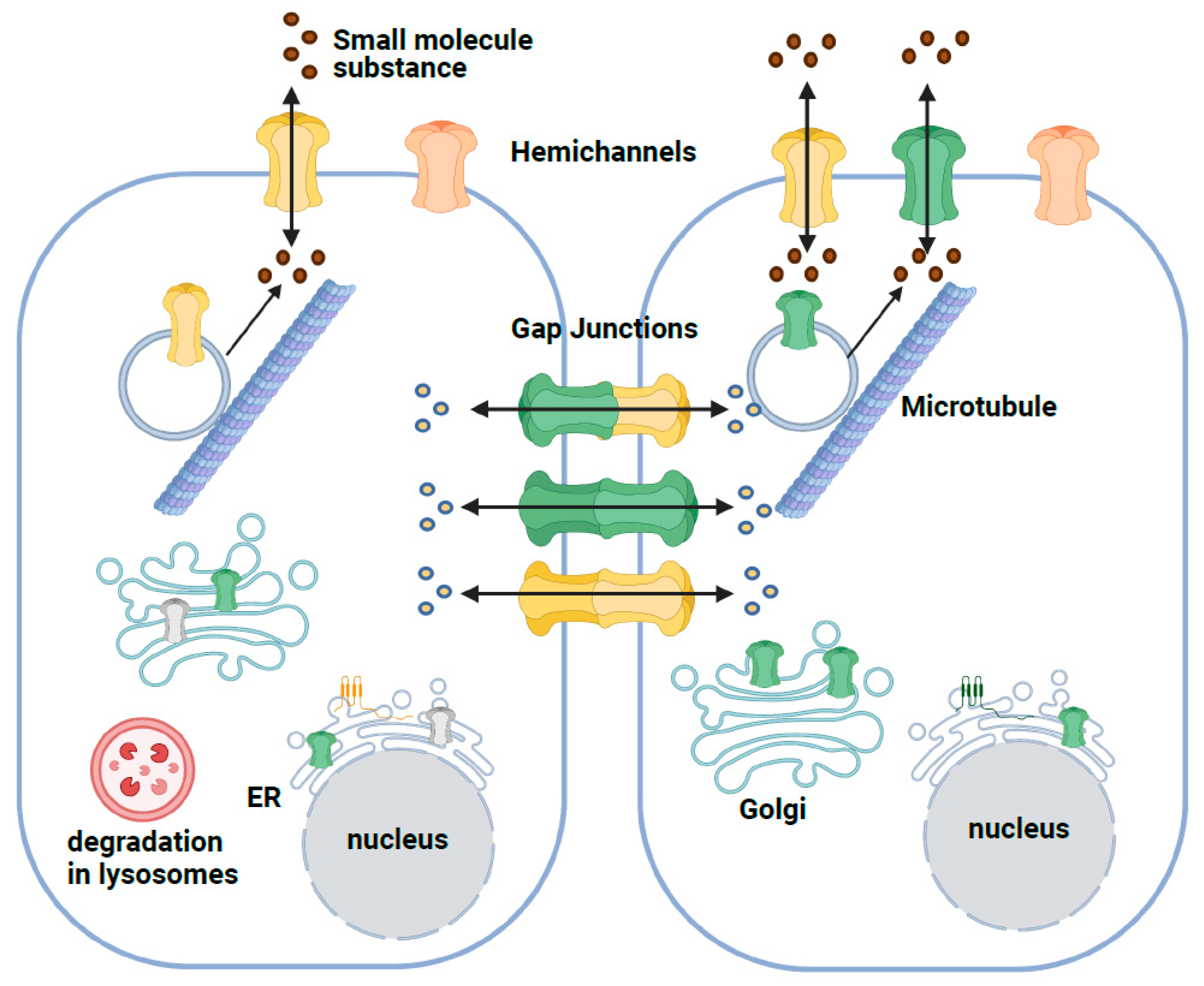
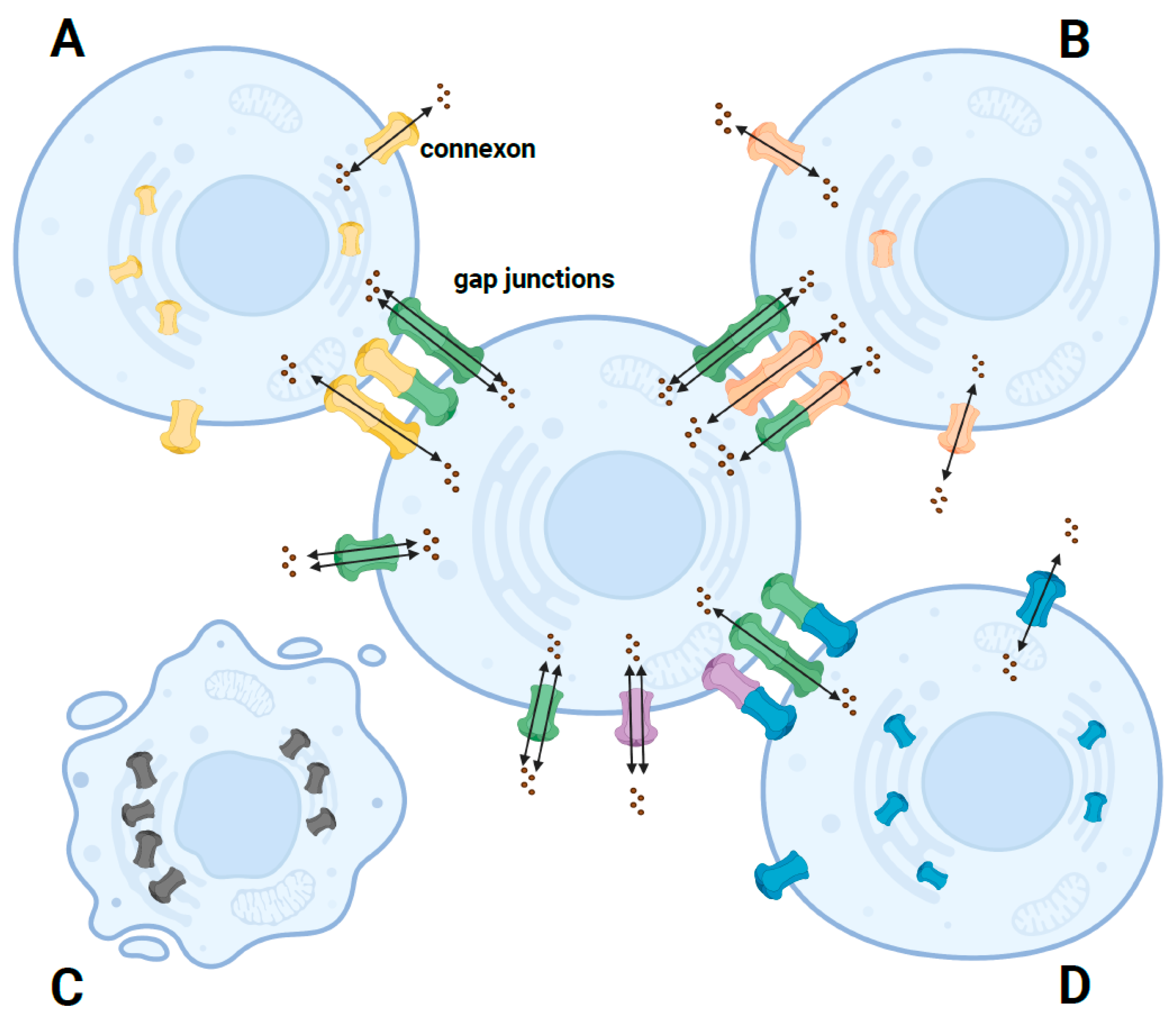
2. Structure and Function of the GJB2 Gene
3. Effect of GJB2 Missense Variants on the Cx26 Protein
4. Association between GJB2 Missense Variants with Clinical Phenotypes
4.1. Nonsyndromic HL Caused by GJB2 Missense Variants
4.1.1. Nonsyndromic HL Caused by GJB2 Recessive Missense Variants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variants | Locations | Inheritance Patterns | Trafficking | Hemichannels | Gap Junction Channel | Clinical Phenotype | References |
|---|---|---|---|---|---|---|---|
| M1V | NT | AR | defect | / | / | severe/profound | [35,36] |
| T8M | NT | AR | normal | / | impaired | severe/profound | [37,38,39] |
| L10P | NT | AR | normal | impaired | impaired | profound | [39,40] |
| G12V | NT | AR | defect | / | / | mild/moderate | [33,41] |
| N14D | NT | AR | normal | impaired | impaired | moderate | [42] |
| S19T | NT | AR | impaired | / | defect | profound | [33] |
| R32C | TM1 | AR | defect | / | / | / | [43] |
| R32H | TM1 | AR | defect | / | / | severe/profound | [44,45] |
| I33T | TM1 | AR | normal | / | defect | severe/profound | [46] |
| M34T | TM1 | AD | normal | impaired | impaired | mild/moderate/severe | [35,41] |
| I35S | TM1 | AR | defect | / | / | severe/profound | [46] |
| V37I | TM1 | AR | normal | defect | impaired | mild | [41,47,48] |
| A40G | TM1 | AR | normal | defect | defect | profound | [48,49] |
| W44C | EC1 | AD | normal | / | Impaired | severe/profound | [50,51] |
| W44S | EC1 | AD | impaired | impaired | impaired | / | [52,53] |
| G45R | EC1 | AD | normal | impaired | impaired | mild | [54] |
| D46E | EC1 | AD | normal | impaired | defect | moderate | [34] |
| E47K | EC1 | AR | normal | impaired | defect | / | [55,56] |
