
Multielectrode Arrays for Functional Phenotyping of Neurons from Induced Pluripotent Stem Cell Models of Neurodevelopmental Disorders

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Historical Perspectives
2.1. Developing Long-Term Culture Methods for Nervous Tissue
2.2. Early MEA Technology
2.3. Qualitative Descriptions of Network Activity
2.4. Quantitative Descriptions of Network Activity
3. Generating Neurons and Glia for MEA Phenotyping Assays
3.1. Neuronal Differentiation Methods for Generating Functional Neurons
3.2. iPSC-Derived Astrocytes and Genotype-Matched Co-Cultures
4. Challenges with Current Approaches to MEA Phenotyping
Appropriate Selection of Phenotyping Metrics
5. Expanding the MEA Analysis Toolkit in iPSC Disease Modeling
5.1. Computational Modeling Approaches and Analysis Methods
5.2. Spike Sorting for Improved Firing Rate Statistics
6. Standardizing MEA Data Reporting
7. Conclusions and Recommendations
7.1. Considerations for Experimental Design
7.2. Considerations for MEA Data Analysis
7.3. Considerations for Data Reporting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomas, C.A.; Springer, P.A.; Loeb, G.E.; Berwald-Netter, Y.; Okun, L.M. A miniature microelectrode array to monitor the bioelectric activity of cultured cells. Exp. Cell Res. 1972, 74, 61–66. [Google Scholar] [CrossRef]
- Shtark, M.B.; Voskresenskaya, L.V.; Olenev, S.N. A Multtielectrode Perfusion Chamber For Tissue Culture Research. J. Phys. A Math. Theor. 1974, 44, 1689–1699. [Google Scholar] [CrossRef]
- Gross, G.W.; Rieske, E.; Kreutzberg, G.W.; Meyer, A. A new fixed-array multi-microelectrode system designed for long-term monitoring of extracellular single unit neuronal activity in vitro. Neurosci. Lett. 1977, 6, 101–105. [Google Scholar] [CrossRef]
- Varon, S.S. In Vitro Study of Developing Neural Tissue and Cells: Past and Prospective Contributions. In The Neurosciences, Second Study Program; Schmitt, F.O., Ed.; The Rockefeller University Press: New York, NY, USA, 1970; pp. 83–99. ISBN 0874700140. [Google Scholar]
- Varon, S.S. The Investigation of Neural Development by Experimental In Vitro Techniques. In Cellular Aspects of Neural Growth and Differentiation; Pease, D.C., Ed.; University of California Press: Los Angeles, CA, USA, 1971; pp. 223–252. ISBN 9780520331884. [Google Scholar]
- Hild, W.; Ichiji, T. Morphological and Physiological Properties of Neurons and Glial Cells in Tissue Culture. J. Neurophys. 1962, 25, 227–304. [Google Scholar] [CrossRef]
- Crain, S.M. Neurophysiologic Studies in Tissue Culture; Raven Press: New York, NY, USA, 1976; ISBN 0890040486. [Google Scholar]
- Crain, S.M.; Bornstein, M.B. Organotypic bioelectric activity in cultured reaggregates of dissociated rodent brain cells. Science 1972, 176, 182–184. [Google Scholar] [CrossRef]
- Baer, S.C.; Crain, S.M. Magnetically coupled micromanipulator for use within a sealed chamber. J. Appl. Physiol. 1971, 31, 926–929. [Google Scholar] [CrossRef]
- Gross, G.W. Simultaneous Single Unit Recording in vitro with a Photoetched Laser Deinsulated Gold Multimicroelectrode Surface. IEEE Trans. Biomed. Eng. 1979, 5, 273–279. [Google Scholar] [CrossRef]
- Gross, G.W.; Lucas, J.H. Long-term monitoring of spontaneous single unit activity from neuronal monolayer networks cultured on photoetched multielectrode surfaces. J. Electrophysiol. Tech. 1982, 9, 55–67. [Google Scholar]
- Droge, M.H.; Gross, G.W.; Hightower, M.H.; Czisny, L.E. Multielectrode analysis of coordinated, multisite, rhythmic bursting in cultured CNS monolayer networks. J. Neurosci. 1986, 6, 1583–1592. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.W.; Kowalski, J.M. Experimental and Theoretical Analysis of Random Nerve Cells Network Dynamics. In Neural Networks: Concepts, Applications, and Implementations, Volume 4; Paolo Antognetti, V.M., Ed.; Prentice Hall: Englewood, NJ, USA, 1991; pp. 47–110. ISBN 0136125166. [Google Scholar]
