
Disease Modeling of Mitochondrial Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells


Abstract
:Simple Summary
Abstract
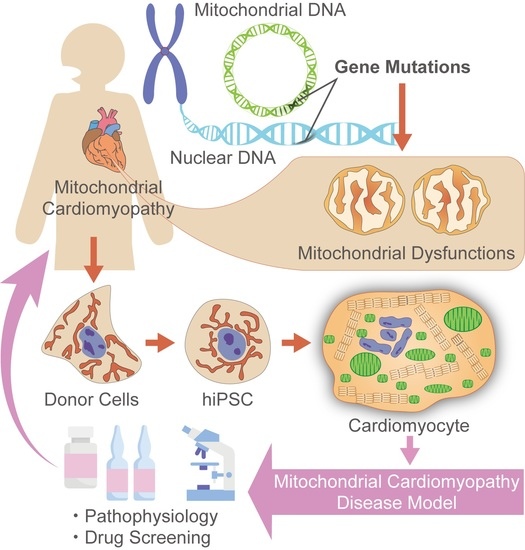
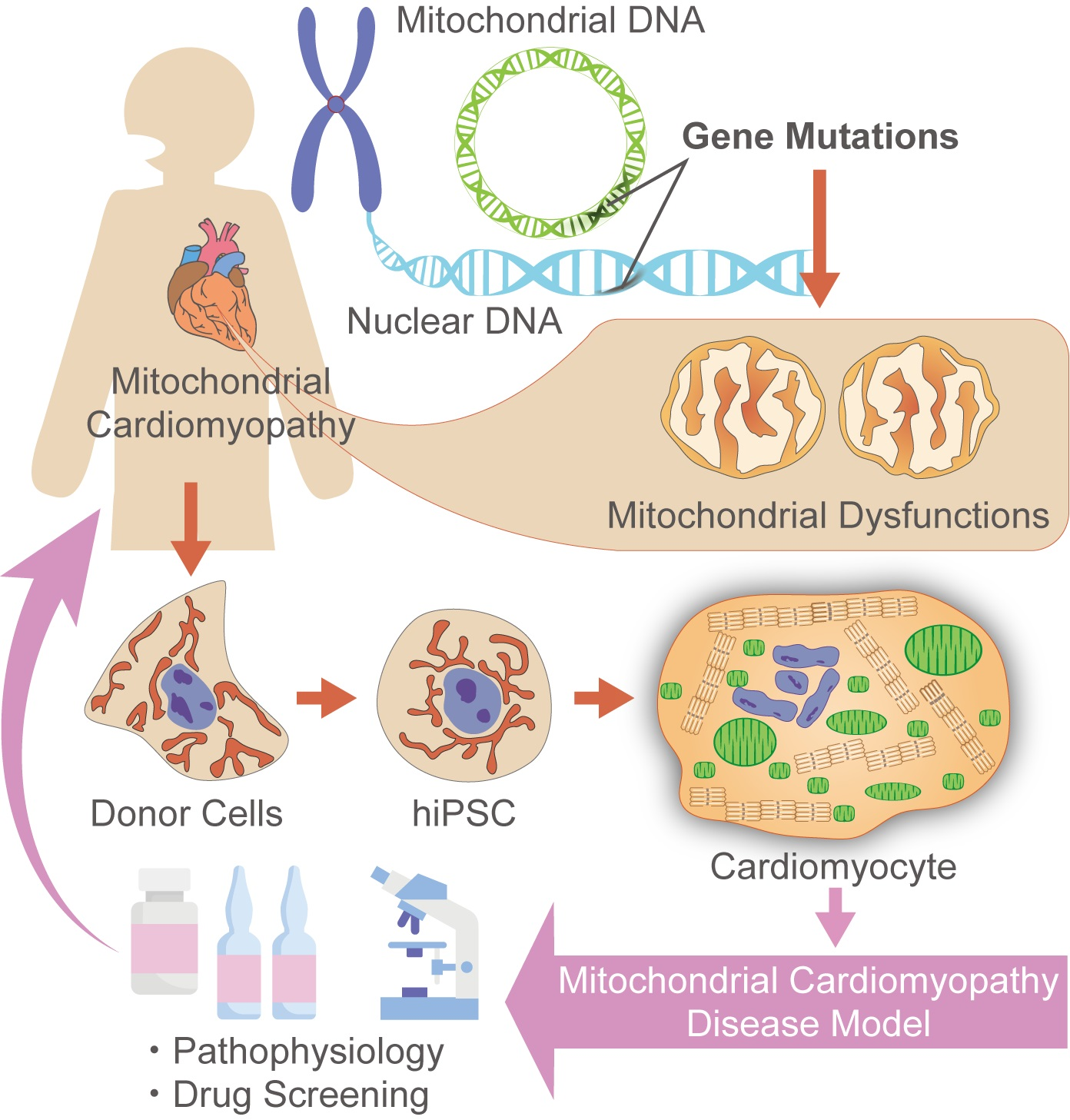
1. Introduction
2. What Is Mitochondrial Cardiomyopathy (MCM)?
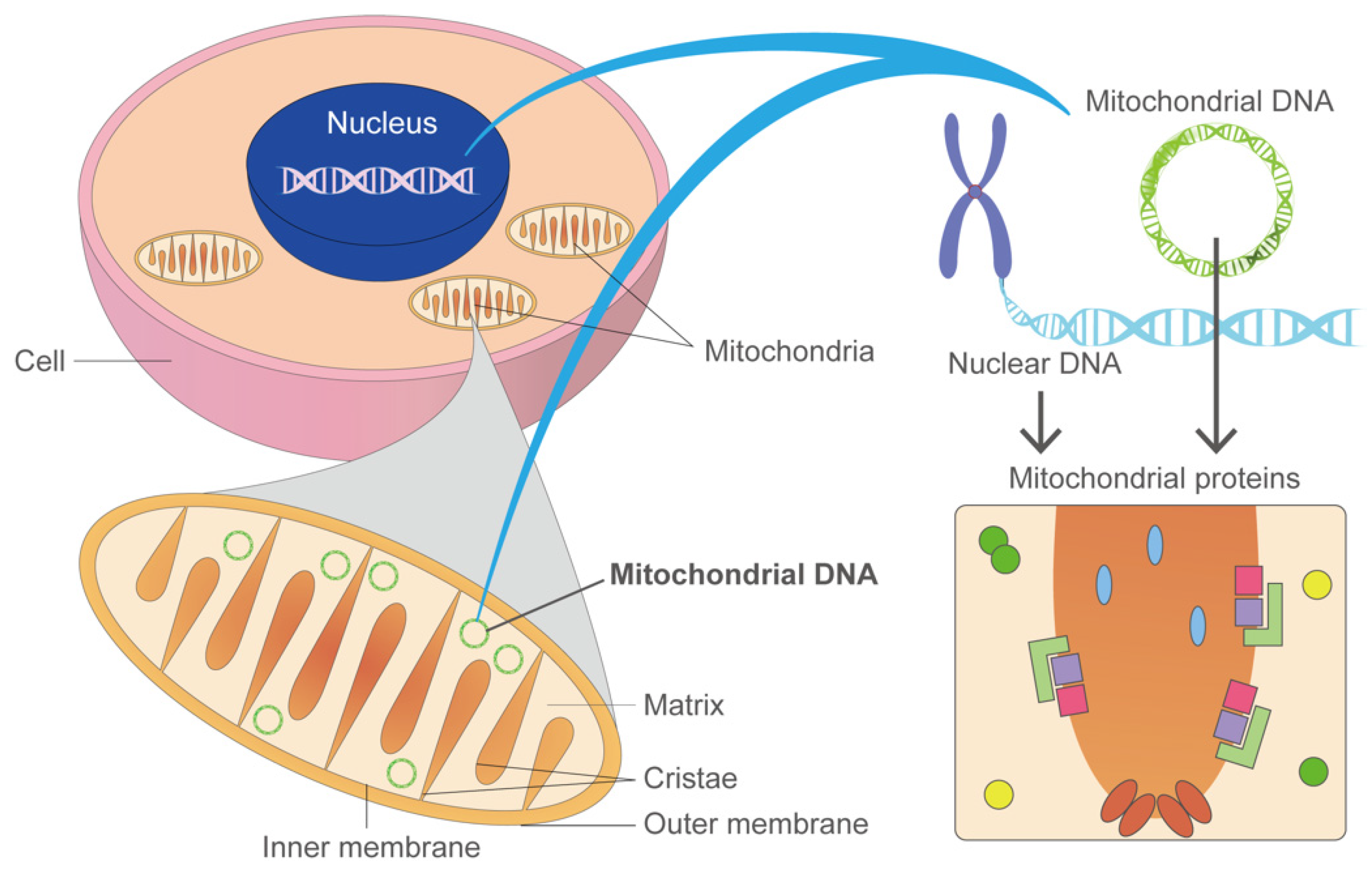
2.1. Genetic Variants Associated with Mitochondrial Dysfunction
2.1.1. Mitochondrial DNA Deletion
2.1.2. Variants in Nuclear and Mitochondrial DNA
2.2. Mechanisms Linking Mitochondrial Dysfunction to Cardiac Dysfunction and the Phenotypes
3. Disease Modeling with Patient-Specific iPSCs
3.1. Perspectives from iPSC Studies to Study Human Cells than Mice
3.2. Cardiomyocyte Differentiation from iPSCs
3.3. Cardiac Disease Models Using Patient-Derived IPSCs
3.4. MCM Disease Model Using Human iPSC-CMs
4. Limitations of iPSC-CMs
4.1. Characteristics of Adult Cardiomyocytes and iPSC-CMs
4.1.1. Morphology and Structure of Cardiomyocytes
4.1.2. Physical and Electrophysiological Properties
4.1.3. Calcium Signaling
4.1.4. Metabolism
4.1.5. Gene Expression
4.2. Approaches for iPSC-CMs Maturation
5. Future Research on MCM Using iPSC-CMs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trounson, A.; McDonald, C. Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell 2015, 17, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbrel, E.A.; Lanza, R. Current status of pluripotent stem cells: Moving the first therapies to the clinic. Nat. Rev. Drug Discov. 2015, 14, 681–692. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, K.; Tsuji, D.; Kudoh, K.; Satomura, K.; Muto, T.; Itoh, K.; Noma, T. Generation of human induced pluripotent stem cells from oral mucosa. J. Biosci. Bioeng. 2010, 110, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. A fresh look at iPS cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrowsmith, J.; Miller, P. Phase II and Phase III attrition rates 2011–2012. Nat. Rev. Drug. Discov. 2013, 12, 569. [Google Scholar] [CrossRef]
