
Abnormal Enhancement of Protein Disulfide Isomerase-like Activity of a Cyclic Diselenide Conjugated with a Basic Amino Acid by Inserting a Glycine Spacer


Abstract
:Simple Summary
Abstract
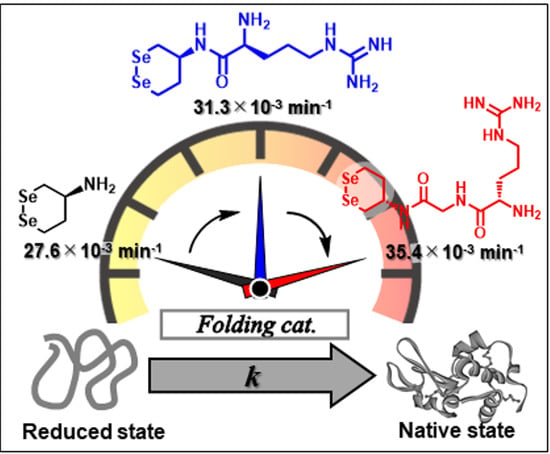
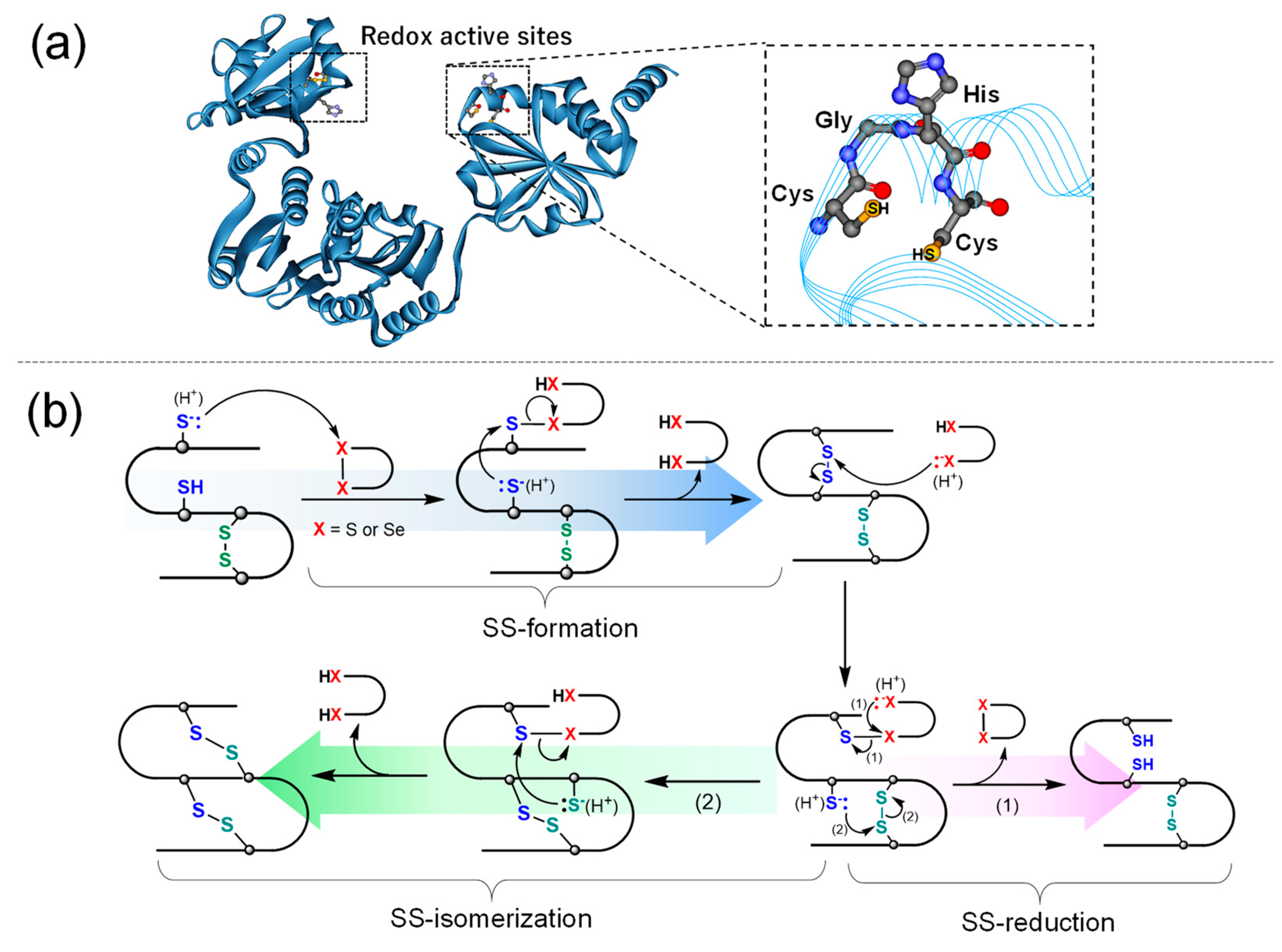
1. Introduction
2. Materials and Methods
2.1. General
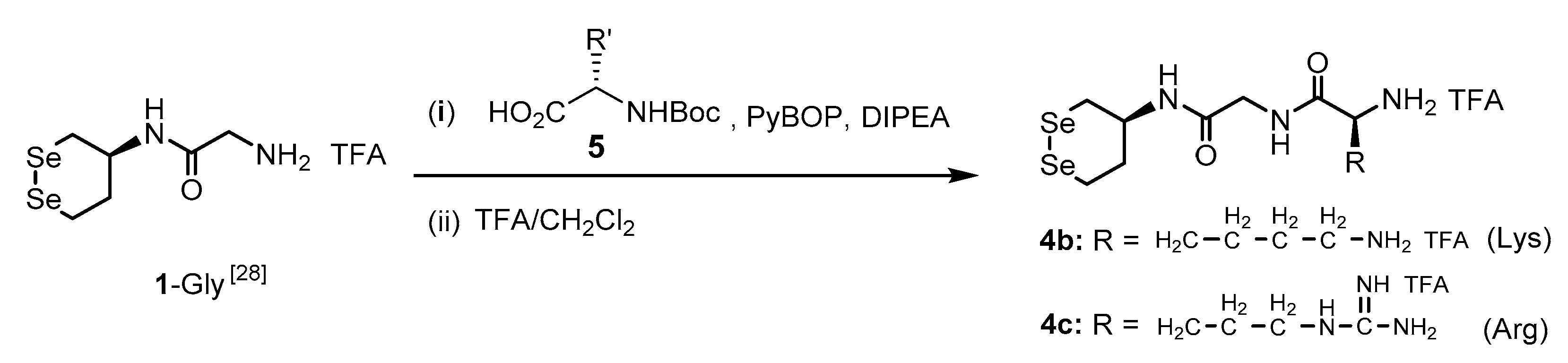
2.2. General Procedure for Synthesis of 4b and 4c
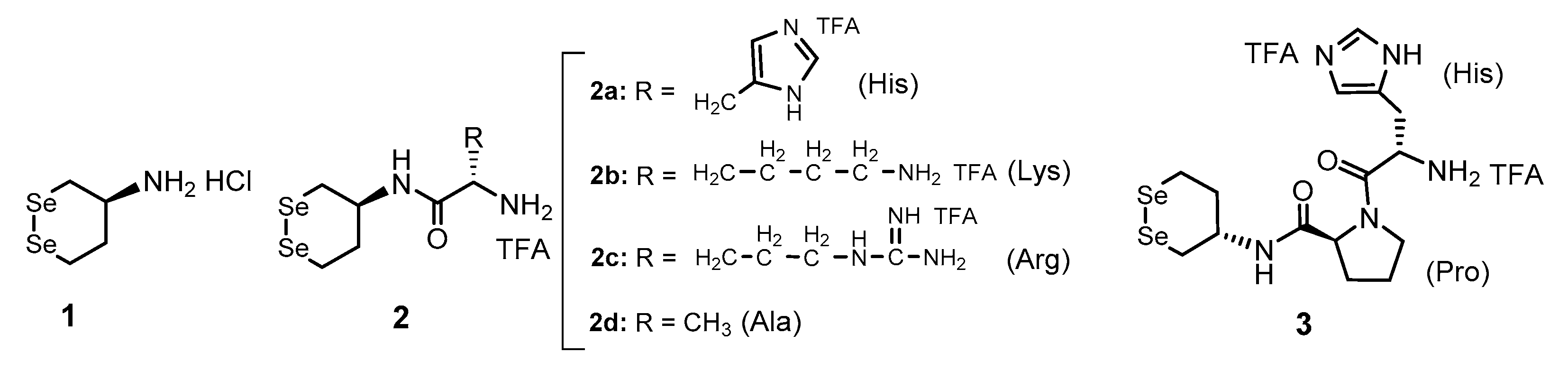
2.3. (. S)-N-(2-(((S)-1,2-Diselenan-4-Yl)Amino)-2-Oxoethyl)-2,6-Diaminohexanamide di-TFA Salt (4b)
2.4. (. S)-N-(2-(((S)-1,2-Diselenan-4-Yl)Amino)-2-Oxoethyl)-2-Amino-5-Guanidinopentanamide di-TFA Salt (4c)
2.5. Heat-Denaturation-Induced Aggregation of Hen Egg-White Lysozyme
3. Results and Discussion
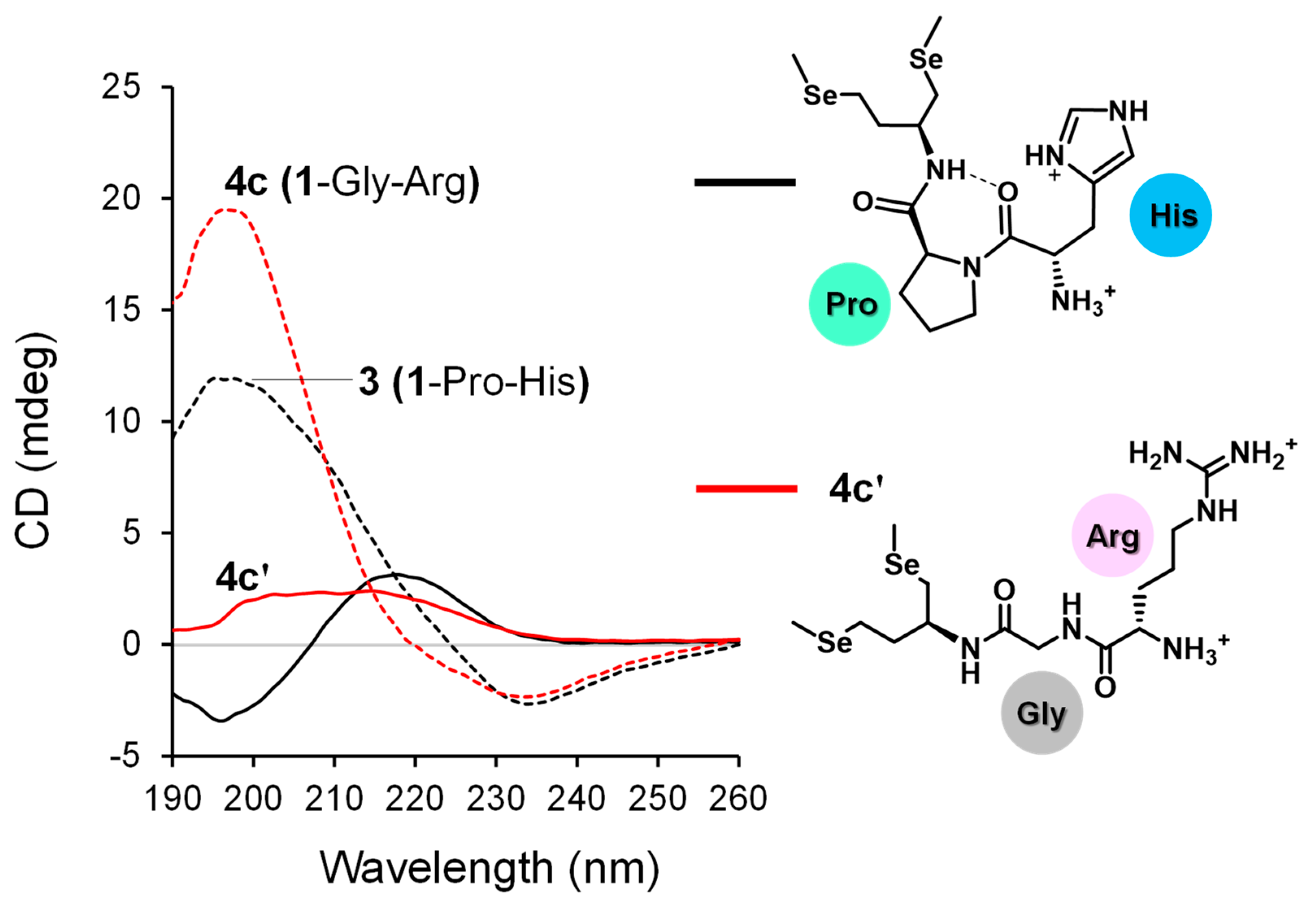
3.1. Synthesis of Gly-Xaa Dipeptide-Conjugated 1 (1-Gly-Xaa)
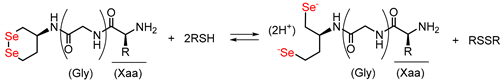
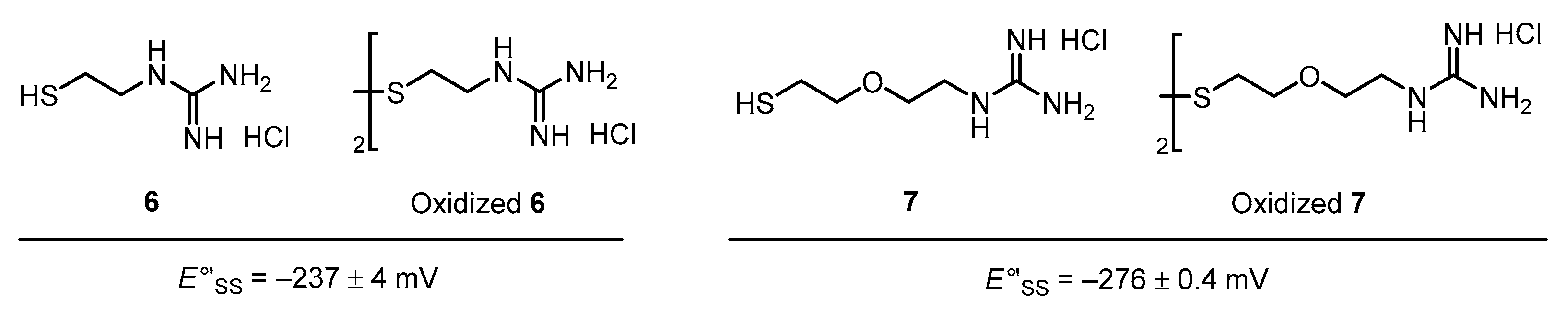
3.2. Diselenide Reduction Potential of Compounds
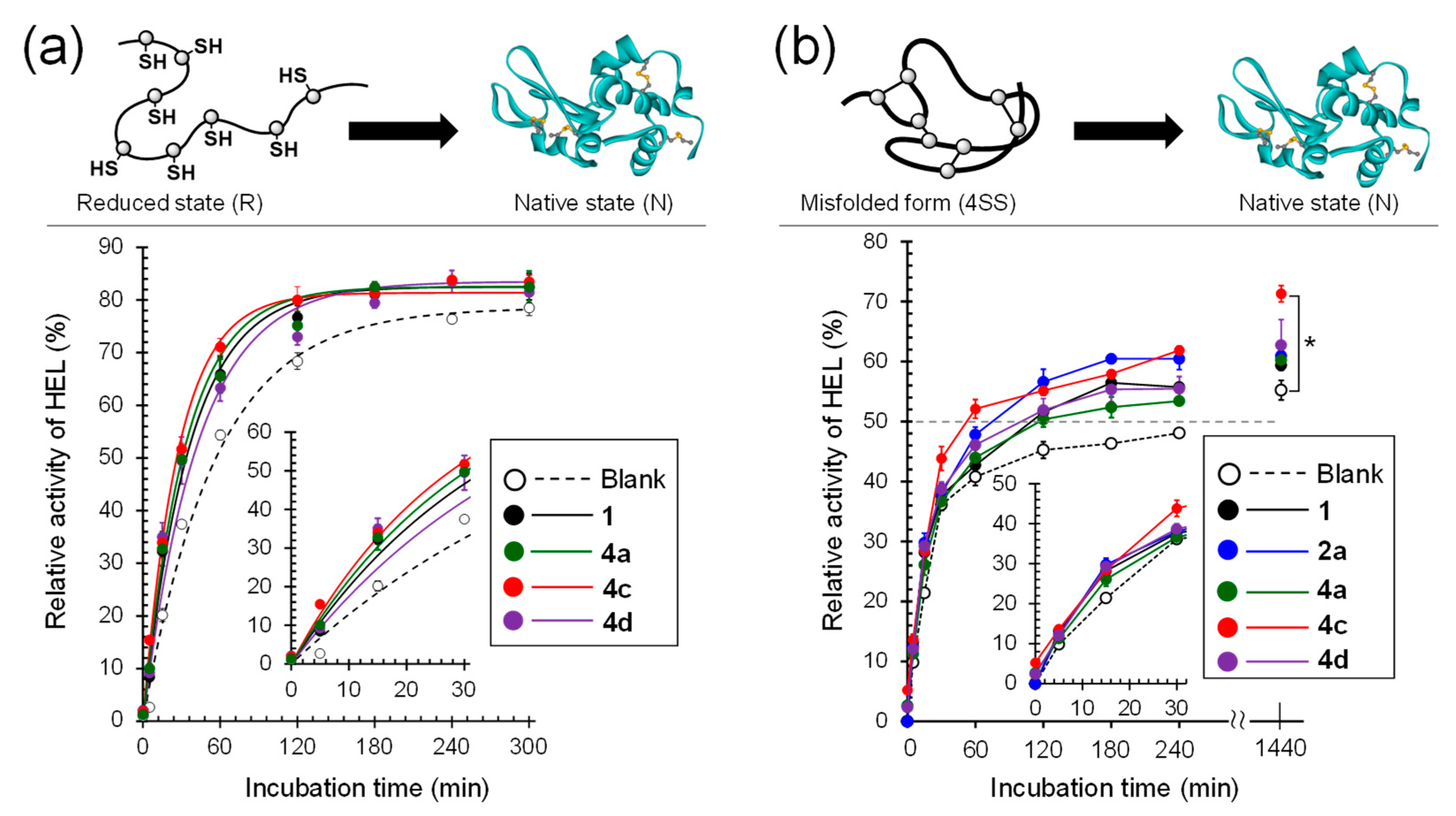
3.3. Oxidative Folding of Lysozyme in the Presence of Diselenide Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | E°′SeSe [mV] b | v0BPIns [μM min−1] c | RHEL → NHEL | 4SS → NHEL d | ||||
| Gly (or Pro) | Xaa | Amax [%] d | k [×10−3 min−1] e | Amax [%] d | t50 [min] f | |||