- Gross, G.W.; Rhoades, B.K.; Kowalski, J.M. Dynamics of Burst Patterns Generated by Monolayer Networks in Culture. In Neurobionics; Bothe, H.-W., Samii, M., Eckmiller, R., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; pp. 89–121. ISBN 0444899588. [Google Scholar]
- Gross, G.W. Internal Dynamics of Randomized Mammalian Neuronal Networks in Culture. In Enabling Technologies for Cultured Neural Networks; Stenger, D.A., McKenna, T.M., Eds.; Academic Press: San Diego, CA, USA, 1994; pp. 227–317. ISBN 9780126659702. [Google Scholar]
- Kowalski, J.M.; Albert, G.L.; Rhoades, B.K.; Gross, G.W. Neuronal networks with spontaneous, correlated bursting activity: Theory and simulations. Neural Netw. 1992, 5, 805–822. [Google Scholar] [CrossRef]
- Gramowski, A.; Jügelt, K.; Weiss, D.G.; Gross, G.W. Substance identification by quantitative characterization of oscillatory activity in murine spinal cord networks on microelectrode arrays. Eur. J. Neurosci. 2004, 19, 2815–2825. [Google Scholar] [CrossRef]
- Shafer, T.J. Application of Microelectrode Array Approaches to Neurotoxicity Testing and Screening BT—In Vitro. In Neuronal Networks: From Culturing Methods to Neuro-Technological Applications; Chiappalone, M., Pasquale, V., Frega, M., Eds.; Springer International Publishing: Cham, Swiztherland, 2019; pp. 275–297. ISBN 978-3-030-11135-9. [Google Scholar]
- Johnstone, A.F.M.; Gross, G.W.; Weiss, D.G.; Schroeder, O.H.U.; Gramowski, A.; Shafer, T.J. Microelectrode arrays: A physiologically based neurotoxicity testing platform for the 21st century. Neurotoxicology 2010, 31, 331–350. [Google Scholar] [CrossRef]
- Pelkonen, A.; Pistono, C.; Klecki, P.; Gómez-Budia, M.; Dougalis, A.; Konttinen, H.; Stanová, I.; Fagerlund, I.; Leinonen, V.; Korhonen, P.; et al. Functional Characterization of Human Pluripotent Stem Cell-Derived Models of the Brain with Microelectrode Arrays. Cells 2022, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.P.; Stice, S.L. Electrophysiological Analysis of Brain Organoids: Current Approaches and Advancements. Front. Neurosci. 2021, 14, 622137. [Google Scholar] [CrossRef] [PubMed]
- Russo, F.B.; Freitas, B.C.; Pignatari, G.C.; Fernandes, I.R.; Sebat, J.; Muotri, A.R.; Beltrão-Braga, P.C.B. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol. Psychiatry 2018, 83, 569–578. [Google Scholar] [CrossRef]
- Deneault, E.; Faheem, M.; White, S.H.; Rodrigues, D.C.; Sun, S.; Wei, W.; Piekna, A.; Thompson, T.; Howe, J.L.; Chalil, L.; et al. CNTN5-/+or EHMT2-/+human iPSC-derived neurons from individuals with autism develop hyperactive neuronal networks. Elife 2019, 8, e40092. [Google Scholar] [CrossRef]
- Deneault, E.; White, S.H.; Rodrigues, D.C.; Ross, P.J.; Faheem, M.; Zaslavsky, K.; Wang, Z.; Alexandrova, R.; Pellecchia, G.; Wei, W.; et al. Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Rep. 2018, 11, 1211–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, M.C.; Belinson, H.; Tian, Y.; Freitas, B.C.; Fu, C.; Vadodaria, K.C.; Beltrao-Braga, P.C.; Trujillo, C.A.; Mendes, A.P.D.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2017, 22, 820–835. [Google Scholar] [CrossRef]
- DeRosa, B.A.; El Hokayem, J.; Artimovich, E.; Garcia-Serje, C.; Phillips, A.W.; Van Booven, D.; Nestor, J.E.; Wang, L.; Cuccaro, M.L.; Vance, J.M.; et al. Convergent Pathways in Idiopathic Autism Revealed by Time Course Transcriptomic Analysis of Patient-Derived Neurons. Sci. Rep. 2018, 8, 8423. [Google Scholar] [CrossRef] [Green Version]
- Amatya, D.N.; Linker, S.B.; Mendes, A.P.D.; Santos, R.; Erikson, G.; Shokhirev, M.N.; Zhou, Y.; Sharpee, T.; Gage, F.H.; Marchetto, M.C.; et al. Dynamical Electrical Complexity Is Reduced during Neuronal Differentiation in Autism Spectrum Disorder. Stem Cell Rep. 2019, 13, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Winden, X.K.D.; Sundberg, X.M.; Yang, C.; Wafa, X.S.M.A.; Dwyer, S.; Chen, P.; Buttermore, X.E.D.; Sahin, X.M. Biallelic Mutations in TSC2 Lead to Abnormalities Associated with Cortical Tubers in Human iPSC-Derived Neurons. J. Neurosci. 2019, 39, 9294–9305. [Google Scholar] [CrossRef] [PubMed]
