
Dopaminergic Input Regulates the Sensitivity of Indirect Pathway Striatal Spiny Neurons to Brain-Derived Neurotrophic Factor

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Housing
2.1.1. Mice
2.1.2. Rats
2.1.3. Unilateral 6-OHDA Lesion
2.2. Cell Culture
2.3. Flow Cytometry Cell Sorting
2.4. Stimulation of DRD2-eGFP Neurons
2.5. Immunocytochemistry
2.6. Preparation of Tissue for Immunostaining
2.7. Immunohistochemistry
2.8. The Cell Surface Biotinylation Assay
2.9. SDS-PAGE and Western Blotting
2.10. Western Blot Analyses
2.11. Image Acquisition
Quantification of TrkB Clusters
2.12. Quantification and Statistical Analysis
3. Results
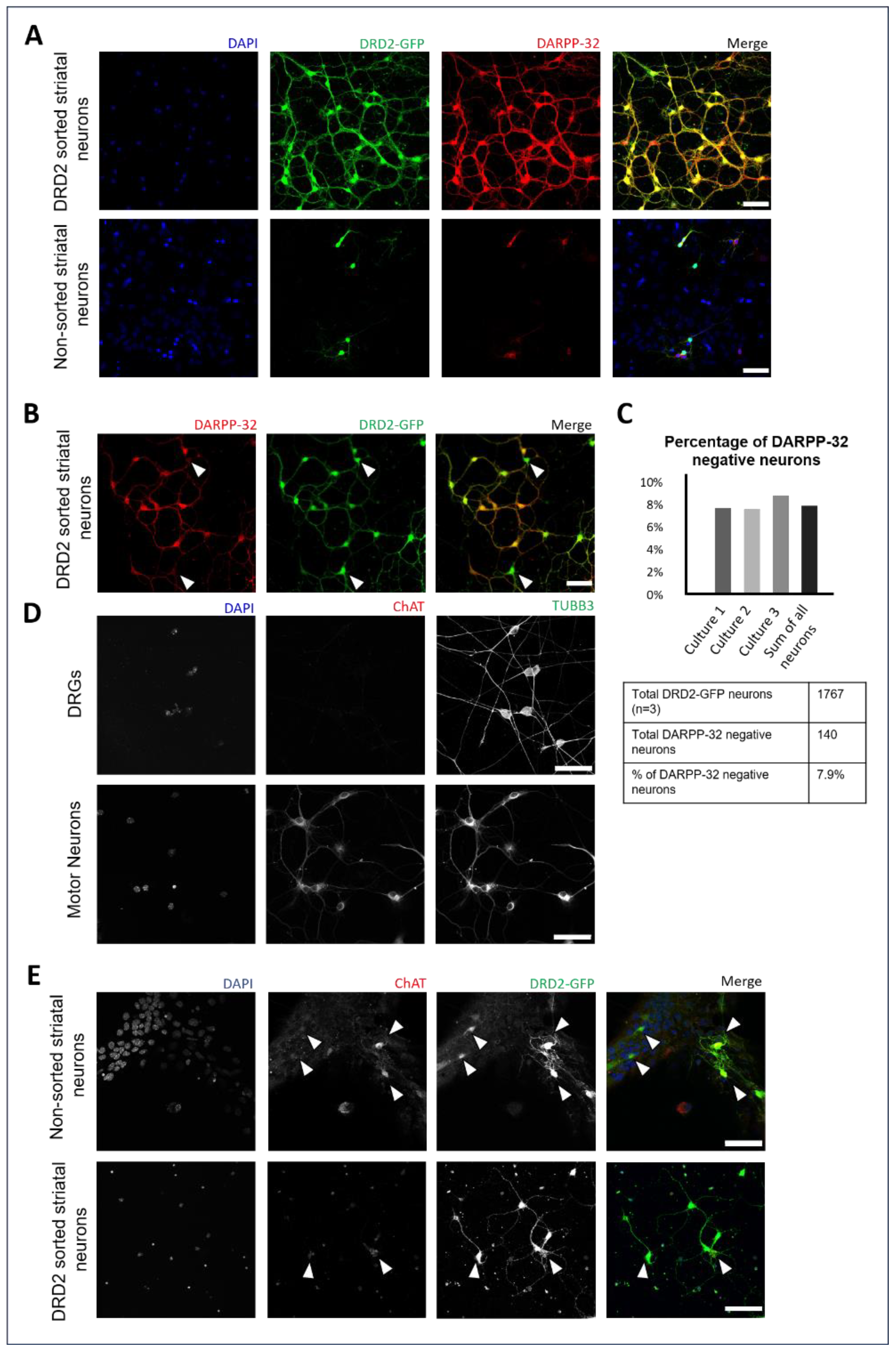
3.1. Enrichment of DRD2 Spiny Projection Neurons and Identification of DRD2-Expressing Cholinergic Interneurons
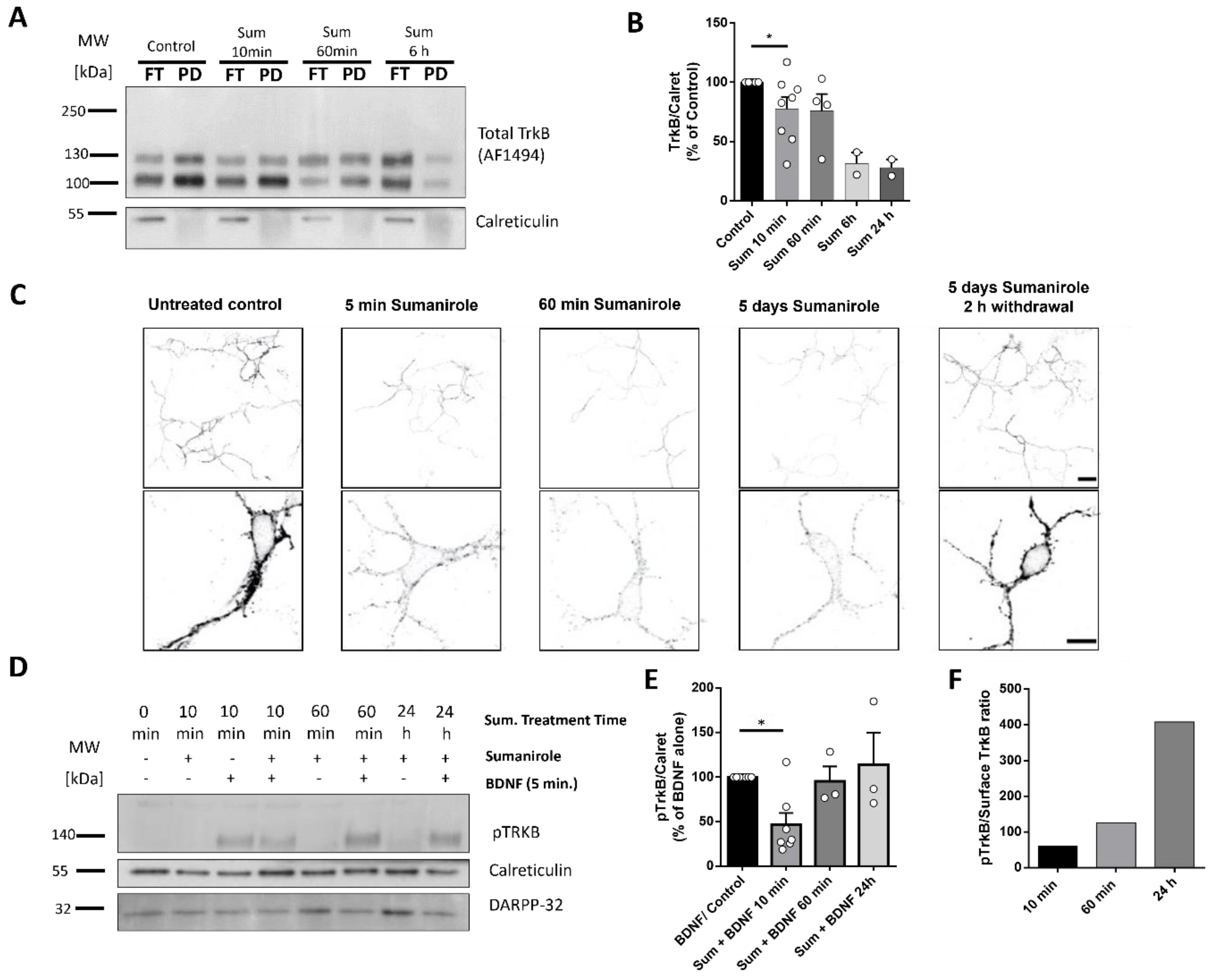
3.2. DRD2 Activation in iSPNs Decreases TrkB Cell Surface Translocation
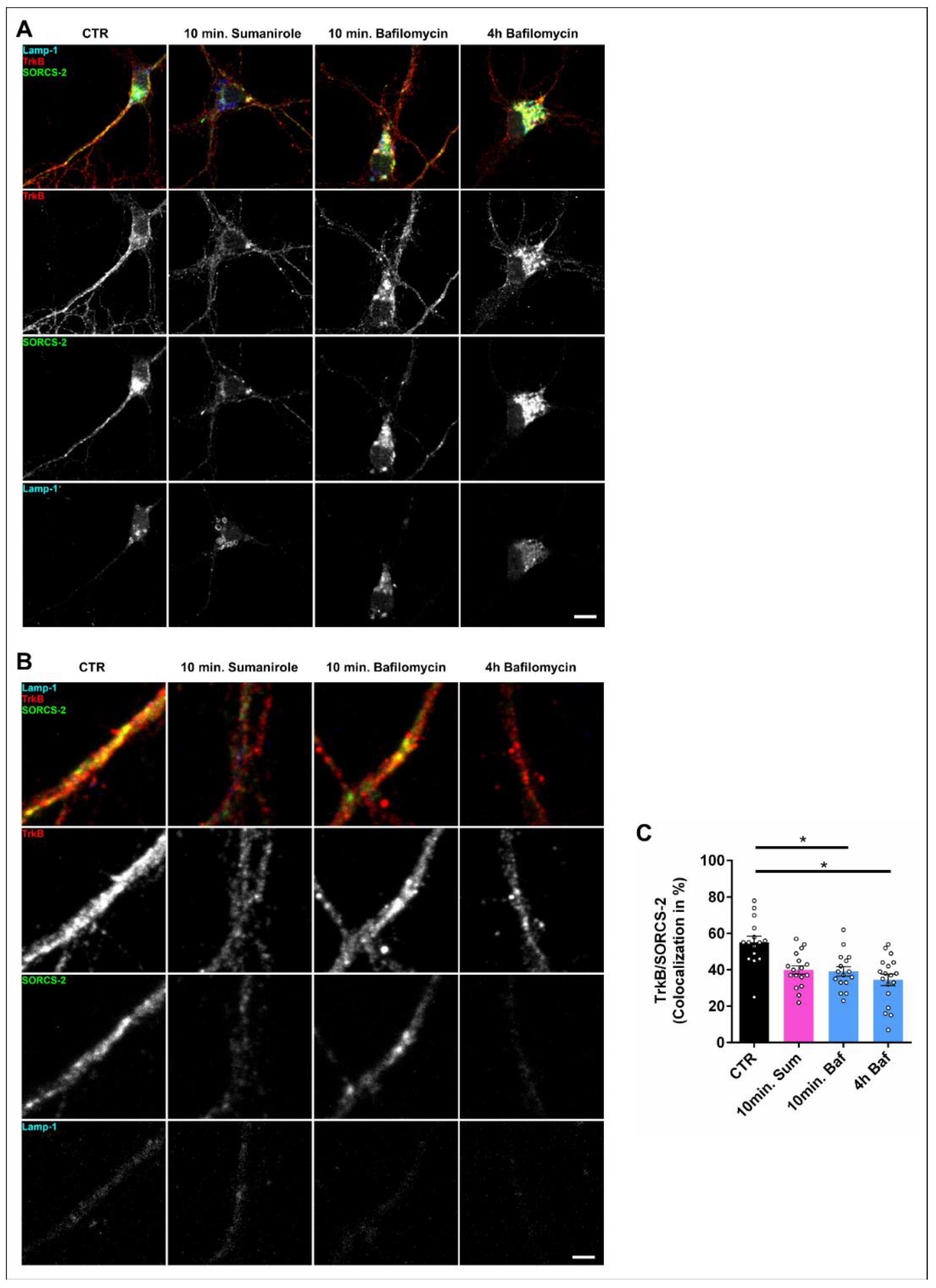
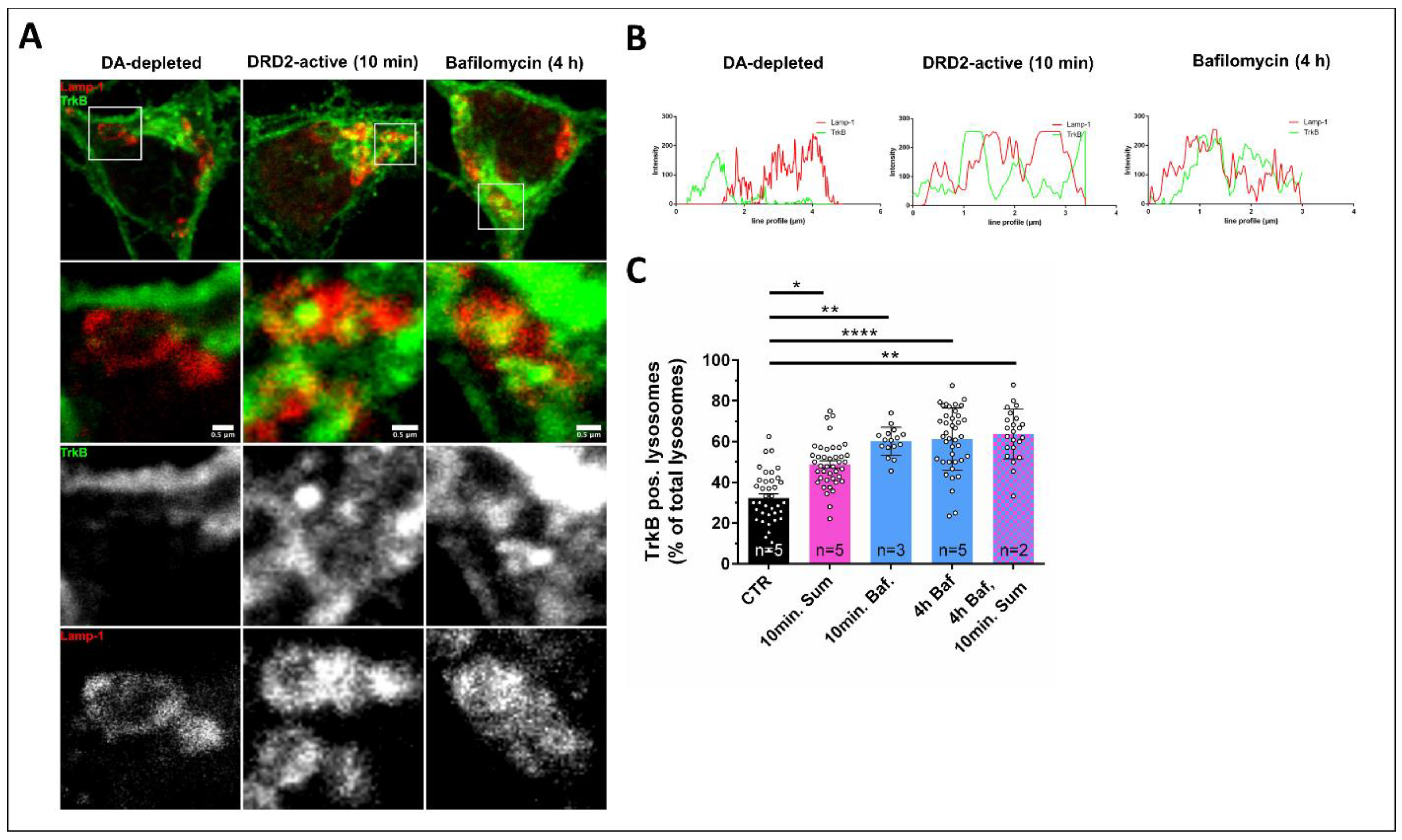
3.3. DRD2 Activation Leads to the Removal of TrkB from the Cell Surface and Subsequent Lysosomal Degradation within the Cell Body
3.4. Dopamine Depletion Causes TrkB Cluster Formation in Pitx3−/− Mice
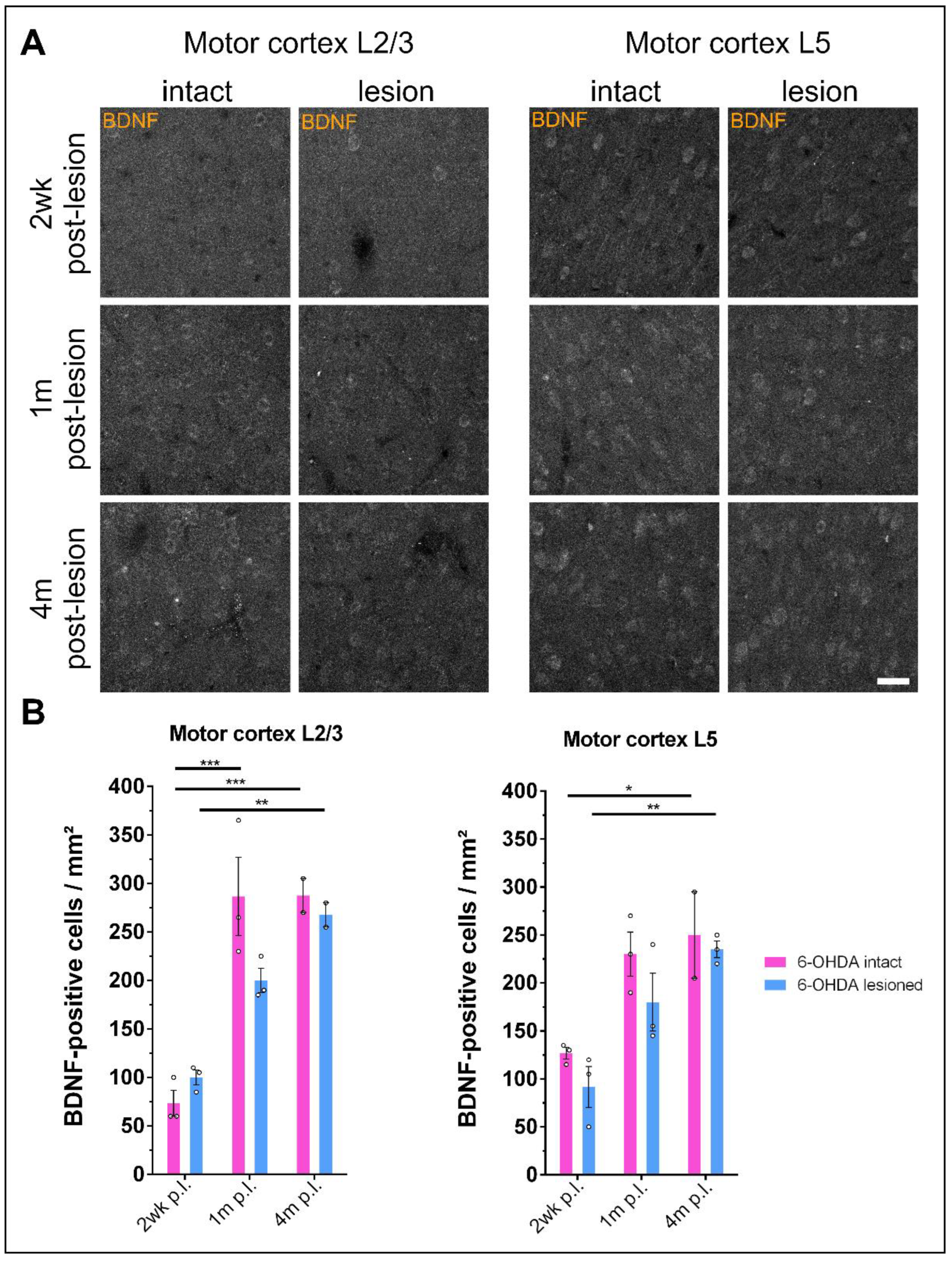
3.5. Prolonged Dopamine Depletion Increases BDNF Expression in the Motor Cortex of 6-OHDA-Treated Rats
4. Discussion
5. Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Checkoway, H.; Lundin, J.I.; Kelada, S.N. Neurodegenerative diseases. IARC Sci. Publ. 2011, 163, 407–419. [Google Scholar]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Morrish, P.K.; Rakshi, J.S.; Bailey, D.L.; Sawle, G.V.; Brooks, D.J. Measuring the rate of progression and estimating the preclinical period of Parkinson’s disease with 18F-dopa PET. J. Neurol. Neurosurg. Amp. Psychiatry 1998, 64, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-Y.; Lang, A.E. The nonmotor symptoms of Parkinson’s disease—An overview. Mov. Disord. 2010, 25, S123–S130. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Lew, M. Overview of Parkinson’s Disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2007, 27, 155S–160S. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Ding, J.; Day, M.; Wang, Z.; Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007, 30, 228–235. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Surmeier, D.J. Modulation of Striatal Projection Systems by Dopamine. Annu. Rev. Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Flajolet, M.; Greengard, P.; Surmeier, D.J. Dichotomous Dopaminergic Control of Striatal Synaptic Plasticity. Science 2008, 321, 848–851. [Google Scholar] [CrossRef]
- Boccalaro, I.L.; Schwerdel, C.; Cristiá-Lara, L.; Fritschy, J.-M.; Rubi, L. Dopamine depletion induces neuron-specific alterations of GABAergic transmission in the mouse striatum. Eur. J. Neurosci. 2020, 52, 3353–3374. [Google Scholar] [CrossRef]