| 1g: | None | None | −368 ± 5 | 1.25 ± 0.03 | 81.1 ± 0.7 | 27.6 ± 0.6 | 59.4 ± 0.9 | 109 ± 4 |
| 2ag: | None | His | −304 ± 7 | 2.45 ± 0.06 | 83.0 ± 1.3 | 32.7 ± 0.7 | 60.9 ± 0.7 | 74 ± 8 |
| 3: | Pro | His | −289 ± 8 h | ND i | 82.8 ± 0.7 h | 32.8 ± 1.4 h | 60.1 ± 1.1 | 122 ± 17 |
| 4a: | Gly | His | −356 ± 2 h | 2.00 ± 0.14 | 82.5 ± 1.0 h | 30.6 ± 0.9 h | 60.3 ± 0.6 | 127.0 ± 26.2 |
| 2bg: | None | Lys | −352 ± 2 | 1.94 ± 0.08 | 82.7 ± 3.2 | 29.8 ± 0.6 | 59.1 ± 0.1 | 107 ± 10 |
| 4b: | Gly | Lys | −352 ± 1 | 2.14 ± 0.03 | 83.2 ± 0.1 | 31.3 ± 1.8 | 53.7± 1.6 | 167 ± 9 |
| 2c g: | None | Arg | −353 ± 1 | 2.64 ± 0.06 | 79.8 ± 0.4 | 30.9 ± 1.2 | 57.7 ± 0.1 | 105 ± 5 |
| 4c: | Gly | Arg | −331 ± 3 | 2.65 ± 0.05 | 81.4 ± 1.0 | 35.4 ± 1.8 | 72.1 ± 1.4 | 54 ± 9 |
| 2dg: | None | Ala | −340 ± 2 | 2.22 ± 0.07 | 80.6 ± 0.3 | 28.3 ± 1.4 | 59.0 ± 0.7 | 101 ± 5 |
| 4d: | Gly | Ala | −361 ± 2 h | 2.12 ± 0.05 | 83.5 ± 2.1 h | 23.5 ± 2.3 h | 62.8 ± 4.2 | 91 ± 6 |
| No compound g | − | 0.36 ± 0.02 | 78.6 ± 1.5 | 17.9 ± 0.6 | 55.2 ± 1.6 | >240 | ||
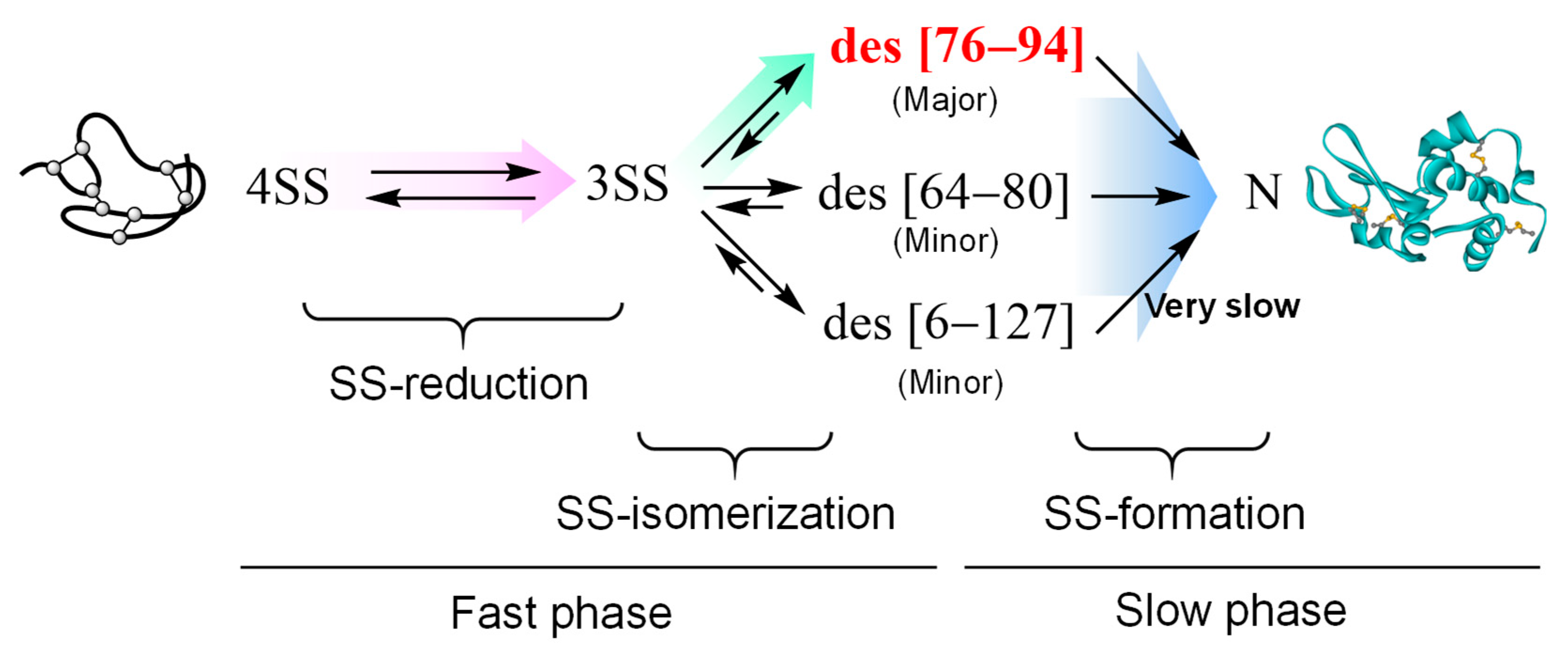
3.4. Repairing of Scrambled Lysozyme in the Presence of Diselenide Catalysts
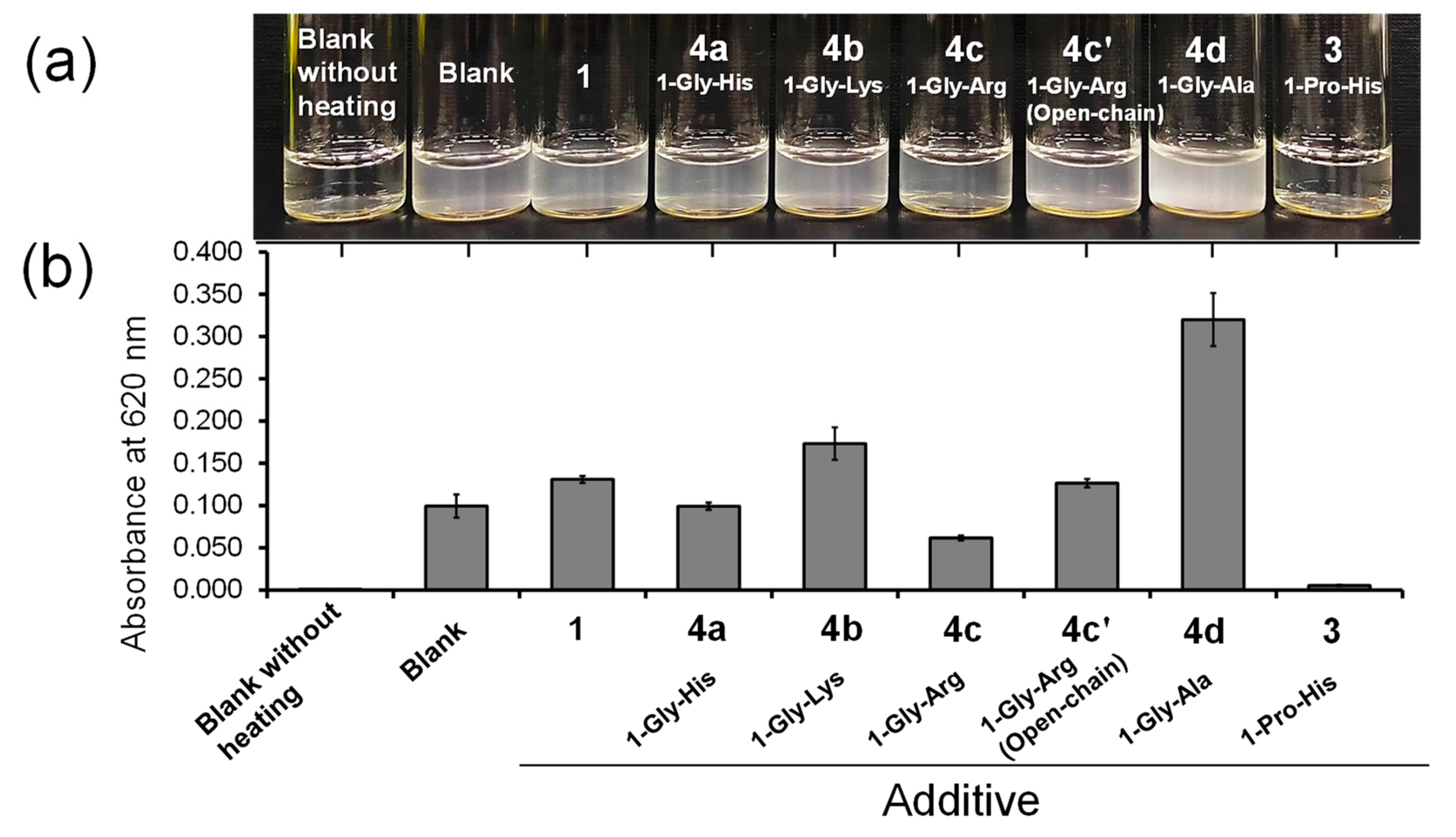
3.5. Suppressive Capbility of Diselenide Compounds against Protein Aggregation