- Davis, R.P.; van den Berg, C.W.; Casini, S.; Braam, S.R.; Mummery, C.L. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol. Med. 2011, 17, 475–484. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-Specific Induced Pluripotent Stem Cells as a Model for Familial Dilated Cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra147. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drawnel, F.M.; Boccardo, S.; Prummer, M.; Delobel, F.; Graff, A.; Weber, M.; Gérard, R.; Badi, L.; Kam-Thong, T.; Bu, L.; et al. Disease Modeling and Phenotypic Drug Screening for Diabetic Cardiomyopathy using Human Induced Pluripotent Stem Cells. Cell Rep. 2014, 9, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.-S.; Schaniel, C.; Lee, D.-F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malan, D.; Zhang, M.; Stallmeyer, B.; Müller, J.; Fleischmann, B.K.; Schulze-Bahr, E.; Sasse, P.; Greber, B. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res. Cardiol. 2016, 111, 14. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef]
- Dominic, E.A.; Ramezani, A.; Anker, S.D.; Verma, M.; Mehta, N.; Rao, M. Mitochondrial cytopathies and cardiovascular disease. Heart 2014, 100, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Jarreta, D.; Orús, J.; Barrientos, A.; Miró, O.; Roig, E.; Heras, M.; Moraes, C.T.; Cardellach, F.; Casademont, J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2000, 45, 860–865. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef]
- Johns, D.R. Mitochondrial DNA and Disease. N. Engl. J. Med. 1995, 333, 638–644. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Smeitink, J.A. Mitochondrial disorders: A proposal for consensus diagnostic criteria in infants and children. Neurology 2002, 59, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [Green Version]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Nesbitt, V.; Pitceathly, R.D.; Turnbull, D.M.; Taylor, R.W.; Sweeney, M.G.; Mudanohwo, E.E.; Rahman, S.; Hanna, M.G.; McFarland, R. The UK MRC Mitochondrial Disease Patient Cohort Study: Clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management. J. Neurol. Neurosurg. Psychiatry 2013, 84, 936–938. [Google Scholar] [CrossRef]
- Bates, M.G.; Bourke, J.P.; Giordano, C.; d’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Honzik, T.; Tesarova, M.; Magner, M.; Mayr, J.; Jesina, P.; Vesela, K.; Wenchich, L.; Szentivanyi, K.; Hansikova, H.; Sperl, W.; et al. Neonatal onset of mitochondrial disorders in 129 patients: Clinical and laboratory characteristics and a new approach to diagnosis. J. Inherit. Metab. Dis. 2012, 35, 749–759. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Wahbi, K.; Bougouin, W.; Béhin, A.; Stojkovic, T.; Bécane, H.M.; Jardel, C.; Berber, N.; Mochel, F.; Lombès, A.; Eymard, B.; et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur. Heart J. 2015, 36, 2886–2893. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, D.; Wåhlander, H.; Eriksson, B.O.; Oldfors, A.; Holme, E.; Tulinius, M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur. Heart J. 2003, 24, 280–288. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Matsunaga, A.; Yatsuka, Y.; Nitta, K.R.; Kishita, Y.; Sugiura, A.; Sugiyama, Y.; Fushimi, T.; Shimura, M.; Ichimoto, K.; et al. Long-term prognosis and genetic background of cardiomyopathy in 223 pediatric mitochondrial disease patients. Int. J. Cardiol. 2021, 341, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Yatsuka, Y.; Hirata, T.; Mizuno, Y.; Harashima, H.; Hirono, K.; Ichida, F.; Noguchi, A.; et al. Barth Syndrome: Different Approaches to Diagnosis. J. Pediatr. 2018, 193, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M. Oxidative phosphorylation at the fin de siècle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Elliott, H.R.; Hudson, G.; Samuels, D.C.; Relton, C.L. Epigenetics, epidemiology and mitochondrial DNA diseases. Int. J. Epidemiol. 2012, 41, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallevelt, S.C.; de Die-Smulders, C.E.; Hendrickx, A.T.; Hellebrekers, D.M.; de Coo, I.F.; Alston, C.L.; Knowles, C.; Taylor, R.W.; McFarland, R.; Smeets, H.J. De novo mtDNA point mutations are common and have a low recurrence risk. J. Med. Genet. 2017, 54, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Rötig, A.; Cormier, V.; Blanche, S.; Bonnefont, J.P.; Ledeist, F.; Romero, N.; Schmitz, J.; Rustin, P.; Fischer, A.; Saudubray, J.M.; et al. Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J. Clin. Investig. 1990, 86, 1601–1608. [Google Scholar] [CrossRef]
- Chinnery, P.F.; DiMauro, S.; Shanske, S.; Schon, E.A.; Zeviani, M.; Mariotti, C.; Carrara, F.; Lombes, A.; Laforet, P.; Ogier, H.; et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet 2004, 364, 592–596. [Google Scholar] [CrossRef]