- Nadadhur, A.G.; Alsaqati, M.; Gasparotto, L.; Cornelissen-Steijger, P.; van Hugte, E.; Dooves, S.; Harwood, A.J.; Heine, V.M. Neuron-Glia Interactions Increase Neuronal Phenotypes in Tuberous Sclerosis Complex Patient iPSC-Derived Models. Stem Cell Rep. 2019, 12, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quraishi, I.H.; Stern, S.; Mangan, K.P.; Zhang, Y.; Ali, S.R.; Mercier, M.R.; Marchetto, M.C.; McLachlan, M.J.; Jones, E.M.; Gage, F.H.; et al. An Epilepsy-Associated KCNT1 Mutation Enhances Excitability of Human iPSC-Derived Neurons by Increasing Slack KNa Currents. J. Neurosci. 2019, 39, 7438–7449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graef, J.D.; Wu, H.; Ng, C.; Sun, C.; Villegas, V.; Qadir, D.; Jesseman, K.; Warren, S.T.; Jaenisch, R.; Cacace, A.; et al. Partial FMRP expression is sufficient to normalize neuronal hyperactivity in Fragile X neurons. Eur. J. Neurosci. 2020, 51, 2143–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utami, K.H.; Skotte, N.H.; Colaço, A.R.; Amirah, N.; Mohammad, B.; Sim, B.; Yeo, X.Y.; Bae, H.; Garcia-miralles, M.; Radulescu, C.I.; et al. Archival Report Integrative Analysis Identi fi es Key Molecular Signatures Underlying Neurodevelopmental De fi cits in Fragile X Syndrome. Biol. Psychiatry 2020, 88, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Kathuria, A.; Lopez-lengowski, K.; Watmuff, B.; Mcphie, D.; Cohen, B.M. Synaptic de fi cits in iPSC-derived cortical interneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Transl. Psychiatry 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Shtrahman, M.; Jin, X.; Gage, F.H.; Sarkar, A.; Mei, A.; Paquola, A.C.M.; Stern, S.; Bardy, C.; Klug, J.R.; Kim, S. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell 2018, 22, 684–697.e9. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Ishikawa, M.; Fujimori, K.; Maeda, T.; Kushima, I.; Arioka, Y.; Mori, D.; Nakatake, Y.; Yamagata, B.; Nio, S.; et al. In Vitro Modeling of the Bipolar Disorder and Schizophrenia Using Patient-Derived Induced Pluripotent Stem Cells with Copy Number Variations of PCDH15 and RELN. eNeuro 2019, 6, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Mesci, P.; Carromeu, C.; McClatchy, D.R.; Schiapparelli, L.; Yates, J.R.; Muotri, A.R.; Cline, H.T. Exosomes regulate neurogenesis and circuit assembly. Proc. Natl. Acad. Sci. USA 2019, 116, 16086–16094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frega, M.; Linda, K.; Keller, J.M.; Gümüş-Akay, G.; Mossink, B.; van Rhijn, J.-R.; Negwer, M.; Klein Gunnewiek, T.; Foreman, K.; Kompier, N.; et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat. Commun. 2019, 10, 4928. [Google Scholar] [CrossRef] [Green Version]
- Mossink, B.; Verboven, A.H.A.; van Hugte, E.J.H.; Klein Gunnewiek, T.M.; Parodi, G.; Linda, K.; Schoenmaker, C.; Kleefstra, T.; Kozicz, T.; van Bokhoven, H.; et al. Human neuronal networks on micro-electrode arrays are a highly robust tool to study disease-specific genotype-phenotype correlations in vitro. Stem Cell Rep. 2021, 16, 2182–2196. [Google Scholar] [CrossRef] [PubMed]
- Alsaqati, M.; Heine, V.M.; Harwood, A.J. Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol. Autism 2020, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global Transcriptional and Translational Repression in Human-Embryonic-Stem-Cell-Derived Rett Syndrome Neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundberg, M.; Pinson, H.; Smith, R.S.; Winden, K.D.; Venugopal, P.; Tai, D.J.C.; Gusella, J.F.; Talkowski, M.E.; Walsh, C.A.; Tegmark, M.; et al. 16p11.2 deletion is associated with hyperactivation of human iPSC-derived dopaminergic neuron networks and is rescued by RHOA inhibition in vitro. Nat. Commun. 2021, 12, 2897. [Google Scholar] [CrossRef]