| E47Q | EC1 | AR | normal | impaired | defect | / | [56] |
| T55N | EC1 | AD(YES) | defect | / | / | severe/profound | [57] |
| G59V | EC1 | AD | normal | / | defect | profound | [58,59] |
| I71N | EC1 | AR | impaired | impaired | / | severe/profound | [60] |
| W77R | TM2 | AR | defect | / | / | severe/profound | [38,50,61] |
| I82M | TM2 | AR | normal | defect | / | profound | [38,59] |
| V84L | TM2 | AR | normal | normal | impaired | profound | [41,62,63] |
| S85Y | TM2 | AR | defect | / | / | / | [56] |
| T86R | TM2 | AR | defect | / | / | profound | [34] |
| A88S | TM2 | AR | normal | normal | impaired | profound | [63,64] |
| L90P | TM2 | AR | impaired | impaired | / | severe/profound | [33,35,38] |
| V95M | TM2 | AR | normal | normal | impaired | profound | [49,62,63] |
| H100L | CL | AR | normal | impaired | impaired | profound | [49,56] |
| H100Y | CL | AR | normal | impaired | impaired | / | [56] |
| G109V | CL | AR | normal | impaired | defect | profound | [39,65] |
| S113R | CL | AR | / | / | defect | [47] | |
| R127H | CL | AR | normal | impaired | impaired | severe/profound | [33,45,56] |
| R127L | CL | AR | normal | impaired | impaired | / | [56] |
| R143Q | TM3 | AD | normal | / | defect | profound | [53,66] |
| R143W | TM3 | AR | / | / | defect | profound | [37,41] |
| V153I | TM3 | AR | / | / | defect | severe/profound | [37,38] |
| F161S | EC2 | AR | impaired | / | impaired | / | [35] |
| M163L | EC2 | AD(YES) | defect | / | / | mild/moderate | [67] |
| M163V | EC2 | AD | normal | / | impaired | severe/profound | [24,38] |
| W172C | EC2 | AR | impaired | impaired | / | profound | [20,68] |
| W172R | EC2 | AR | normal | / | impaired | / | [46] |
| P173R | EC2 | AR | defect | / | / | severe/profound | [35,69] |
| D179N | EC2 | AR | normal | / | impaired | severe | [53,70] |