- de Laat, P.; Koene, S.; van den Heuvel, L.P.; Rodenburg, R.J.; Janssen, M.C.; Smeitink, J.A. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A > G mutation. J. Inherit. Metab. Dis. 2012, 35, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- White, S.L.; Collins, V.R.; Wolfe, R.; Cleary, M.A.; Shanske, S.; DiMauro, S.; Dahl, H.H.; Thorburn, D.R. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am. J. Hum. Genet. 1999, 65, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A.; et al. Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet. 2016, 99, 860–876. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Wang, J.; Lee, N.C.; Milone, M.; Halberg, M.C.; Schmitt, E.S.; Craigen, W.J.; Zhang, W.; Wong, L.J. Mitochondrial DNA polymerase gamma mutations: An ever expanding molecular and clinical spectrum. J. Med. Genet. 2011, 48, 669–681. [Google Scholar] [CrossRef]
- Sperl, W.; Fleuren, L.; Freisinger, P.; Haack, T.B.; Ribes, A.; Feichtinger, R.G.; Rodenburg, R.J.; Zimmermann, F.A.; Koch, J.; Rivera, I.; et al. The spectrum of pyruvate oxidation defects in the diagnosis of mitochondrial disorders. J. Inherit. Metab. Dis. 2015, 38, 391–403. [Google Scholar] [CrossRef]
- Berger, I.; Hershkovitz, E.; Shaag, A.; Edvardson, S.; Saada, A.; Elpeleg, O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann. Neurol. 2008, 63, 405–408. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Freisinger, P.; Karall, D.; Makowski, C.; Koch, J.; Feichtinger, R.G.; Zimmermann, F.A.; Rolinski, B.; Ahting, U.; et al. Spectrum of combined respiratory chain defects. J. Inherit. Metab. Dis. 2015, 38, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Nouws, J.; Nijtmans, L.; Houten, S.M.; van den Brand, M.; Huynen, M.; Venselaar, H.; Hoefs, S.; Gloerich, J.; Kronick, J.; Hutchin, T.; et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010, 12, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.V.; Schapira, A.H. Mitochondrial respiratory chain disorders I: Mitochondrial DNA defects. Lancet 2000, 355, 299–304. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, D.M.; Crawford, M.; Cleary, M.A.; Dahl, H.H.; Dennett, X.; Thorburn, D.R. Respiratory chain complex I deficiency: An underdiagnosed energy generation disorder. Neurology 1999, 52, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Howard, C.; Oláhová, M.; Hardy, S.A.; He, L.; Murray, P.G.; O’Sullivan, S.; Doherty, G.; Shield, J.P.; Hargreaves, I.P.; et al. A recurrent mitochondrial p.Trp22Arg NDUFB3 variant causes a distinctive facial appearance, short stature and a mild biochemical and clinical phenotype. J. Med. Genet. 