- Calabresi, P.; Maj, R.; Pisani, A.; Mercuri, N.; Bernardi, G. Long-term synaptic depression in the striatum: Physiological and pharmacological characterization. J. Neurosci. 1992, 12, 4224–4233. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Saiardi, A.; Pisani, A.; Baik, J.-H.; Centonze, D.; Mercuri, N.B.; Bernardi, G.; Borrelli, E. Abnormal Synaptic Plasticity in the Striatum of Mice Lacking Dopamine D2 Receptors. J. Neurosci. 1997, 17, 4536–4544. [Google Scholar] [CrossRef]
- Centonze, D.; Gubellini, P.; Picconi, B.; Calabresi, P.; Giacomini, P.; Bernardi, G. Unilateral Dopamine Denervation Blocks Corticostriatal LTP. J. Neurophysiol. 1999, 82, 3575–3579. [Google Scholar] [CrossRef]
- Pisani, A.; Centonze, D.; Bernardi, G.; Calabresi, P. Striatal synaptic plasticity: Implications for motor learning and Parkinson’s disease. Mov. Disord. 2005, 20, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Grande, C.; Saulle, E.; Martín, A.B.; Gubellini, P.; Pavón, N.; Pisani, A.; Bernardi, G.; Moratalla, R.; Calabresi, P. Distinct Roles of D1 and D5 Dopamine Receptors in Motor Activity and Striatal Synaptic Plasticity. J. Neurosci. 2003, 23, 8506–8512. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.S.; Vincent, S.R.; Fibiger, H.C. D1 and D2 dopamine receptors differentially regulate c-fos expression in striatonigral and striatopallidal neurons. Neuroscience 1992, 49, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Le Moine, C.; Bloch, B. D1 and D2 dopamine receptor gene expression in the rat striatum: Sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAS in distinct neuronal populations of the dorsal and ventral striatum. J. Comp. Neurol. 1995, 355, 418–426. [Google Scholar] [CrossRef]
- Harrison, M.B.; Wiley, R.G.; Wooten, G.F. Selective localization of striatal D1 receptors to striatonigral neurons. Brain Res. 1990, 528, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.B.; Kreitzer, A.C. Reassessing Models of Basal Ganglia Function and Dysfunction. Annu. Rev. Neurosci. 2014, 37, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.; Padungchaichot, P.; Wong, J.Y.F.; Lawrence, A.J.; McManus, J.F.; Sumarsono, S.H.; Natoli, A.L.; Lakso, M.; Wreford, N.; Westphal, H.; et al. Targeted Expression of a Toxin Gene to D1 Dopamine Receptor Neurons by Cre-Mediated Site-Specific Recombination. J. Neurosci. 1998, 18, 9845–9857. [Google Scholar] [CrossRef]
- Hikida, T.; Kimura, K.; Wada, N.; Funabiki, K.; Nakanishi, S. Distinct Roles of Synaptic Transmission in Direct and Indirect Striatal Pathways to Reward and Aversive Behavior. Neuron 2010, 66, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Kravitz, A.V.; Freeze, B.S.; Parker, P.R.L.; Kay, K.; Thwin, M.T.; Deisseroth, K.; Kreitzer, A.C. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 2010, 466, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.N.D.; Wickens, J.R. Dopamine D-1/D-5 Receptor Activation Is Required for Long-Term Potentiation in the Rat Neostriatum In Vitro. J. Neurophysiol. 2001, 85, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Picconi, B.; Gubellini, P.; Bernardi, G.; Calabresi, P. Dopaminergic control of synaptic plasticity in the dorsal striatum. Eur. J. Neurosci. 2001, 13, 1071–1077. [Google Scholar] [CrossRef]