3.6. Future Prospects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feige, M.J. (Ed.) Oxidative Folding of Proteins; Royal Society of Chemistry: Cambridge, UK, 2018; ISBN 978-1-78262-990-0. [Google Scholar]
- Arai, K.; Iwaoka, M. Flexible Folding: Disulfide-Containing Peptides and Proteins Choose the Pathway Depending on the Environments. Molecules 2021, 26, 195. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Lee, S.-J.; Lim, H.-S.; Masliah, E.; Lee, H.-J. Protein Aggregate Spreading in Neurodegenerative Diseases: Problems and Perspectives. Neurosci. Res. 2011, 70, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Protein Aggregation and Neurodegenerative Diseases: From Theory to Therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef]
- Bäuerlein, F.J.B.; Fernández-Busnadiego, R.; Baumeister, W. Investigating the Structure of Neurotoxic Protein Aggregates Inside Cells. Trends Cell Biol. 2020, 30, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Molecular Mechanisms of Nitrosative Stress-Mediated Protein Misfolding in Neurodegenerative Diseases. Cell. Mol. Life Sci. 2007, 64, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Nakamura, T.; Lipton, S.A. Redox Reactions Induced by Nitrosative Stress Mediate Protein Misfolding and Mitochondrial Dysfunction in Neurodegenerative Diseases. Mol. Neurobiol. 