- Kim, D.-S.; Ross, P.J.; Zaslavsky, K.; Ellis, J. Optimizing neuronal differentiation from induced pluripotent stem cells to model ASD. Front. Cell. Neurosci. 2014, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Zhang, W.-B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef]
- Djuric, U.; Cheung, A.Y.L.; Zhang, W.; Mok, R.S.; Lai, W.; Piekna, A.; Hendry, J.A.; Ross, P.J.; Pasceri, P.; Kim, D.-S.; et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol. Dis. 2015, 76, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pak, C.H.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.-H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain. Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Penzes, P. Common Mechanisms of Excitatory and Inhibitory Imbalance in Schizophrenia and Autism Spectrum Disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef]
- Mossink, B.; van Rhijn, J.-R.; Wang, S.; Linda, K.; Vitale, M.R.; Zöller, J.E.M.; van Hugte, E.J.H.; Bak, J.; Verboven, A.H.A.; Selten, M.; et al. Cadherin-13 is a critical regulator of GABAergic modulation in human stem-cell-derived neuronal networks. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Barres, B.A. Synaptic efficacy enhanced by glial cells in vitro. Science 1997, 277, 1684–1687. [Google Scholar] [CrossRef] [Green Version]
- Odawara, A.; Saitoh, Y.; Alhebshi, A.H.; Gotoh, M.; Suzuki, I. Long-term electrophysiological activity and pharmacological response of a human induced pluripotent stem cell-derived neuron and astrocyte co-culture. Biochem. Biophys. Res. Commun. 2014, 443, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Barres, B.A. The Mystery and Magic of Glia: A Perspective on Their Roles in Health and Disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, S.A.; Barres, B.A. Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr. Opin. Neurobiol. 2014, 27, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbar, L.; Jain, T.; Zimmer, M.; Kruglikov, I.; Sadick, J.S.; Wang, M.; Kalpana, K.; Rose, I.V.L.; Burstein, S.R.; Rusielewicz, T.; et al. CD49f Is a Novel Marker of Functional and Reactive Human iPSC-Derived Astrocytes. Neuron 2020, 107, 436–453. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Bartels, T.; Holmqvist, S.; Kleshchevnikov, V.; Martirosyan, A.; Polioudakis, D.; Ben Haim, L.; Young, A.M.H.; Batiuk, M.Y.; Prakash, K.; et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 2020, 23, 500–509. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [Green Version]
- Taga, A.; Dastgheyb, R.; Habela, C.; Joseph, J.; Richard, J.P.; Gross, S.K.; Lauria, G.; Lee, G.; Haughey, N.; Maragakis, N.J. Role of Human-Induced Pluripotent Stem Cell-Derived Spinal Cord Astrocytes in the Functional Maturation of Motor Neurons in a Multielectrode Array System. Stem Cells Transl. Med. 2019, 8, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Roybon, L.; Lamas, N.J.; Garcia-Diaz, A.; Yang, E.J.; Sattler, R.; Jackson-Lewis, V.; Kim, Y.A.; Kachel, C.A.; Rothstein, J.D.; Przedborski, S.; et al. Human Stem Cell-Derived Spinal Cord Astrocytes with Defined Mature or Reactive Phenotypes. Cell Rep. 2013, 4, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Szczesna, K.; Ochalek, A.; Kobolák, J.; Varga, E.; Nemes, C.; Chandrasekaran, A.; Rasmussen, M.; Cirera, S.; Hyttel, P.; et al. Neurosphere based differentiation of human IPSC improves astrocyte differentiation. Stem Cells Int. 2016, 2016, 4937689. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Paşca, S.P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790.e6. [Google Scholar] [CrossRef] [PubMed]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R.; Rowitch, D.H. Evolving concepts of gliogenesis: A look way back and ahead to the next 25 years. Neuron 2013, 80, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.C.; Zhong, X.; Mohamed, A.; Li, R.; Liu, Y.; Dong, Q.; Ananiev, G.E.; Choongmok, J.C.; Lin, B.R.; Lu, J.; et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wildtype neurons. Hum. Mol. Genet. 2014, 23, 2968–2980. [Google Scholar] [CrossRef]