- Andreska, T.; Rauskolb, S.; Schukraft, N.; Lüningschrör, P.; Sasi, M.; Signoret-Genest, J.; Behringer, M.; Blum, R.; Sauer, M.; Tovote, P.; et al. Induction of BDNF Expression in Layer II/III and Layer V Neurons of the Motor Cortex Is Essential for Motor Learning. J. Neurosci. 2020, 40, 6289–6308. [Google Scholar] [CrossRef]
- West, M.O.; Carelli, R.M.; Pomerantz, M.; Cohen, S.M.; Gardner, J.P.; Chapin, J.K.; Woodward, D.J. A region in the dorsolateral striatum of the rat exhibiting single-unit correlations with specific locomotor limb movements. J. Neurophysiol. 1990, 64, 1233–1246. [Google Scholar] [CrossRef]
- Sheng, M.-j.; Lu, D.; Shen, Z.-m.; Poo, M.-m. Emergence of stable striatal D1R and D2R neuronal ensembles with distinct firing sequence during motor learning. Proc. Natl. Acad. Sci. USA 2019, 116, 11038–11047. [Google Scholar] [CrossRef]
- Suarez, L.M.; Solis, O.; Aguado, C.; Lujan, R.; Moratalla, R. L-DOPA Oppositely Regulates Synaptic Strength and Spine Morphology in D1 and D2 Striatal Projection Neurons in Dyskinesia. Cereb. Cortex 2016, 26, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Suarez, L.M.; Solis, O.; Sanz-Magro, A.; Alberquilla, S.; Moratalla, R. Dopamine D1 Receptors Regulate Spines in Striatal Direct-Pathway and Indirect-Pathway Neurons. Mov. Disord. 2020, 35, 1810–1821. [Google Scholar] [CrossRef]
- Harrison, M.B.; Wiley, R.G.; Wooten, G.F. Changes in D2 but not D1 receptor binding in the striatum following a selective lesion of striatopallidal neurons. Brain Res. 1992, 590, 305–310. [Google Scholar] [CrossRef]
- Durieux, P.F.; Bearzatto, B.; Guiducci, S.; Buch, T.; Waisman, A.; Zoli, M.; Schiffmann, S.N.; de Kerchove d’Exaerde, A. D2R striatopallidal neurons inhibit both locomotor and drug reward processes. Nat. Neurosci. 2009, 12, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Yasoshima, Y.; Matsushita, N.; Kaneko, T.; Kohno, K.; Pastan, I.; Kobayashi, K. Conditional Ablation of Striatal Neuronal Types Containing Dopamine D2 Receptor Disturbs Coordination of Basal Ganglia Function. J. Neurosci. 2003, 23, 9078–9088. [Google Scholar] [CrossRef]
- Neve, K.A.; Seamans, J.K.; Trantham-Davidson, H. Dopamine Receptor Signaling. J. Recept. Signal Transduct. 2004, 24, 165–205. [Google Scholar] [CrossRef] [PubMed]
- Stoof, J.C.; Kebabian, J.W. Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature 1981, 294, 366–368. [Google Scholar] [CrossRef]
- Herve, D.; Levi-Strauss, M.; Marey-Semper, I.; Verney, C.; Tassin, J.; Glowinski, J.; Girault, J. G(olf) and Gs in rat basal ganglia: Possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J. Neurosci. 1993, 13, 2237–2248. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Belluscio, L.; Hen, R. GOLFα Mediates Dopamine D1 Receptor Signaling. J. Neurosci. 2000, 20, RC91. [Google Scholar] [CrossRef]