| R184Q | EC2 | AD | normal | / | defect | profound | [51,53] |
| R184P | EC2 | AR | defect | / | / | severe/profound | [35,44,49] |
| K188R | EC2 | AR | defect | / | / | / | [71] |
| M195V | TM4 | AD(YES) | defect | / | / | severe/profound | [56,72] |
| M195T | TM4 | AR | normal | / | impaired | [71] | |
| M195L | TM4 | AR | normal | impaired | impaired | / | [56] |
| A197S | TM4 | AD | normal | / | impaired | profound | [71,73] |
| S199F | TM4 | AR | defect | / | / | severe/profound | [71,74] |
| G200R | TM4 | AR | defect | / | / | severe | [71,75] |
| C202F | TM4 | AD | normal | impaired | impaired | mild/moderate | [71,76] |
| I203T | TM4 | AR | normal | impaired | impaired | profound | [71,77] |
| I203K | TM4 | AR | defect | / | / | profound | [71,73] |
| L205V | TM4 | AR | normal | impaired | impaired | profound | [71,78] |
| L205P | TM4 | AR | defect | / | / | moderate/severe/profound | [71,79] |
| N206S | TM4 | AR | normal | impaired | impaired | mild/moderate/severe | [71,77] |
| T208P | TM4 | AR | defect | / | / | / | [71] |
| L214P | TM4 | AR | / | / | defect | profound | [37,73] |
4.1.2. Nonsyndromic HL Caused by GJB2 Dominant Variants
4.1.3. Relationship between Variants Causing Nonsyndromic HL and Clinical Phenotype
4.2. Syndromic HL Caused by GJB2 Missense Variants
4.2.1. Syndromic HL Caused by GJB2 Dominant Variants
4.2.2. Relationship between Pathogenic Variants Causing Syndromic HL and Clinical Phenotypes
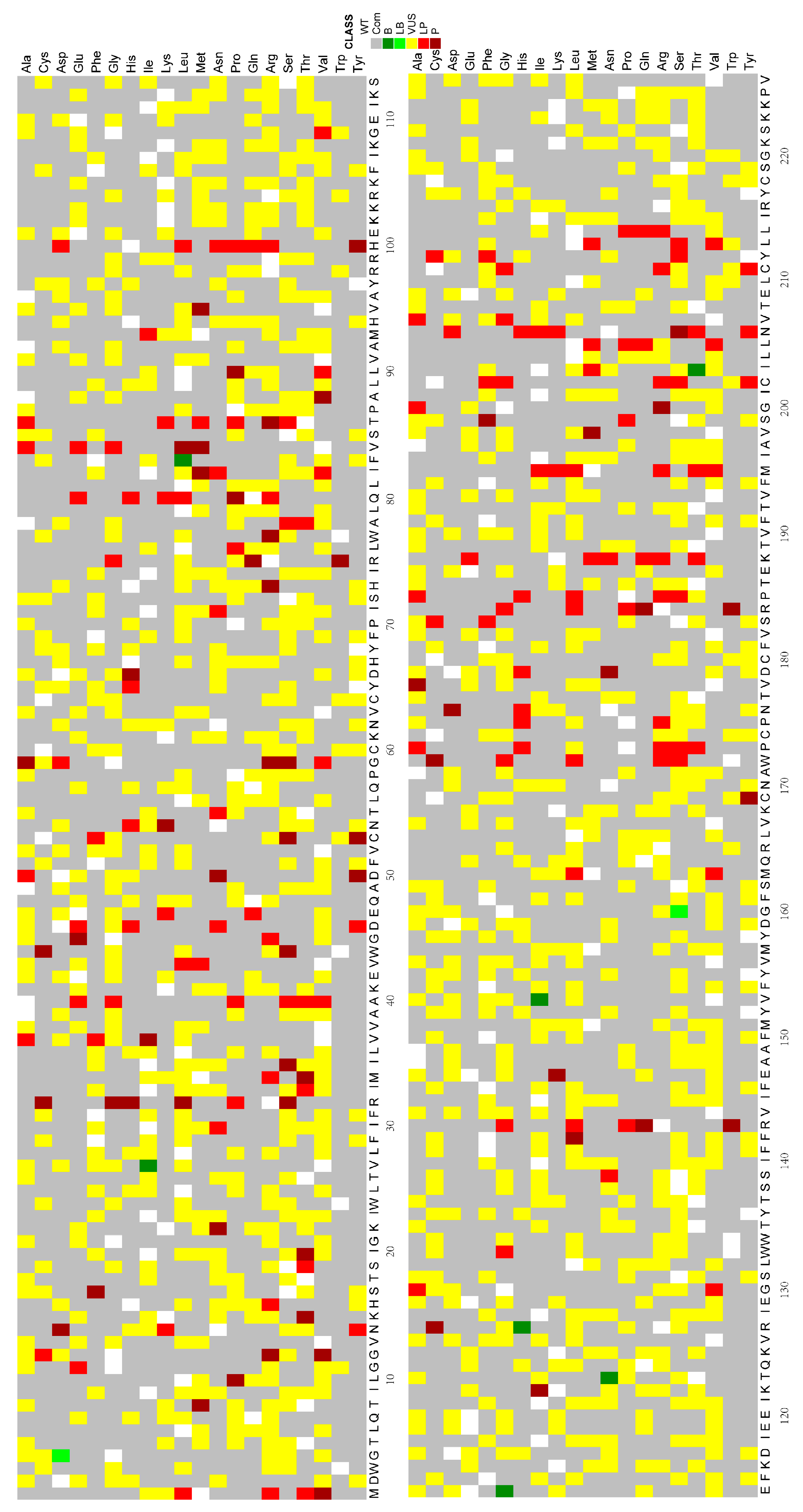
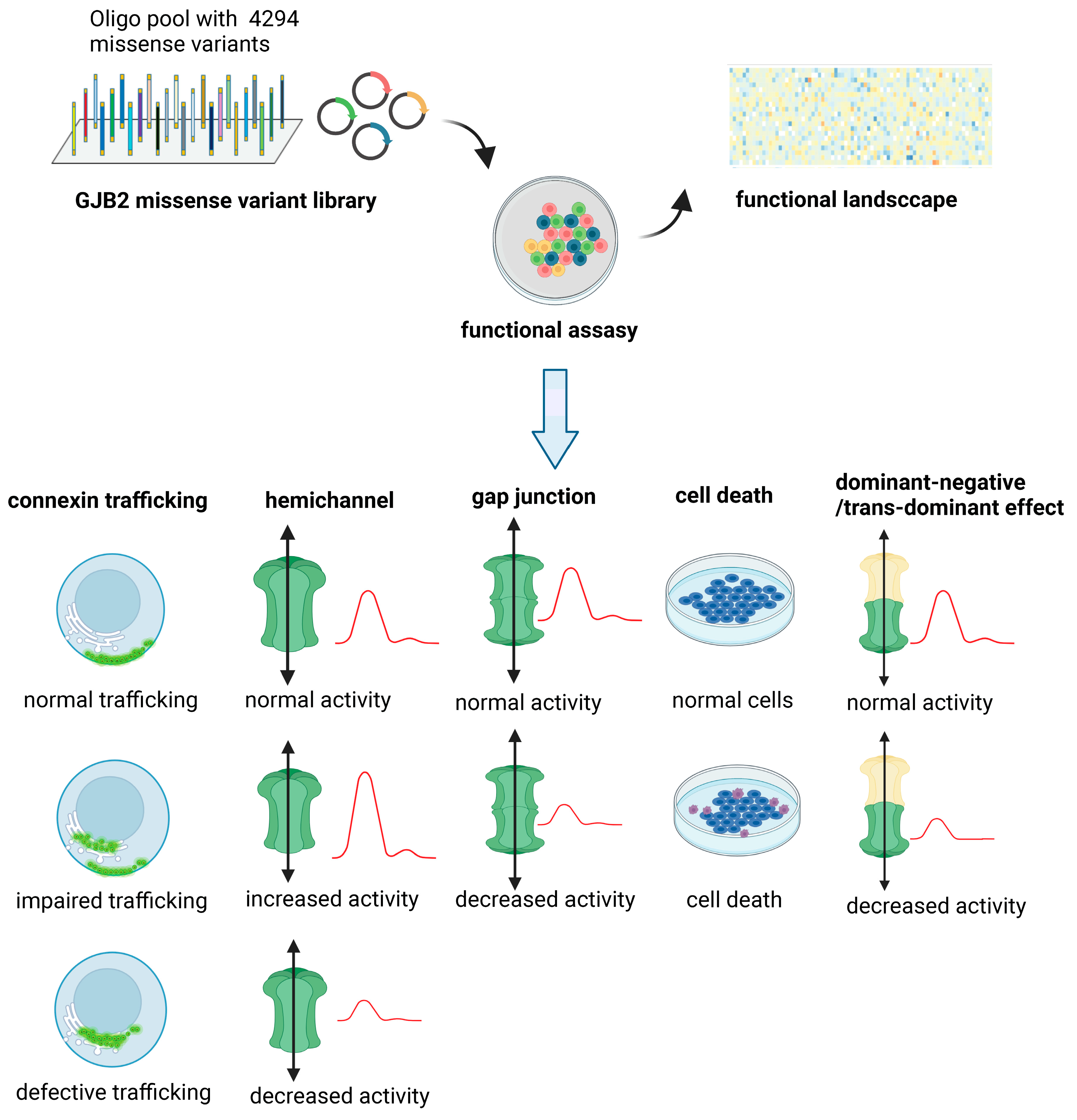
5. The Gap between Possible and Functionally Studied Missense Variants of the GJB2 Gene
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Deafness and Hearing Loss. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 31 December 2021).