2010, 41, 55–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegeder, I. Nitric Oxide Mediated Redox Regulation of Protein Homeostasis. Cell. Signal. 2019, 53, 348–356. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Lees, W.J. Small-Molecule Catalysts of Oxidative Protein Folding. Curr. Opin. Chem. Biol. 2008, 12, 740–745. [Google Scholar] [CrossRef]
- Madar, D.J.; Patel, A.S.; Lees, W.J. Comparison of the Oxidative Folding of Lysozyme at a High Protein Concentration Using Aromatic Thiols versus Glutathione. J. Biotechnol. 2009, 142, 214–219. [Google Scholar] [CrossRef]
- Potempa, M.; Hafner, M.; Frech, C. Mechanism of Gemini Disulfide Detergent Mediated Oxidative Refolding of Lysozyme in a New Artificial Chaperone System. Protein J. 2010, 29, 457–465. [Google Scholar] [CrossRef]
- Iii, J.C.L.; Andersen, K.A.; Wallin, K.K.; Raines, R.T. Organocatalysts of Oxidative Protein Folding Inspired by Protein Disulfide Isomerase. Org. Biomol. Chem. 2014, 12, 8598–8602. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.S.; Metanis, N. Small Molecule Diselenide Additives for in Vitro Oxidative Protein Folding. Chem. Commun. 2016, 52, 3336–3339. [Google Scholar] [CrossRef] [Green Version]