- Corvol, J.C.; Studler, J.M.; Schonn, J.S.; Girault, J.A.; Hervé, D. Gαolf is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J. Neurochem. 2001, 76, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-DeDiego, I.; Naranjo, J.R.; Hervé, D.; Moratalla, R. Dopaminergic regulation of olfactory type G-protein α subunit expression in the striatum. Mov. Disord. 2015, 30, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W.; Chen-Goodspeed, M.; Chen, J. Mechanism of Gαi-mediated Inhibition of Type V Adenylyl Cyclase. J. Biol. Chem. 2002, 277, 28823–28829. [Google Scholar] [CrossRef] [PubMed]
- Huff, R.M.; Chio, C.L.; Lajiness, M.E.; Goodman, L.V. Signal Transduction Pathways Modulated by D2-Like Dopamine Receptors. Adv. Pharmacol. 1997, 42, 454–457. [Google Scholar] [CrossRef]
- Gazi, L.; Nickolls, S.A.; Strange, P.G. Functional coupling of the human dopamine D2 receptor with Gαi1, Gαi2, Gαi3 and Gαo G proteins: Evidence for agonist regulation of G protein selectivity. Br. J. Pharmacol. 2003, 138, 775–786. [Google Scholar] [CrossRef]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.-P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942.e918. [Google Scholar] [CrossRef]
- Thiele, S.L.; Chen, B.; Lo, C.; Gertler, T.S.; Warre, R.; Surmeier, J.D.; Brotchie, J.M.; Nash, J.E. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiol. Dis. 2014, 71, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Figurov, A.; Pozzo-Miller, L.D.; Olafsson, P.; Wang, T.; Lu, B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 1996, 381, 706–709. [Google Scholar] [CrossRef]
- Kang, H.; Welcher, A.A.; Shelton, D.; Schuman, E.M. Neurotrophins and Time: Different Roles for TrkB Signaling in Hippocampal Long-Term Potentiation. Neuron 1997, 19, 653–664. [Google Scholar] [CrossRef]
- Korte, M.; Kang, H.; Bonhoeffer, T.; Schuman, E. A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology 1998, 37, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, E.; Ying, S.-W.; Kanhema, T.; Croll, S.D.; Bramham, C.R. Brain-Derived Neurotrophic Factor Triggers Transcription-Dependent, Late Phase Long-Term Potentiation In Vivo. J. Neurosci. 2002, 22, 7453–7461. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Nawa, H.; Sora, I.; Chao, M.V. Dopamine D1 Receptor-induced Signaling through TrkB Receptors in Striatal Neurons. J. Biol. Chem. 2008, 283, 15799–15806. [Google Scholar] [CrossRef]
- Jia, Y.; Gall, C.M.; Lynch, G. Presynaptic BDNF Promotes Postsynaptic Long-Term Potentiation in the Dorsal Striatum. J. Neurosci. 2010, 30, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Gangarossa, G.; Perez, S.; Dembitskaya, Y.; Prokin, I.; Berry, H.; Venance, L. BDNF Controls Bidirectional Endocannabinoid Plasticity at Corticostriatal Synapses. Cereb. Cortex 2019, 30, 197–214. [Google Scholar] [CrossRef]