- Wang, Q.; Xiang, J.; Sun, J.; Yang, Y.; Guan, J.; Wang, D.; Song, C.; Guo, L.; Wang, H.; Chen, Y.; et al. Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet. Med. 2019, 21, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Huang, L.H.; Wang, G.J.; Gao, X.; Qu, C.Y.; Chen, X.W.; Ma, F.R.; Zhang, J.; Xing, W.L.; Xi, S.Y.; et al. Concurrent Hearing and Genetic Screening of 180,469 Neonates with Follow-up in Beijing, China. Am. J. Hum. Genet. 2019, 105, 803–812. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Current concepts: Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Michalski, N.; Petit, C. Genes Involved in the Development and Physiology of Both the Periphera l and Central Auditory Systems. Annu. Rev. Neurosci. 2019, 42, 67–86. [Google Scholar] [CrossRef]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.D.; Acuña, R.; Figueroa, V.; Maripillan, J.; Nicholson, B. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox Signal. 2009, 11, 309–322. [Google Scholar] [CrossRef]
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kamasawa, N.; Ciolofan, C.; Olson, C.O.; Lu, S.; Davidson, K.G.; Yasumura, T.; Shigemoto, R.; Rash, J.E.; Nagy, J.I. Connexin45-containing neuronal gap junctions in rodent retina also contain connexin36 in both apposing hemiplaques, forming bihomotypic gap junctions, with scaffolding contributed by zonula occludens-1. J. Neurosci. 2008, 28, 9769–9789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koval, M.; Molina, S.A.; Burt, J.M. Mix and match: Investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 2014, 588, 1193–1204. [Google Scholar] [CrossRef]
- Wei, C.J.; Xu, X.; Lo, C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 811–838. [Google Scholar] [CrossRef] [PubMed]
- Van Campenhout, R.; Gomes, A.R.; De Groof, T.W.M.; Muyldermans, S.; Devoogdt, N.; Vinken, M. Mechanisms Underlying Connexin Hemichannel Activation in Disease. Int. J. Mol. Sci. 2021, 22, 3503. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.D.; Jung, D.; Bützler, C.; Temme, A.; Traub, O.; Winterhager, E.; Willecke, K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 1998, 140, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Fu, X.; Li, Y.; Hong, G.; Li, P.; Lin, J.; Xun, Y.; Fang, L.; Weng, W.; Yue, R.; et al. Deletion of Kcnj16 in Mice Does Not Alter Auditory Function. Front. Cell Dev. Biol. 2021, 9, 630361. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Chen, S.; Xu, K.; Cao, H.Y.; Du, A.N.; Bai, X.; Sun, Y.; Kong, W.J. Reduced postnatal expression of cochlear Connexin26 induces hearing loss and affects the developmental status of pillar cells in a dose-dependent manner. Neurochem. Int. 2019, 128, 196–205. [Google Scholar] [CrossRef]
- Chen, P.; Wu, W.; Zhang, J.; Chen, J.; Li, Y.; Sun, L.; Hou, S.; Yang, J. Pathological mechanisms of connexin26-related hearing loss: Potassium recycling, ATP-calcium signaling, or energy supply? Front. Mol. Neurosci. 2022, 15, 976388. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Maslova, E.A.; Orishchenko, K.E.; Posukh, O.L. Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss. Biomolecules 2021, 11, 61. [Google Scholar] [CrossRef]
- Laird, D.W.; Naus, C.C.; Lampe, P.D. SnapShot: Connexins and Disease. Cell 2017, 170, 1260–1260. [Google Scholar] [CrossRef] [PubMed]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- Wingard, J.C.; Zhao, H.B. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss—A Common Hereditary Deafness. Front. Cell Neurosci. 2015, 9, 202. [Google Scholar] [CrossRef]
- Press, E.R.; Shao, Q.; Kelly, J.J.; Chin, K.; Alaga, A.; Laird, D.W. Induction of cell death and gain-of-function properties of connexin26 mutants predict severity of skin disorders and hearing loss. J. Biol. Chem. 2017, 292, 9721–9732. [Google Scholar] [CrossRef] [PubMed]
- Iossa, S.; Marciano, E.; Franze, A. GJB2 Gene Mutations in Syndromic Skin Diseases with Sensorineural Hearing Loss. Curr. Genom. 2011, 12, 475–785. [Google Scholar] [CrossRef] [PubMed]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Heathcote, K.; Syrris, P.; Carter, N.D.; Patton, M.A. A connexin 26 mutation causes a syndrome of sensorineural hearing loss and palmoplantar hyperkeratosis (MIM 148350). J. Med. Genet. 2000, 37, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Maestrini, E.; Korge, B.P.; Ocana-Sierra, J.; Calzolari, E.; Cambiaghi, S.; Scudder, P.M.; Hovnanian, A.; Monaco, A.P.; Munro, C.S. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families. Hum. Mol. Genet. 1999, 8, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Rouan, F.; Willoughby, C.E.; Brown, N.; Chung, P.; Ryynänen, M.; Jabs, E.W.; Bale, S.J.; DiGiovanna, J.J.; Uitto, J.; et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 2002, 70, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- van Geel, M.; van Steensel, M.A.; Küster, W.; Hennies, H.C.; Happle, R.; Steijlen, P.M.; König, A. HID and KID syndromes are associated with the same connexin 26 mutation. Br. J. Derm. 2002, 146, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Brown, N.; Ishida-Yamamoto, A.; Krol, A. Expanding the phenotypic spectrum of Cx26 disorders: Bart-Pumphrey syndrome is caused by a novel missense mutation in GJB2. J. Investig. Derm. 2004, 123, 856–863. [Google Scholar] [CrossRef]
- Avshalumova, L.; Fabrikant, J.; Koriakos, A. Overview of skin diseases linked to connexin gene mutations. Int J. Derm. 2014, 53, 192–205. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, P.; Veronesi, V.; Bicego, M.; Melchionda, S.; Zelante, L.; Di Iorio, E.; Bruzzone, R.; Gasparini, P. Hearing loss: Frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 2002, 296, 685–691. [Google Scholar] [CrossRef]