- Arai, K.; Ueno, H.; Asano, Y.; Chakrabarty, G.; Shimodaira, S.; Mugesh, G.; Iwaoka, M. Protein Folding in the Presence of Water-Soluble Cyclic Diselenides with Novel Oxidoreductase and Isomerase Activities. ChemBioChem 2018, 19, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Matsusaki, M.; Arai, K.; Hidaka, Y.; Inaba, K.; Okumura, M.; Muraoka, T. Coupling Effects of Thiol and Urea-Type Groups for Promotion of Oxidative Protein Folding. Chem. Commun. 2019, 55, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Tsukagoshi, S.; Mikami, R.; Arai, K. Basic Amino Acid Conjugates of 1,2-Diselenan-4-Amine with Protein Disulfide Isomerase-like Functions as a Manipulator of Protein Quality Control. Chem. Asian J. 2020, 15, 2646–2652. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Matsusaki, M.; Okumura, M.; Muraoka, T. Conjugate of Thiol and Guanidyl Units with Oligoethylene Glycol Linkage for Manipulation of Oxidative Protein Folding. Molecules 2021, 26, 879. [Google Scholar] [CrossRef]
- Wilkinson, B.; Gilbert, H.F. Protein Disulfide Isomerase. Biochim. Biophys. Acta Proteins Proteom. 2004, 1699, 35–44. [Google Scholar] [CrossRef]
- Gruber, C.W.; Čemažar, M.; Heras, B.; Martin, J.L.; Craik, D.J. Protein Disulfide Isomerase: The Structure of Oxidative Folding. Trends Biochem. Sci. 2006, 31, 455–464. [Google Scholar] [CrossRef]