- Krencik, R.; Weick, J.P.; Liu, Y.; Zhang, Z.J.; Zhang, S.C. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat. Biotechnol. 2011, 29, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Hedegaard, A.; Monzón-Sandoval, J.; Newey, S.E.; Whiteley, E.S.; Webber, C.; Akerman, C.J. Pro-maturational Effects of Human iPSC-Derived Cortical Astrocytes upon iPSC-Derived Cortical Neurons. Stem Cell Rep. 2020, 15, 38–51. [Google Scholar] [CrossRef]
- Gupta, K.; Patani, R.; Baxter, P.; Serio, A.; Story, D.; Tsujita, T.; Hayes, J.D.; Pedersen, R.A.; Hardingham, G.E.; Chandran, S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 2012, 19, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Shaltouki, A.; Peng, J.; Liu, Q.; Rao, M.S.; Zeng, X. Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells 2013, 31, 941–952. [Google Scholar] [CrossRef]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [Green Version]
- TCW, J.; Wang, M.; Pimenova, A.A.; Bowles, K.R.; Hartley, B.J.; Lacin, E.; Machlovi, S.I.; Abdelaal, R.; Karch, C.M.; Phatnani, H.; et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 600–614. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.; Vadodaria, K.C.; Jaeger, B.N.; Mei, A.; Lefcochilos-Fogelquist, S.; Mendes, A.P.D.; Erikson, G.; Shokhirev, M.; Randolph-Moore, L.; Fredlender, C.; et al. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 1757–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Tao, Y.; Bradley, R.; Du, Z.; Tao, Y.; Kong, L.; Dong, Y.; Jones, J.; Yan, Y.; Harder, C.R.K.; et al. Fast Generation of Functional Subtype Astrocytes from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 11, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.S.; Lee, C.O.; Oh, M.; Cha, D.; Kim, W.K.; Oh, K.J.; Bae, K.H.; Lee, S.C.; Han, B.S. Rapid differentiation of astrocytes from human embryonic stem cells. Neurosci. Lett. 2020, 716, 134681. [Google Scholar] [CrossRef] [PubMed]
- Caiazzo, M.; Giannelli, S.; Valente, P.; Lignani, G.; Carissimo, A.; Sessa, A.; Colasante, G.; Bartolomeo, R.; Massimino, L.; Ferroni, S.; et al. Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Rep. 2015, 4, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef]
- Tchieu, J.; Calder, E.L.; Guttikonda, S.R.; Gutzwiller, E.M.; Aromolaran, K.A.; Steinbeck, J.A.; Goldstein, P.A.; Studer, L. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat. Biotechnol. 2019, 37, 267–275. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Engle, S.J.; Blaha, L.; Kleiman, R.J. Best Practices for Translational Disease Modeling Using Human iPSC-Derived Neurons. Neuron 2018, 100, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Fardet, T.; Ballandras, M.; Bottani, S.; Métens, S.; Monceau, P. Understanding the Generation of Network Bursts by Adaptive Oscillatory Neurons. Front. Neurosci. 2018, 12, 41. [Google Scholar] [CrossRef]
- Penn, Y.; Segal, M.; Moses, E. Network synchronization in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2016, 113, 3341–3346. [Google Scholar] [CrossRef] [Green Version]
- Suresh, J.; Radojicic, M.; Pesce, L.L.; Bhansali, A.; Wang, J.; Tryba, A.K.; Marks, J.D.; van Drongelen, W. Network burst activity in hippocampal neuronal cultures: The role of synaptic and intrinsic currents. J. Neurophysiol. 2016, 115, 3073–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, E.; Robinson, H.P.C.; Kawana, A. The mechanisms of generation and propagation of synchronized bursting in developing networks of cortical neurons. J. Neurosci. 1995, 15, 6834–6845. [Google Scholar] [CrossRef] [PubMed]