- Choi, S.Y.; Park, H.J.; Lee, K.Y.; Dinh, E.H.; Chang, Q.; Ahmad, S.; Lee, S.H.; Bok, J.; Lin, X.; Kim, U.K. Different functional consequences of two missense mutations in the GJB2 gene associated with non-syndromic hearing loss. Hum. Mutat. 2009, 30, E716–E727. [Google Scholar] [CrossRef] [PubMed]
- Thönnissen, E.; Rabionet, R.; Arbonès, M.L.; Estivill, X.; Willecke, K.; Ott, T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 2002, 111, 190–197. [Google Scholar] [CrossRef]
- Bajaj, Y.; Sirimanna, T.; Albert, D.M.; Qadir, P.; Jenkins, L.; Bitner-Glindzicz, M. Spectrum of GJB2 mutations causing deafness in the British Bangladeshi population. Clin. Otolaryngol. 2008, 33, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Meşe, G.; Londin, E.; Mui, R.; Brink, P.R.; White, T.W. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum. Genet. 2004, 115, 191–199. [Google Scholar] [CrossRef]
- Dalamon, V.; Beheran, A.; Diamante, F.; Pallares, N.; Diamante, V.; Elgoyhen, A.B. Prevalence of GJB2 mutations and the del(GJB6-D13S1830) in Argentinean non-syndromic deaf patients. Hear. Res. 2005, 207, 43–49. [Google Scholar] [CrossRef]
- Dalamon, V.; Fiori, M.C.; Figueroa, V.A.; Oliva, C.A.; Del Rio, R.; Gonzalez, W.; Canan, J.; Elgoyhen, A.B.; Altenberg, G.A.; Retamal, M.A. Gap-junctional channel and hemichannel activity of two recently identified connexin 26 mutants associated with deafness. Pflug. Arch. 2016, 468, 909–918. [Google Scholar] [CrossRef]
- Dalamón, V.; Lotersztein, V.; Béhèran, A.; Lipovsek, M.; Diamante, F.; Pallares, N.; Francipane, L.; Frechtel, G.; Paoli, B.; Mansilla, E.; et al. GJB2 and GJB6 genes: Molecular study and identification of novel GJB2 mutations in the hearing-impaired Argentinean population. Audiol. Neurootol. 2010, 15, 194–202. [Google Scholar] [CrossRef]
- Kenna, M.A.; Wu, B.L.; Cotanche, D.A.; Korf, B.R.; Rehm, H.L. Connexin 26 studies in patients with sensorineural hearing loss. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 1037–1042. [Google Scholar] [CrossRef][Green Version]
- Haack, B.; Schmalisch, K.; Palmada, M.; Bohmer, C.; Kohlschmidt, N.; Keilmann, A.; Zechner, U.; Limberger, A.; Beckert, S.; Zenner, H.P.; et al. Deficient membrane integration of the novel p.N14D-GJB2 mutant associated with non-syndromic hearing impairment. Hum. Mutat. 2006, 27, 1158–1159. [Google Scholar] [CrossRef]
- Li, J.-Z.; Hu, Y.-Q.; Wang, S.-H.; Cheng, H.-S.; Pan, Q.; Xia, K.; Hu, Z.-M.; Feng, Y. Mutations of Cx26 gene in patients with NSHL and intracellular distrib ution of two mutants. Yi Chuan = Hereditas 2009, 31, 705–712. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beach, R.; Abitbol, J.M.; Allman, B.L.; Esseltine, J.L.; Shao, Q.; Laird, D.W. GJB2 Mutations Linked to Hearing Loss Exhibit Differential Trafficking and Functional Defects as Revealed in Cochlear-Relevant Cells. Front. Cell Dev. Biol. 2020, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Mahdieh, N.; Mahmoudi, H.; Ahmadzadeh, S.; Bakhtiyari, S. GJB2 mutations in deaf population of Ilam (Western Iran): A different pattern of mutation distribution. Eur. Arch. Otorhinolaryngol. 2016, 273, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.S.; Ganapathy, A.; Jalvi, R.; Srikumari Srisailapathy, C.R.; Malhotra, V.; Chadha, S.; Agarwal, A.; Ramesh, A.; Rangasayee, R.R.; Anand, A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 2009, 17, 502–509. [Google Scholar] [CrossRef]
- Bruzzone, R.; Veronesi, V.; Gomès, D.; Bicego, M.; Duval, N.; Marlin, S.; Petit, C.; D’Andrea, P.; White, T.W. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003, 533, 79–88. [Google Scholar] [CrossRef]
- Jara, O.; Acuña, R.; García, I.E.; Maripillán, J.; Figueroa, V.; Sáez, J.C.; Araya-Secchi, R.; Lagos, C.F.; Pérez-Acle, T.; Berthoud, V.M.; et al. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol. Biol. Cell 2012, 23, 3299–3311. [Google Scholar] [CrossRef]
- Primignani, P.; Trotta, L.; Castorina, P.; Lalatta, F.; Sironi, F.; Radaelli, C.; Degiorgio, D.; Curcio, C.; Travi, M.; Ambrosetti, U.; et al. Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: Frequencies, novel mutations, genotypes, and degree of hearing loss. Genet. Test. Mol. Biomark. 2009, 13, 209–217. [Google Scholar] [CrossRef]
- Martin, P.E.; Coleman, S.L.; Casalotti, S.O.; Forge, A.; Evans, W.H. Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness. Hum. Mol. Genet. 1999, 8, 2369–2376. [Google Scholar] [CrossRef]
- Welch, K.O.; Marin, R.S.; Pandya, A.; Arnos, K.S. Compound heterozygosity for dominant and recessive GJB2 mutations: Effect on phenotype and review of the literature. Am. J. Med. Genet. A 2007, 143, 1567–1573. [Google Scholar] [CrossRef]
- Marziano, N.K.; Casalotti, S.O.; Portelli, A.E.; Becker, D.L.; Forge, A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 2003, 12, 805–812. [Google Scholar] [CrossRef]
- Yum, S.W.; Zhang, J.; Scherer, S.S. Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30. Neurobiol. Dis. 2010, 38, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paris, J.; Waldhaus, J.; Gordhandas, J.A.; Pique, L.; Schrijver, I. Comparative functional characterization of novel non-syndromic GJB2 gene variant p.Gly45Arg and lethal syndromic variant p.Gly45Glu. PeerJ 2016, 4, e2494. [Google Scholar] [CrossRef] [PubMed]
- Stong, B.C.; Chang, Q.; Ahmad, S.; Lin, X. A novel mechanism for connexin 26 mutation linked deafness: Cell death caused by leaky gap junction hemichannels. Laryngoscope 2006, 116, 2205–2210. [Google Scholar] [CrossRef]