- Andreska, T.; Lüningschrör, P.; Sendtner, M. Regulation of TrkB cell surface expression—A mechanism for modulation of neuronal responsiveness to brain-derived neurotrophic factor. Cell Tissue Res. 2020, 382, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef]
- Baquet, Z.C.; Bickford, P.C.; Jones, K.R. Brain-Derived Neurotrophic Factor Is Required for the Establishment of the Proper Number of Dopaminergic Neurons in the Substantia Nigra Pars Compacta. J. Neurosci. 2005, 25, 6251–6259. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Franke, A.; Wilkinson, G.A.; Kruttgen, A.; Hu, M.; Munro, E.; Hanson, M.G.; Reichardt, L.F.; Barres, B.A. Depolarization and cAMP Elevation Rapidly Recruit TrkB to the Plasma Membrane of CNS Neurons. Neuron 1998, 21, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.; Poo, M.-m. Gating of BDNF-Induced Synaptic Potentiation by cAMP. Science 1999, 284, 1982–1984. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Pang, P.T.; Feng, L.; Lu, B. Cyclic AMP controls BDNF-induced TrkB phosphorylation and dendritic spine formation in mature hippocampal neurons. Nat. Neurosci. 2005, 8, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Lüningschrör, P.; Andreska, T.; Veh, A.; Wolf, D.; Giridhar, N.J.; Moradi, M.; Denzel, A.; Sendtner, M. Calnexin controls TrkB cell surface transport and ER-phagy in mouse cerebral cortex development. Dev. Cell 2023, 58, 1733–1747.e6. [Google Scholar] [CrossRef]
- Puehringer, D.; Orel, N.; Lüningschrör, P.; Subramanian, N.; Herrmann, T.; Chao, M.V.; Sendtner, M. EGF transactivation of Trk receptors regulates the migration of newborn cortical neurons. Nat. Neurosci. 2013, 16, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Andreska, T.; Lüningschrör, P.; Wolf, D.; McFleder, R.L.; Ayon-Olivas, M.; Rattka, M.; Drechsler, C.; Perschin, V.; Blum, R.; Aufmkolk, S.; et al. DRD1 signaling modulates TrkB turnover and BDNF sensitivity in direct pathway striatal medium spiny neurons. Cell Rep. 2023, 42, 112575. [Google Scholar] [CrossRef] [PubMed]
- Rauskolb, S.; Zagrebelsky, M.; Dreznjak, A.; Deogracias, R.; Matsumoto, T.; Wiese, S.; Erne, B.; Sendtner, M.; Schaeren-Wiemers, N.; Korte, M.; et al. Global Deprivation of Brain-Derived Neurotrophic Factor in the CNS Reveals an Area-Specific Requirement for Dendritic Growth. J. Neurosci. 2010, 30, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Baydyuk, M.; Russell, T.; Liao, G.-Y.; Zang, K.; An, J.J.; Reichardt, L.F.; Xu, B. TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Baydyuk, M.; Xu, B. BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 2014, 8, 254. [Google Scholar] [CrossRef]