- Oka, O.B.V.; Bulleid, N.J. Forming Disulfides in the Endoplasmic Reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, X.; Wang, C. Protein Disulfide–Isomerase, a Folding Catalyst and a Redox-Regulated Chaperone. Free Radic. Biol. Med. 2015, 83, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Matsusaki, M.; Kanemura, S.; Kinoshita, M.; Lee, Y.-H.; Inaba, K.; Okumura, M. The Protein Disulfide Isomerase Family: From Proteostasis to Pathogenesis. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129338. [Google Scholar] [CrossRef]
- Lundstroem, J.; Holmgren, A. Determination of the Reduction-Oxidation Potential of the Thioredoxin-like Domains of Protein Disulfide-Isomerase from the Equilibrium with Glutathione and Thioredoxin. Biochemistry 1993, 32, 6649–6655. [Google Scholar] [CrossRef] [PubMed]
- Lukesh, J.C.; VanVeller, B.; Raines, R.T. Thiols and Selenols as Electron-Relay Catalysts for Disulfide-Bond Reduction. Angew. Chem. Int. Ed. 2013, 52, 12901–12904. [Google Scholar] [CrossRef] [Green Version]
- Arai, K.; Matsunaga, T.; Ueno, H.; Akahoshi, N.; Sato, Y.; Chakrabarty, G.; Mugesh, G.; Iwaoka, M. Modeling Thioredoxin Reductase-Like Activity with Cyclic Selenenyl Sulfides: Participation of an NH⋅⋅⋅Se Hydrogen Bond through Stabilization of the Mixed Se−S Intermediate. Chem. Eur. J. 2019, 25, 12751–12760. [Google Scholar] [CrossRef] [PubMed]