- Kim, H.R.; Oh, S.K.; Lee, E.S.; Choi, S.Y.; Roh, S.E.; Kim, S.J.; Tsukihara, T.; Lee, K.Y.; Jeon, C.J.; Kim, U.K. The pathological effects of connexin 26 variants related to hearing loss by in silico and in vitro analysis. Hum. Genet. 2016, 135, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, S.; Bicego, M.; Marciano, E.; Franze, A.; Morgutti, M.; Bortone, G.; Zelante, L.; Carella, M.; D’Andrea, P. Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 2005, 337, 799–805. [Google Scholar] [CrossRef]
- Toth, T.; Kupka, S.; Haack, B.; Riemann, K.; Braun, S.; Fazakas, F.; Zenner, H.P.; Muszbek, L.; Blin, N.; Pfister, M.; et al. GJB2 mutations in patients with non-syndromic hearing loss from Northeastern Hungary. Hum. Mutat. 2004, 23, 631–632. [Google Scholar] [CrossRef]
- Palmada, M.; Schmalisch, K.; Böhmer, C.; Schug, N.; Pfister, M.; Lang, F.; Blin, N. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol. Dis. 2006, 22, 112–118. [Google Scholar] [CrossRef]
- Mohamed, M.R.; Alesutan, I.; Föller, M.; Sopjani, M.; Bress, A.; Baur, M.; Salama, R.H.; Bakr, M.S.; Mohamed, M.A.; Blin, N.; et al. Functional analysis of a novel I71N mutation in the GJB2 gene among Southern Egyptians causing autosomal recessive hearing loss. Cell Physiol. Biochem. 2010, 26, 959–966. [Google Scholar] [CrossRef]
- Richard, G.; White, T.W.; Smith, L.E.; Bailey, R.A.; Compton, J.G.; Paul, D.L.; Bale, S.J. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 1998, 103, 393–399. [Google Scholar] [CrossRef]
- Wang, H.L.; Chang, W.T.; Li, A.H.; Yeh, T.H.; Wu, C.Y.; Chen, M.S.; Huang, P.C. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J. Neurochem. 2003, 84, 735–742. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, W.; Ahmad, S.; Sipp, J.A.; Chen, P.; Lin, X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc. Natl. Acad. Sci. USA 2005, 102, 15201–15206. [Google Scholar] [CrossRef] [PubMed]
- Koenighofer, M.; Lucas, T.; Parzefall, T.; Ramsebner, R.; Schoefer, C.; Frei, K. The promoter mutation c.-259C>T (-3438C>T) is not a common cause of non-syndromic hearing impairment in Austria. Eur. Arch. Otorhinolaryngol. 2015, 272, 229–232. [Google Scholar] [CrossRef]
- Dalamon, V.; Lotersztein, V.; Lipovsek, M.; Beheran, A.; Mondino, M.E.; Diamante, F.; Pallares, N.; Diamante, V.; Elgoyhen, A.B. Performance of speech perception after cochlear implantation in DFNB1 patients. Acta Otolaryngol. 2009, 129, 395–398. [Google Scholar] [CrossRef]
- Riahi, Z.; Zainine, R.; Mellouli, Y.; Hannachi, R.; Bouyacoub, Y.; Laroussi, N.; Beltaief, N.; Kefi, R.; Romdhane, L.; Bonnet, C.; et al. Compound heterozygosity for dominant and recessive GJB2 mutations in a Tunisian family and association with successful cochlear implant outcome. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.D.; Caria, H.; Simoes-Teixeira, H.; Aasen, T.; Dias, O.; Andrea, M.; Kelsell, D.P.; Fialho, G. A novel M163L mutation in connexin 26 causing cell death and associated with autosomal dominant hearing loss. Hear. Res. 2008, 240, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Posukh, O.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.; Claustres, M.; Roux, A.F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, R.; Zelante, L.; López-Bigas, N.; D’Agruma, L.; Melchionda, S.; Restagno, G.; Arbonés, M.L.; Gasparini, P.; Estivill, X. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum. Genet. 2000, 106, 40–44. [Google Scholar] [PubMed]
- Primignani, P.; Castorina, P.; Sironi, F.; Curcio, C.; Ambrosetti, U.; Coviello, D.A. A novel dominant missense mutation--D179N--in the GJB2 gene (Connexin 26) associated with non-syndromic hearing loss. Clin. Genet. 2003, 63, 516–521. [Google Scholar] [CrossRef]
- Ambrosi, C.; Walker, A.E.; Depriest, A.D.; Cone, A.C.; Lu, C.; Badger, J.; Skerrett, I.M.; Sosinsky, G.E. Analysis of trafficking, stability and function of human connexin 26 gap junction channels with deafness-causing mutations in the fourth transmembrane helix. PLoS ONE 2013, 8, e70916. [Google Scholar] [CrossRef]
- Dai, P.; Yu, F.; Han, B.; Liu, X.; Wang, G.; Li, Q.; Yuan, Y.; Liu, X.; Huang, D.; Kang, D.; et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 2009, 7, 26. [Google Scholar] [CrossRef]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef]
- Tamayo, M.L.; Olarte, M.; Gelvez, N.; Gómez, M.; Frías, J.L.; Bernal, J.E.; Florez, S.; Medina, D. Molecular studies in the GJB2 gene (Cx26) among a deaf population from Bogotá, Colombia: Results of a screening program. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bakhchane, A.; Bousfiha, A.; Charoute, H.; Salime, S.; Detsouli, M.; Snoussi, K.; Nadifi, S.; Kabine, M.; Rouba, H.; Dehbi, H.; et al. Update of the spectrum of GJB2 gene mutations in 152 Moroccan families with autosomal recessive nonsyndromic hearing loss. Eur. J. Med. Genet. 2016, 59, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Morlé, L.; Bozon, M.; Alloisio, N.; Latour, P.; Vandenberghe, A.; Plauchu, H.; Collet, L.; Edery, P.; Godet, J.; Lina-Granade, G. A novel C202F mutation in the connexin26 gene (GJB2) associated with autosomal dominant isolated hearing loss. J. Med. Genet. 2000, 37, 368–370. [Google Scholar] [CrossRef]
- Wu, B.L.; Lindeman, N.; Lip, V.; Adams, A.; Amato, R.S.; Cox, G.; Irons, M.; Kenna, M.; Korf, B.; Raisen, J.; et al. Effectiveness of sequencing connexin 26 (GJB2) in cases of familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet. Med. 2002, 4, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Menevse, S.; Bayazit, Y.; Karamert, R.; Ergin, V.; Menevse, A. Two novel missense mutations in the connexin 26 gene in Turkish patients with nonsyndromic hearing loss. Biochem. Genet. 2010, 48, 248–256. [Google Scholar] [CrossRef]