- Li, Y.; Yui, D.; Luikart, B.W.; McKay, R.M.; Li, Y.; Rubenstein, J.L.; Parada, L.F. Conditional ablation of brain-derived neurotrophic factor-TrkB signaling impairs striatal neuron development. Proc. Natl. Acad. Sci. USA 2012, 109, 15491–15496. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Functional analysis of brain derived neurotrophic factor (BDNF) in Huntington’s disease. Aging 2021, 13, 6103–6114. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.P.; Wong, T.; Bricker-Anthony, C.; Deng, B.; Reiner, A. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington’s disease mice. Neurobiol. Dis. 2013, 60, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Samadi, P.; Boutet, A.; Rymar, V.V.; Rawal, K.; Maheux, J.; Kvann, J.-C.; Tomaszewski, M.; Beaubien, F.; Cloutier, J.F.; Levesque, D.; et al. Relationship between BDNF expression in major striatal afferents, striatum morphology and motor behavior in the R6/2 mouse model of Huntington’s disease. Genes Brain Behav. 2013, 12, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Surmeier, D.J. Corticostriatal synaptic adaptations in Huntington’s disease. Curr. Opin. Neurobiol. 2015, 33, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB Receptor Signaling Underlies Corticostriatal Dysfunction in Huntington’s Disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Villalba, R.M.; Lee, H.; Smith, Y. Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Exp. Neurol. 2009, 215, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Suárez, L.M.; Solís, O.; Caramés, J.M.; Taravini, I.R.; Solís, J.M.; Murer, M.G.; Moratalla, R. L-DOPA Treatment Selectively Restores Spine Density in Dopamine Receptor D2–Expressing Projection Neurons in Dyskinetic Mice. Biol. Psychiatry 2014, 75, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Le Moine, C.; Tison, F.; Bloch, B. D2 dopamine receptor gene expression by cholinergic neurons in the rat striatum. Neurosci. Lett. 1990, 117, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, A.A.; Chen, V.; Herring, B.E.; Mendenhall, J.M.; Berlanga, M.L. Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res. 2003, 986, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.K.; Smith, Y. Cholinergic interneurons in the dorsal and ventral striatum: Anatomical and functional considerations in normal and diseased conditions. Ann. N. Y. Acad. Sci. 2015, 1349, 1–45. [Google Scholar] [CrossRef]
- Ztaou, S.; Maurice, N.; Camon, J.; Guiraudie-Capraz, G.; Goff, L.K.-L.; Beurrier, C.; Liberge, M.; Amalric, M. Involvement of Striatal Cholinergic Interneurons and M1 and M4 Muscarinic Receptors in Motor Symptoms of Parkinson’s Disease. J. Neurosci. 2016, 36, 9161–9172. [Google Scholar] [CrossRef]
- Shen, W.; Plotkin, J.L.; Francardo, V.; Ko, W.K.D.; Xie, Z.; Li, Q.; Fieblinger, T.; Wess, J.; Neubig, R.R.; Lindsley, C.W.; et al. M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron 2015, 88, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.-Y.; Ardayfio, P.; Kang, U.J.; Semina, E.V.; Kim, K.-S. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Mol. Brain Res. 2003, 114, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.-Y.; Fleming, S.M.; Ardayfio, P.; Moran-Gates, T.; Kim, H.; Tarazi, F.I.; Chesselet, M.-F.; Kim, K.