- Muramoto, K.; Ichikawa, M.; Kawahara, M.; Kobayashi, K.; Kuroda, Y. Frequency of synchronous oscillations of neuronal activity increases during development and is correlated to the number of synapses in cultured cortical neuron networks. Neurosci. Lett. 1993, 163, 163–165. [Google Scholar] [CrossRef]
- Bosl, W.; Tierney, A.; Tager-Flusberg, H.; Nelson, C. EEG complexity as a biomarker for autism spectrum disorder risk. BMC Med. 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catarino, A.; Churches, O.; Baron-Cohen, S.; Andrade, A.; Ring, H. Atypical EEG complexity in autism spectrum conditions: A multiscale entropy analysis. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2011, 122, 2375–2383. [Google Scholar] [CrossRef]
- Lipsitz, L.A. Physiological Complexity, Aging, and the Path to Frailty. Sci. Aging Knowl. Environ. 2004, 2004, pe16. [Google Scholar] [CrossRef]
- Costa, M.; Goldberger, A.L.; Peng, C.-K. Multiscale entropy analysis of biological signals. Phys. Rev. E 2005, 71, 21906. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological reversal of synaptic and network pathology in human MECP2-KO neurons and cortical organoids. EMBO Mol. Med. 2021, 13, e12523. [Google Scholar] [CrossRef]
- Mok, R.S.; Zhang, W.; Sheikh, T.I.; Pradeepan, K.; Fernandes, I.R.; DeJong, L.C.; Benigno, G.; Hildebrandt, M.R.; Mufteev, M.; Rodrigues, D.C.; et al. Wide spectrum of neuronal and network phenotypes in human stem cell-derived excitatory neurons with Rett syndrome-associated MECP2 mutations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bateup, H.; Denefrio, C.; Johnson, C.; Saulnier, J.; Sabatini, B. Temporal dynamics of a homeostatic pathway controlling neural network activity. Front. Mol. Neurosci. 2013, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Vogt, N. Machine learning in neuroscience. Nat. Methods 2018, 15, 33. [Google Scholar] [CrossRef]
- Poli, D.; Pastore, V.P.; Massobrio, P. Functional connectivity in in vitro neuronal assemblies. Front. Neural Circuits 2015, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, M.C.; Downes, J.H.; Xydas, D.; Hammond, M.W.; Becerra, V.M.; Warwick, K.; Whalley, B.J.; Nasuto, S.J. Multiscale Evolving Complex Network Model of Functional Connectivity in Neuronal Cultures. IEEE Trans. Biomed. Eng. 2012, 59, 30–34. [Google Scholar] [CrossRef]
- Massobrio, P.; Tessadori, J.; Chiappalone, M.; Ghirardi, M. In Vitro Studies of Neuronal Networks and Synaptic Plasticity in Invertebrates and in Mammals Using Multielectrode Arrays. Neural Plast. 2015, 2015, 196195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenaar, D.A.; Pine, J.; Potter, S.M. Searching for plasticity in dissociated cortical cultures on multi-electrode arrays. J. Negat. Results Biomed. 2006, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Feber, J.; Stegenga, J.; Rutten, W.L.C. The Effect of Slow Electrical Stimuli to Achieve Learning in Cultured Networks of Rat Cortical Neurons. PLoS ONE 2010, 5, e8871. [Google Scholar] [CrossRef] [PubMed]
- Le Feber, J.; Witteveen, T.; van Veenendaal, T.M.; Dijkstra, J. Repeated stimulation of cultured networks of rat cortical neurons induces parallel memory traces. Learn. Mem. 2015, 22, 594–603. [Google Scholar] [CrossRef] [Green Version]
- Johnson, H.A.; Goel, A.; Buonomano, D. V Neural dynamics of in vitro cortical networks reflects experienced temporal patterns. Nat. Neurosci. 2010, 13, 917–919. [Google Scholar] [CrossRef] [Green Version]
- Jimbo, Y.; Robinson, H.P.; Kawana, A. Strengthening of synchronized activity by tetanic stimulation in cortical cultures: Application of planar electrode arrays. IEEE Trans. Biomed. Eng. 1998, 45, 1297–1304. [Google Scholar] [CrossRef]
- Chiappalone, M.; Massobrio, P.; Martinoia, S. Network plasticity in cortical assemblies. Eur. J. Neurosci. 2008, 28, 221–237. [Google Scholar] [CrossRef]