- Leshinsky-Silver, E.; Berman, Z.; Vinkler, C.; Yannov-Sharav, M.; Lev, D. A novel missense mutation in the Connexin 26 gene associated with autosomal recessive sensorineural deafness. Hear. Res. 2005, 202, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ying, Z.; Cai, Z.; Sun, D.; He, Z.; Gao, Y.; Zhang, T.; Zhu, Y.; Chen, Y.; Guan, M.X. GJB2 Mutation Spectrum and Genotype-Phenotype Correlation in 1067 Han Chinese Subjects with Non-Syndromic Hearing Loss. PLoS ONE 2015, 10, e0128691. [Google Scholar] [CrossRef]
- Shen, J.; Oza, A.M.; Del Castillo, I.; Duzkale, H.; Matsunaga, T.; Pandya, A.; Kang, H.P.; Mar-Heyming, R.; Guha, S.; Moyer, K.; et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet. Med. 2019, 21, 2442–2452. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Z.; Jiang, Y.; Lin, Y.; Wang, X.; Wang, Z.; Tang, Z.; Wang, Y.; Wang, J.; Gao, Y.; et al. Biallelic p.V37I variant in GJB2 is associated with increasing incidence of hearing loss with age. Genet. Med. 2022, 24, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Terrinoni, A.; Codispoti, A.; Serra, V.; Didona, B.; Bruno, E.; Nistico, R.; Giustizieri, M.; Alessandrini, M.; Campione, E.; Melino, G. Connexin 26 (GJB2) mutations, causing KID Syndrome, are associated with cell death due to calcium gating deregulation. Biochem. Biophys. Res. Commun. 2010, 394, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikov, O.; Kralina, S.; Yurasova, Y.; Kenis, V.; Kircher, S.G.; Al Kaissi, A. Progressive Deformity of the Lower Limbs in a Patient with KID (Keratitis-Ichthyosis-Deafness) Syndrome. Case Rep. Orthop. 2020, 2020, 8747392. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Derosa, A.M.; White, T.W. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J. Investig. Derm. 2009, 129, 870–878. [Google Scholar] [CrossRef] [PubMed]
- García, I.E.; Maripillán, J.; Jara, O.; Ceriani, R.; Palacios-Muñoz, A.; Ramachandran, J.; Olivero, P.; Perez-Acle, T.; González, C.; Sáez, J.C.; et al. Keratitis-ichthyosis-deafness syndrome-associated Cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type Cx43. J. Investig. Derm. 2015, 135, 1338–1347. [Google Scholar] [CrossRef]
- Albuloushi, A.; Lovgren, M.L.; Steel, A.; Yeoh, Y.; Waters, A.; Zamiri, M.; Martin, P.E. A heterozygous mutation in GJB2 (Cx26F142L) associated with deafness and recurrent skin rashes results in connexin assembly deficiencies. Exp. Derm. 2020, 29, 970–979. [Google Scholar] [CrossRef]
- Arita, K.; Akiyama, M.; Aizawa, T.; Umetsu, Y.; Segawa, I.; Goto, M.; Sawamura, D.; Demura, M.; Kawano, K.; Shimizu, H. A novel N14Y mutation in Connexin26 in keratitis-ichthyosis-deafness syndrome: Analyses of altered gap junctional communication and molecular structure of N terminus of mutated Connexin26. Am. J. Pathol. 2006, 169, 416–423. [Google Scholar] [CrossRef]
- Aypek, H.; Bay, V.; Meşe, G. Altered cellular localization and hemichannel activities of KID syndrome associated connexin26 I30N and D50Y mutations. BMC Cell Biol. 2016, 17, 5. [Google Scholar] [CrossRef]
- Arndt, S.; Aschendorff, A.; Schild, C.; Beck, R.; Maier, W.; Laszig, R.; Birkenhager, R. A novel dominant and a de novo mutation in the GJB2 gene (connexin-26) cause keratitis-ichthyosis-deafness syndrome: Implication for cochlear implantation. Otol. Neurotol. 2010, 31, 210–215. [Google Scholar] [CrossRef]
- Gerido, D.A.; DeRosa, A.M.; Richard, G.; White, T.W. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol. Cell Physiol. 2007, 293, C337–C345. [Google Scholar] [CrossRef]
- Montgomery, J.R.; White, T.W.; Martin, B.L.; Turner, M.L.; Holland, S.M. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J. Am. Acad. Dermatol. 2004, 51, 377–382. [Google Scholar] [CrossRef]
- Mhaske, P.V.; Levit, N.A.; Li, L.; Wang, H.Z.; Lee, J.R.; Shuja, Z.; Brink, P.R.; White, T.W. The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am. J. Physiol. Cell Physiol. 2013, 304, C1150–C1158. [Google Scholar] [CrossRef]
- de Zwart-Storm, E.A.; van Geel, M.; Veysey, E.; Burge, S.; Cooper, S.; Steijlen, P.M.; Martin, P.E.; van Steensel, M.A. A novel missense mutation in GJB2, p.Tyr65His, causes severe Vohwinkel syndrome. Br. J. Derm. 2011, 164, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Shuja, Z.; Li, L.; Gupta, S.; Mese, G.; White, T.W. Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J. Investig. Derm. 2016, 136, 225–235. [Google Scholar] [CrossRef]
- de Zwart-Storm, E.A.; Hamm, H.; Stoevesandt, J.; Steijlen, P.M.; Martin, P.E.; van Geel, M.; van Steensel, M.A. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J. Med. Genet. 2008, 45, 161–166. [Google Scholar] [CrossRef]
- Guerra, L.; Bergamo, F.; D’Apice, M.R.; Angelucci, F.; di Girolamo, S.; Camerota, L.; Monetta, R.; Annessi, G.; Castiglia, D.; Novelli, G.; et al. Keratoderma-Deafness-Mucocutaneous Syndrome Associated with Phe142Leu in the GJB2 Gene. Acta Derm. Venereol. 2019, 99, 1192–1194. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Xiang, J.; Sun, X.; Song, N.; Ramaswamy, S.; Abou Tayoun, A.N.; Peng, Z. Comprehensive interpretation of single-nucleotide substitutions in GJB2 reveals the genetic and phenotypic landscape of GJB2-related hearing loss. Hum. Genet. 2022. [CrossRef] [PubMed]
- Li, J.; Zhao, T.; Zhang, Y.; Zhang, K.; Shi, L.; Chen, Y.; Wang, X.; Sun, Z. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018, 46, 7793–7804. [Google Scholar] [CrossRef]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Findlay, G.M.; Daza, R.M.; Martin, B.; Zhang, M.D.; Leith, A.P.; Gasperini, M.; Janizek, J.D.; Huang, X.; Starita, L.M.; Shendure, J. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018, 562, 217–222. [Google Scholar] [CrossRef]