-S. 3,4-Dihydroxyphenylalanine Reverses the Motor Deficits in Pitx3-Deficient Aphakia Mice: Behavioral Characterization of a Novel Genetic Model of Parkinson’s Disease. J. Neurosci. 2005, 25, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Smidt, M.P.; Smits, S.M.; Bouwmeester, H.; Hamers, F.P.T.; van der Linden, A.J.A.; Hellemons, A.J.C.G.M.; Graw, J.; Burbach, J.P.H. Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development 2004, 131, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Le, W.; Zhang, L.; Xie, W.; Li, S.; Dani, J.A. Pitx3 deficiency produces decreased dopamine signaling and induces motor deficits in Pitx3(−/−) mice. Neurobiol. Aging 2015, 36, 3314–3320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Varnum, D.S.S.; Leroy, C. Aphakia, a New Mutation in the Mouse. J. Hered. 1968, 59, 147–150. [Google Scholar] [CrossRef]
- Smidt, M.P.; van Schaick, H.S.A.; Lanctôt, C.; Tremblay, J.J.; Cox, J.J.; van der Kleij, A.A.M.; Wolterink, G.; Drouin, J.; Burbach, J.P.H. A homeodomain gene Ptx3 has highly restricted brain expression in mesencephalic dopaminergic neurons. Proc. Natl. Acad. Sci. USA 1997, 94, 13305–13310. [Google Scholar] [CrossRef] [PubMed]
- Alberquilla, S.; Gonzalez-Granillo, A.; Martín, E.D.; Moratalla, R. Dopamine regulates spine density in striatal projection neurons in a concentration-dependent manner. Neurobiol. Dis. 2020, 134, 104666. [Google Scholar] [CrossRef] [PubMed]
- Gallo, E.F.; Greenwald, J.; Yeisley, J.; Teboul, E.; Martyniuk, K.M.; Villarin, J.M.; Li, Y.; Javitch, J.A.; Balsam, P.D.; Kellendonk, C. Dopamine D2 receptors modulate the cholinergic pause and inhibitory learning. Mol. Psychiatry 2022, 27, 1502–1514. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Arnold, S.A.; Habas, A.; Hetman, M.; Hagg, T. Ciliary Neurotrophic Factor Mediates Dopamine D2 Receptor-Induced CNS Neurogenesis in Adult Mice. J. Neurosci. 2008, 28, 2231–2241. [Google Scholar] [CrossRef]
- Liu, G.-Q.; Liu, Z.-X.; Lin, Z.-X.; Chen, P.; Yan, Y.-C.; Lin, Q.-R.; Hu, Y.-J.; Jiang, N.; Yu, B. Effects of Dopamine on stem cells and its potential roles in the treatment of inflammatory disorders: A narrative review. Stem Cell Res. Ther. 2023, 14, 230. [Google Scholar] [CrossRef]
- Anderson, K.D.; Panayotatos, N.; Corcoran, T.L.; Lindsay, R.M.; Wiegand, S.J. Ciliary neurotrophic factor protects striatal output neurons in an animal model of Huntington disease. Proc. Natl. Acad. Sci. USA 1996, 93, 7346–7351. [Google Scholar] [CrossRef] [PubMed]
- Mallet, N.; Delgado, L.; Chazalon, M.; Miguelez, C.; Baufreton, J. Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping Out of the Striatum. Cells 2019, 8, 1005. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, K.; Laurent, B. Dysfunction of the human memory systems: Role of the dopaminergic transmission. Curr. Opin. Neurol. 2003, 16, S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Suarez, L.M.; Alberquilla, S.; García-Montes, J.R.; Moratalla, R. Differential Synaptic Remodeling by Dopamine in Direct and Indirect Striatal Projection Neurons in Pitx3−/− Mice, a Genetic Model of Parkinson’s Disease. J. Neurosci. 2018, 38, 3619–3630. [Google Scholar] [CrossRef] [PubMed]