- Mikami, R.; Tsukagoshi, S.; Oda, Y.; Arai, K. S-Denitrosylase-like Activity of Cyclic Diselenides Conjugated with Xaa-His Dipeptide: Role of Proline Spacer as a Key Activity Booster. ChemBioChem 2021. [Google Scholar] [CrossRef]
- Arai, K.; Shibagaki, W.; Shinozaki, R.; Iwaoka, M. Reinvestigation of the Oxidative Folding Pathways of Hen Egg White Lysozyme: Switching of the Major Pathways by Temperature Control. Int. J. Mol. Sci. 2013, 14, 13194–13212. [Google Scholar] [CrossRef] [Green Version]
- Beld, J.; Woycechowsky, K.J.; Hilvert, D. Selenoglutathione: Efficient Oxidative Protein Folding by a Diselenide. Biochemistry 2007, 46, 5382–5390. [Google Scholar] [CrossRef]
- Biswal, H.S.; Wategaonkar, S. Nature of the N−H···S Hydrogen Bond. J. Phys. Chem. A 2009, 113, 12763–12773. [Google Scholar] [CrossRef]
- Arai, K.; Moriai, K.; Ogawa, A.; Iwaoka, M. An Amphiphilic Selenide Catalyst Behaves Like a Hybrid Mimic of Protein Disulfide Isomerase and Glutathione Peroxidase 7. Chem. Asian J. 2014, 9, 3464–3471. [Google Scholar] [CrossRef] [PubMed]
- Gurbhele-Tupkar, M.C.; Perez, L.R.; Silva, Y.; Lees, W.J. Rate Enhancement of the Oxidative Folding of Lysozyme by the Use of Aromatic Thiol Containing Redox Buffers. Bioorg. Med. Chem. 2008, 16, 2579–2590. [Google Scholar] [CrossRef] [PubMed]