- Majithia, A.R.; Tsuda, B.; Agostini, M.; Gnanapradeepan, K.; Rice, R.; Peloso, G.; Patel, K.A.; Zhang, X.; Broekema, M.F.; Patterson, N.; et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016, 48, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, A.O.; Yang, X.; Lintner, R.E.; McFarland, J.M.; Duby, M.; Kim, J.; Howard, T.P.; Takeda, D.Y.; Ly, S.H.; Kim, E.; et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018, 50, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Suiter, C.C.; Moriyama, T.; Matreyek, K.A.; Yang, W.; Scaletti, E.R.; Nishii, R.; Yang, W.; Hoshitsuki, K.; Singh, M.; Trehan, A.; et al. Massively parallel variant characterization identifies NUDT15 alleles associated with thiopurine toxicity. Proc. Natl. Acad Sci. USA 2020, 117, 5394–5401. [Google Scholar] [CrossRef]
- Jia, X.; Burugula, B.B.; Chen, V.; Lemons, R.M.; Jayakody, S.; Maksutova, M.; Kitzman, J.O. Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am. J. Hum. Genet. 2021, 108, 163–175. [Google Scholar] [CrossRef] [PubMed]
- An, L.E.I.; Wang, Y.; Wu, G.; Wang, Z.; Shi, Z.; Liu, C.; Wang, C.; Yi, M.; Niu, C.; Duan, S.; et al. Defining the Sensitivity Landscape of EGFR Variants to Tyrosine Kinase Inhibitors. Transl. Res. 2022, in press. [Google Scholar] [CrossRef]
- Fowler, D.M.; Stephany, J.J.; Fields, S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc. 2014, 9, 2267–2284. [Google Scholar] [CrossRef]
- Glazer, A.M.; Kroncke, B.M.; Matreyek, K.A.; Yang, T.; Wada, Y.; Shields, T.; Salem, J.E.; Fowler, D.M.; Roden, D.M. Deep Mutational Scan of an SCN5A Voltage Sensor. Circ. Genom. Precis. Med. 2020, 13, e002786. [Google Scholar] [CrossRef]
- Heredia, J.D.; Park, J.; Brubaker, R.J.; Szymanski, S.K.; Gill, K.S.; Procko, E. Mapping Interaction Sites on Human Chemokine Receptors by Deep Mutational Scanning. J. Immunol. 2018, 200, 3825–3839. [Google Scholar] [CrossRef]
- Jones, E.M.; Lubock, N.B.; Venkatakrishnan, A.J.; Wang, J.; Tseng, A.M.; Paggi, J.M.; Latorraca, N.R.; Cancilla, D.; Satyadi, M.; Davis, J.E.; et al. Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. eLife 2020, 9, e54895. [Google Scholar] [CrossRef]
- Coyote-Maestas, W.; Nedrud, D.; He, Y.; Schmidt, D. Determinants of trafficking, conduction, and disease within a K(+) channel revealed through multiparametric deep mutational scanning. eLife 2022, 11, e76903. [Google Scholar] [CrossRef] [PubMed]





| Variants | Locations | Inheritance Patterns | Trafficking | Hemichannels | Gap Junction Channel | Cell Death | Dominant Negtive/Trans-Dominant Effects | Clinical Phenotype | References |
|---|---|---|---|---|---|---|---|---|---|
| G11E | NT | AD | defect | Increased | / | Increased | / | KID | [83,84] |
| G12R | NT | AD | impaired | Increased | impaired | Increased | YES | KID | [29,85,86,87] |
| N14Y | NT | AD | impaired | Increased | impaired | / | / | KID | [86,88] |
| N14K | NT | AD | defect | Increased | impaired | Increased | / | KID | [24,85] |
| S17F | NT | AD | impaired | defect | impaired | / | / | KID | [29,85,86] |
| I30N | TM1 | AD | impaired | Increased | / | / | / | KID | [89,90] |
| A40V | TM1 | AD | / | Increased | impaired | Increased | / | KID | [91,92] |
| G45E | EC1 | AD | impaired | Increased | impaired | Increased | YES | KID | [54,91] |
| D50A | EC1 | AD | impaired | Increased | / | KID | [93] | ||
| D50N | EC1 | AD | defect | Increased | impaired | Increased | / | KID, HID | [30,83,85] |
| D50Y | EC1 | AD | impaired | Increased | / | / | / | KID | [89] |
| N54K | EC1 | AD | defect | impaired | impaired | / | YES | BPS, PPK + deafness | [24,31,44] |
| G59A | EC1 | AD | defect | / | impaired | / | YES | PPK + deafness | [52,53] |
| Y65H | EC1 | AD | impaired | / | impaired | / | / | Vohwinkel | [94] |
| D66H | EC1 | AD | defect | impaired | impaired | / | YES/ | Vohwinkel | [52,87] |
| H73R | EC1 | AD | impaired | / | impaired | / | YES | PPK + deafness | [95,96] |
| R75Q | TM2 | AD | normal | / | impaired | / | YES | PPK + deafness | [53] |
| R75W | TM2 | AD | normal | / | impaired | / | YES | PPK + deafness | [52,53] |
| A88V | TM2 | AD | impaired | Increased | / | / | / | KID | [93] |
| F142L | TM3 | AD | impaired | impaired | impaired | / | YES/ | Keratoderma-Deafness-Mucocutaneous Syndrome | [87,97] |
| S183F | TM4 | AD | impaired | impaired | impaired | / | YES | PPK + deafness | [24] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, L.; Wang, Y.; An, L.; Zeng, B.; Wang, Y.; Frishman, D.; Liu, M.; Chen, Y.; Tang, W.; Xu, H. Molecular Mechanisms and Clinical Phenotypes of GJB2 Missense Variants. Biology 2023, 12, 505. https://doi.org/10.3390/biology12040505
Mao L, Wang Y, An L, Zeng B, Wang Y, Frishman D, Liu M, Chen Y, Tang W, Xu H. Molecular Mechanisms and Clinical Phenotypes of GJB2 Missense Variants. Biology. 2023; 12(4):505. https://doi.org/10.3390/biology12040505
Chicago/Turabian StyleMao, Lu, Yueqiang Wang, Lei An, Beiping Zeng, Yanyan Wang, Dmitrij Frishman, Mengli Liu, Yanyu Chen, Wenxue Tang, and Hongen Xu. 2023. "Molecular Mechanisms and Clinical Phenotypes of GJB2 Missense Variants" Biology 12, no. 4: 505. https://doi.org/10.3390/biology12040505
APA StyleMao, L., Wang, Y., An, L., Zeng, B., Wang, Y., Frishman, D., Liu, M., Chen, Y., Tang, W., & Xu, H. (2023). Molecular Mechanisms and Clinical Phenotypes of GJB2 Missense Variants. Biology, 12(4), 505. https://doi.org/10.3390/biology12040505