- Fieblinger, T.; Graves, S.M.; Sebel, L.E.; Alcacer, C.; Plotkin, J.L.; Gertler, T.S.; Chan, C.S.; Heiman, M.; Greengard, P.; Cenci, M.A.; et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat. Commun. 2014, 5, 5316. [Google Scholar] [CrossRef] [PubMed]
- Gerfen, C.R. Segregation of D1 and D2 dopamine receptors in the striatal direct and indirect pathways: An historical perspective. Front. Synaptic Neurosci. 2023, 14, 1002960. [Google Scholar] [CrossRef]
- Aubert, I.; Ghorayeb, I.; Normand, E.; Bloch, B. Phenotypical characterization of the neurons expressing the D1 and D2 dopamine receptors in the monkey striatum. J. Comp. Neurol. 2000, 418, 22–32. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Song, W.-J.; Yan, Z. Coordinated Expression of Dopamine Receptors in Neostriatal Medium Spiny Neurons. J. Neurosci. 1996, 16, 6579–6591. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, A.; Murphy, A.J.; Ozkan, A.; Davis, P.; Hatch, J.; Kirchner, R.; Macklis, J.D. Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature 2019, 565, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.A.; Adkins, D.L. Motor System Reorganization After Stroke: Stimulating and Training Toward Perfection. Physiology 2015, 30, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, M.; Dirren, E.; Bourgeois, A.; Kleinschmidt, A.; Richiardi, J.; Carrera, E. Periinfarct rewiring supports recovery after primary motor cortex stroke. J. Cereb. Blood Flow Metab. 2021, 41, 2174–2184. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayon-Olivas, M.; Wolf, D.; Andreska, T.; Granado, N.; Lüningschrör, P.; Ip, C.W.; Moratalla, R.; Sendtner, M. Dopaminergic Input Regulates the Sensitivity of Indirect Pathway Striatal Spiny Neurons to Brain-Derived Neurotrophic Factor. Biology 2023, 12, 1360. https://doi.org/10.3390/biology12101360
Ayon-Olivas M, Wolf D, Andreska T, Granado N, Lüningschrör P, Ip CW, Moratalla R, Sendtner M. Dopaminergic Input Regulates the Sensitivity of Indirect Pathway Striatal Spiny Neurons to Brain-Derived Neurotrophic Factor. Biology. 2023; 12(10):1360. https://doi.org/10.3390/biology12101360
Chicago/Turabian StyleAyon-Olivas, Maurilyn, Daniel Wolf, Thomas Andreska, Noelia Granado, Patrick Lüningschrör, Chi Wang Ip, Rosario Moratalla, and Michael Sendtner. 2023. "Dopaminergic Input Regulates the Sensitivity of Indirect Pathway Striatal Spiny Neurons to Brain-Derived Neurotrophic Factor" Biology 12, no. 10: 1360. https://doi.org/10.3390/biology12101360
APA StyleAyon-Olivas, M., Wolf, D., Andreska, T., Granado, N., Lüningschrör, P., Ip, C. W., Moratalla, R., & Sendtner, M. (2023). Dopaminergic Input Regulates the Sensitivity of Indirect Pathway Striatal Spiny Neurons to Brain-Derived Neurotrophic Factor. Biology, 12(10), 1360. https://doi.org/10.3390/biology12101360