- Biffi, E.; Regalia, G.; Menegon, A.; Ferrigno, G.; Pedrocchi, A. The Influence of Neuronal Density and Maturation on Network Activity of Hippocampal Cell Cultures: A Methodological Study. PLoS ONE 2013, 8, e83899. [Google Scholar] [CrossRef] [PubMed]
- Wagenaar, D.A.; Pine, J.; Potter, S.M. An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci. 2006, 7, 11. [Google Scholar] [CrossRef]
- Ivenshitz, M.; Segal, M. Neuronal Density Determines Network Connectivity and Spontaneous Activity in Cultured Hippocampus. J. Neurophysiol. 2010, 104, 1052–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroga, R.Q. Spike sorting. Curr. Biol. 2012, 22, R45–R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, F.; Black, M.J.; Vargas-Irwin, C.; Fellows, M.; Donoghue, J.P. On the variability of manual spike sorting. IEEE Trans. Biomed. Eng. 2004, 51, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Buccino, A.P.; Hurwitz, C.L.; Garcia, S.; Magland, J.; Siegle, J.H.; Hurwitz, R.; Hennig, M.H. SpikeInterface, a unified framework for spike sorting. Elife 2020, 9, e61834. [Google Scholar] [CrossRef]
- Magland, J.; Jun, J.J.; Lovero, E.; Morley, A.J.; Hurwitz, C.L.; Buccino, A.P.; Garcia, S.; Barnett, A.H. SpikeForest, reproducible web-facing ground-truth validation of automated neural spike sorters. Elife 2020, 9, e55167. [Google Scholar] [CrossRef]
- Negri, J.; Menon, V.; Young-Pearse, T.L. Assessment of spontaneous neuronal activity In vitro using multi-well multi-electrode arrays: Implications for assay development. eNeuro 2020, 7, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.D.; Henze, D.A.; Csicsvari, J.; Hirase, H.; Buzsáki, G. Accuracy of tetrode spike separation as determined by simultaneous intracellular and extracellular measurements. J. Neurophysiol. 2000, 84, 401–414. [Google Scholar] [CrossRef]
- Cohen, M.R.; Kohn, A. Measuring and interpreting neuronal correlations. Nat. Neurosci. 2011, 14, 811–819. [Google Scholar] [CrossRef]
- Pazienti, A.; Grün, S. Robustness of the significance of spike synchrony with respect to sorting errors. J. Comput. Neurosci. 2006, 21, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.N.; Mehta, S.B.; Kleinfeld, D. Quality metrics to accompany spike sorting of extracellular signals. J. Neurosci. 2011, 31, 8699–8705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-C.; He, Z.; Ebert, S.; Schörnig, M.; Santel, M.; Nikolova, M.T.; Weigert, A.; Hevers, W.; Kasri, N.N.; Taverna, E.; et al. NGN2 induces diverse neuron types from human pluripotency. Stem Cell Rep. 2021, 16, 2118–2127. [Google Scholar] [CrossRef] [PubMed]

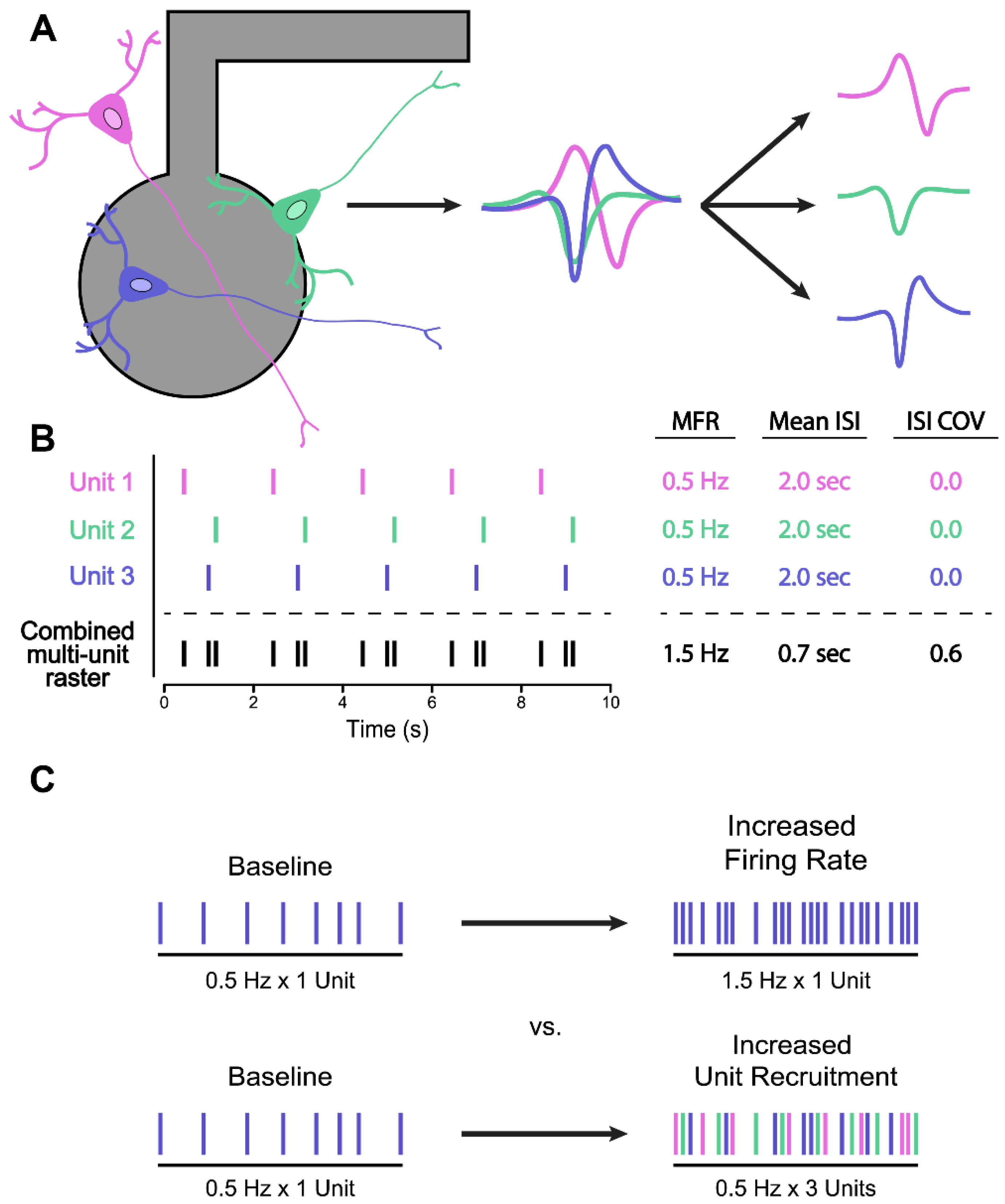
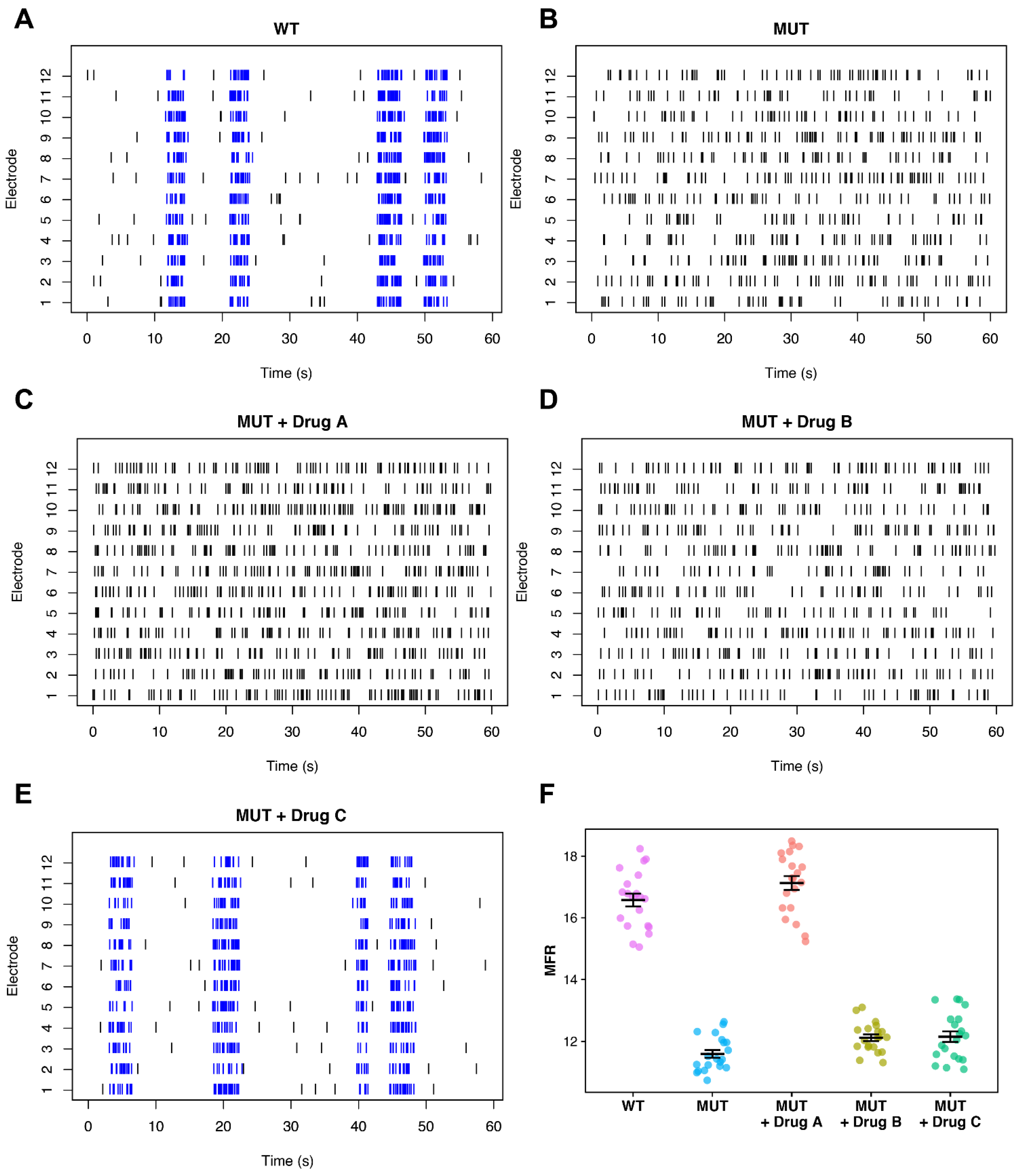

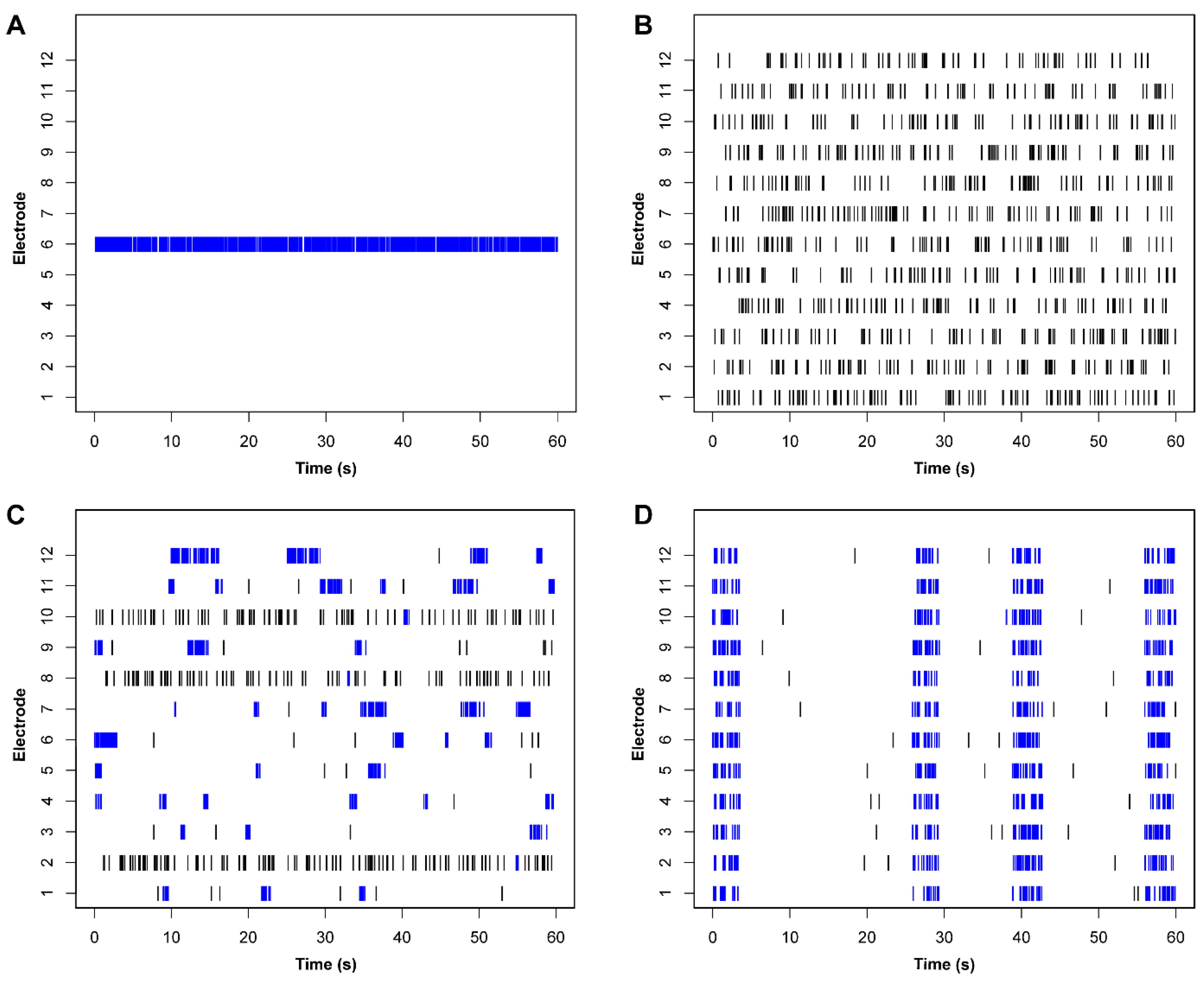{kind=link}
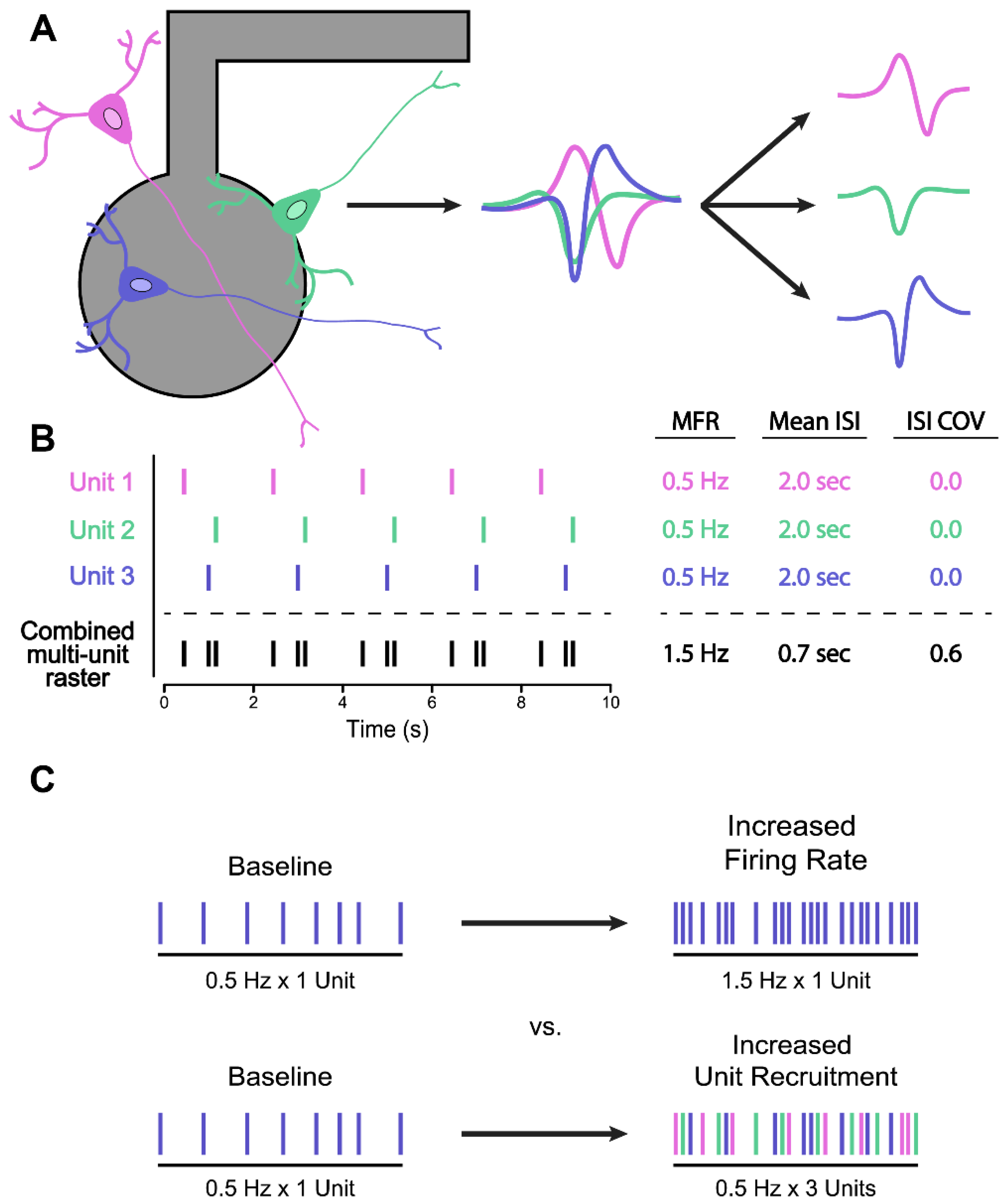{kind=link}
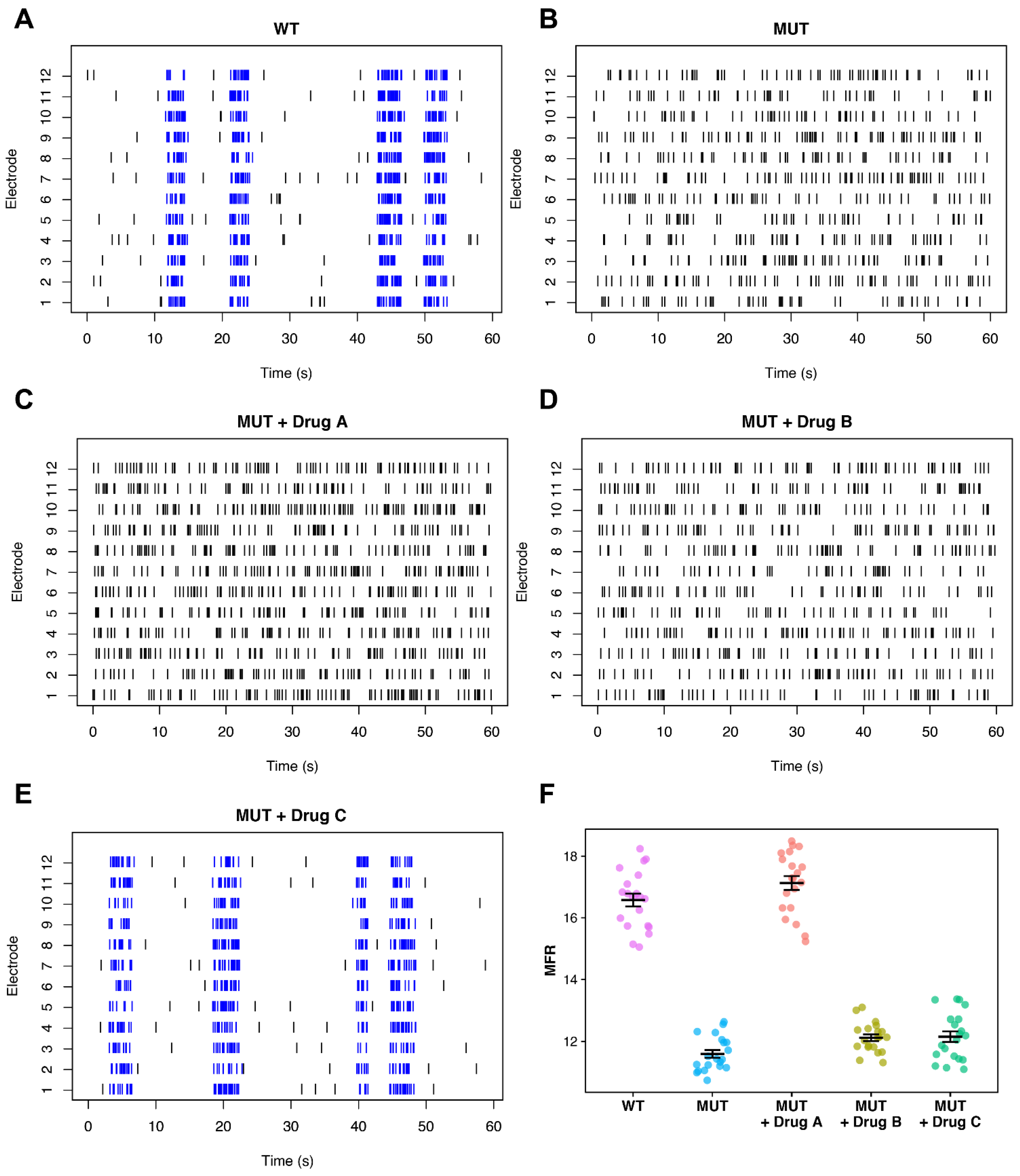{kind=link}
| Reference | Differentiation Type | Disease Model | Associated Mutations | System and Plate Format Used | Number of Electrodes | Reported Metrics | Recording Duration | Replicates per Line |
|---|---|---|---|---|---|---|---|---|
| Russo et al. (2018) [22] | Directed | ASD | SETD5, Idiopathic | Axion Biosystems 12-well | 64 | MFR | 3 min | 6 |
| Deneault et al. (2019) [23] | TF Programming (NGN2) | ASD | CTN5, EHMT2, DLGAP2, CAPRIN1, SET, GLI3, VIP, ANOS1, THRA, NRXN1, AGBL4 | Axion Biosystems 48-well | 16 | wMFR, network burst frequency | 5 min | 9–24 |
| Deneault et al. (2018) [24] | TF Programming (NGN2) | ASD | ATRX, AFF2, KCNQ2N SCN2AM ASTN2 | Axion Biosystems 48-well | 16 | MFR, burst frequency, network burst frequency | 5 min | 21–55 |
| Marchetto et al. (2017) [25] | Directed | ASD | - | Axion Biosystems 12-well | 64 | Number of spikes, network burst frequency | 10 min | 3 |
| DeRosa et al. (2018) [26] | Directed | ASD | - | Axion Biosystems 12-well | 64 | MFR | 10 min | 16 |
| Amatya et al. (2019) [27] | Directed | ASD | - | Axion Biosystems 96-well | 8 | Minimum embedding dimension, ISI COV | 10 min | 6 |