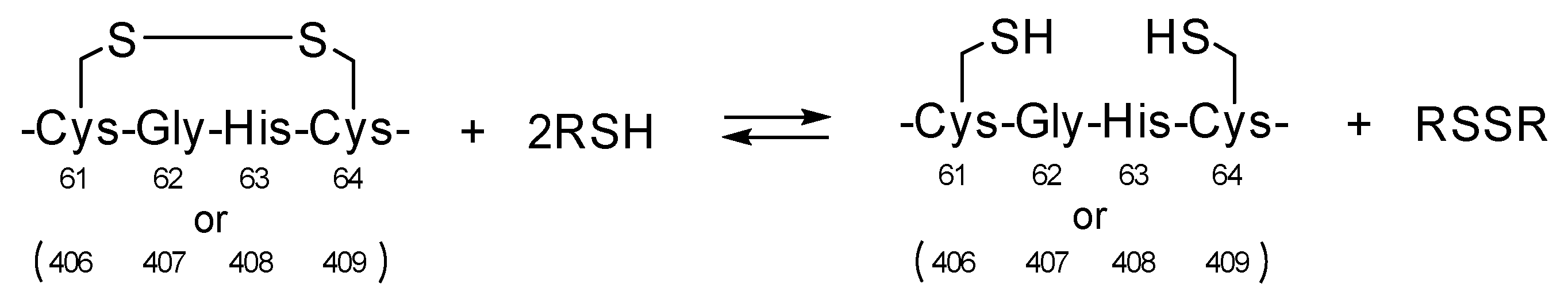

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikami, R.; Tsukagoshi, S.; Arai, K. Abnormal Enhancement of Protein Disulfide Isomerase-like Activity of a Cyclic Diselenide Conjugated with a Basic Amino Acid by Inserting a Glycine Spacer. Biology 2021, 10, 1090. https://doi.org/10.3390/biology10111090
Mikami R, Tsukagoshi S, Arai K. Abnormal Enhancement of Protein Disulfide Isomerase-like Activity of a Cyclic Diselenide Conjugated with a Basic Amino Acid by Inserting a Glycine Spacer. Biology. 2021; 10(11):1090. https://doi.org/10.3390/biology10111090
Chicago/Turabian StyleMikami, Rumi, Shunsuke Tsukagoshi, and Kenta Arai. 2021. "Abnormal Enhancement of Protein Disulfide Isomerase-like Activity of a Cyclic Diselenide Conjugated with a Basic Amino Acid by Inserting a Glycine Spacer" Biology 10, no. 11: 1090. https://doi.org/10.3390/biology10111090
APA StyleMikami, R., Tsukagoshi, S., & Arai, K. (2021). Abnormal Enhancement of Protein Disulfide Isomerase-like Activity of a Cyclic Diselenide Conjugated with a Basic Amino Acid by Inserting a Glycine Spacer. Biology, 10(11), 1090. https://doi.org/10.3390/biology10111090