| Winden et al. (2019) [28] | TF Programming (NGN2) | TSC | TSC2 | Axion Biosystems 48-well | 16 | wMFR, synchrony index | - | 48 |
| Nadadhur et al. (2019) [29] | Directed | TSC | TSC1, TSC2 | Multi Channel Systems single well | 60 | MFR | 10 min | 6–8 |
| Quraishi et al. (2019) [30] | Cellular Dynamics (proprietary) | Epilepsy | KCNT1 | Axion Biosystems 48-well | 16 | MFR, Synchrony Index, burst rate, burst duration, burst intensity | 8 min | 24 |
| Graef et al. (2020) [31] | TF Programming (NGN2) | FXS | FMR1 | Axion Biosystems 48-well | 16 | wMFR | 5 min | 12–24 |
| Liu et al. (2018) [32] | Directed | FXS | FMR1 | Axion Biosystems 12-well | 64 | MFR | 5 min | 2–6 |
| Utami et al. (2020) [33] | Directed | FXS | FMR1 | Axion Biosystems 12-well | 64 | MFR, max firing rate, number of unresponsive | 5 min | 6 |
| Nageshappa et al. (2016) [34] | Directed | MECP2 duplication syndrome | MECP2 | MED64 single well | 64 | Network burst frequency | 5 min | 3 |
| Kathuria et al. (2019) [35] | Directed | SCZ | - | MED64 12-well | 16 | MFR | 1 min | 3 |
| Sarkar et al. (2018) [36] | Directed | SCZ | - | Axion Biosystems 96-well | 8 | Number of spikes, Synchrony Index, Burst Frequency, Network Burst Frequency | 10 min | 6 or 12 |
| Ishii et al. (2019) [37] | TF Programming (NGN2 or ASCL1 + DLX2) | SCZ and Bipolar | idiopathic, PDH15, RELN | Axion Biosystems 48-well | 16 | wMFR, GABA Sensitivity | 5 min | 4–6 |
| Sharma et al. (2019) [38] | Directed | Rett | MECP2 | Axion Biosystems 12-well | 64 | Network burst frequency | 5 min | 3 |
| Frega et al. (2019) [39] | TF Programming (NGN2) | Kleefstra syndrome | EHMT1 | Multi Channel Systems 24-well | 12 | MFR, burst frequency, burst duration, mean IBI, IBI CV, % spikes out of bursts | 20 min | 10–23 |
| Mossink et al. (2021) [40] | TF Programming (NGN2 or ASCL1 + forskolin) | ASD, ADHD | CHD13 | Multi Chanel Systems 24-well | 12 | Network burst duration, number of spikes per network burst | 10 min | 20–49 |
| Alsaqati et al. (2020) [41] | Directed | TSC | TSC2 | Axion Biosystems 24-well | 16 | MFR, network burst frequency, network burst duration, inter-network burst interval, burst frequency, connectivity correlation, % spikes outside network bursts, frequency distribution | - | 3–10 |
| Li et al. (2013) [42] | Directed | Rett | MECP2 | MED64 single well | 64 | MFR | 5 min | - |
| Sundberg et al. (2021) [43] | Directed | 16p11.2 CNV | 16p11.2 dup, 16p11.2 deletion | MaxWell Biosystems single well Axion Biosystems 48-well | 26,400 16 | MFR, fraction of synchronized sensors, burst frequency, burst duration, inter-burst interval, number of spikes per burst | 2 min 5 min | 4–7 6–16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCready, F.P.; Gordillo-Sampedro, S.; Pradeepan, K.; Martinez-Trujillo, J.; Ellis, J. Multielectrode Arrays for Functional Phenotyping of Neurons from Induced Pluripotent Stem Cell Models of Neurodevelopmental Disorders. Biology 2022, 11, 316. https://doi.org/10.3390/biology11020316
McCready FP, Gordillo-Sampedro S, Pradeepan K, Martinez-Trujillo J, Ellis J. Multielectrode Arrays for Functional Phenotyping of Neurons from Induced Pluripotent Stem Cell Models of Neurodevelopmental Disorders. Biology. 2022; 11(2):316. https://doi.org/10.3390/biology11020316
Chicago/Turabian StyleMcCready, Fraser P., Sara Gordillo-Sampedro, Kartik Pradeepan, Julio Martinez-Trujillo, and James Ellis. 2022. "Multielectrode Arrays for Functional Phenotyping of Neurons from Induced Pluripotent Stem Cell Models of Neurodevelopmental Disorders" Biology 11, no. 2: 316. https://doi.org/10.3390/biology11020316
APA StyleMcCready, F. P., Gordillo-Sampedro, S., Pradeepan, K., Martinez-Trujillo, J., & Ellis, J. (2022). Multielectrode Arrays for Functional Phenotyping of Neurons from Induced Pluripotent Stem Cell Models of Neurodevelopmental Disorders. Biology, 11(2), 316. https://doi.org/10.3390/biology11020316