2016, 53, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swalwell, H.; Kirby, D.M.; Blakely, E.L.; Mitchell, A.; Salemi, R.; Sugiana, C.; Compton, A.G.; Tucker, E.J.; Ke, B.X.; Lamont, P.J.; et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur. J. Hum. Genet. 2011, 19, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baysal, B.E. Mutations in SDHD, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Niemann, S.; Müller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1.23.1–1.23.26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordaunt, D.A.; Jolley, A.; Balasubramaniam, S.; Thorburn, D.R.; Mountford, H.S.; Compton, A.G.; Nicholl, J.; Manton, N.; Clark, D.; Bratkovic, D.; et al. Phenotypic variation of TTC19-deficient mitochondrial complex III deficiency: A case report and literature review. Am. J. Med. Genet. A 2015, 167, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A. Cytochrome c oxidase deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Rahman, S.; Wedatilake, Y.; Polke, J.M.; Cirak, S.; Foley, A.R.; Sailer, A.; Hurles, M.E.; Stalker, J.; Hargreaves, I.; et al. NDUFA4 mutations underlie dysfunction of a cytochrome c oxidase subunit linked to human neurological disease. Cell Rep. 2013, 3, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Stroud, D.A.; Maher, M.J.; Lindau, C.; Vögtle, F.N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Bonas, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar] [CrossRef] [Green Version]
- Mourier, A.; Ruzzenente, B.; Brandt, T.; Kühlbrandt, W.; Larsson, N.G. Loss of LRPPRC causes ATP synthase deficiency. Hum. Mol. Genet. 2014, 23, 2580–2592. [Google Scholar] [CrossRef] [Green Version]
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 deficiency: A multi-centre natural history study. Orphanet. J. Rare. Dis. 2013, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Tamiya, G.; Makino, S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N.; et al. A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Hogeveen, M.; Nijtmans, L.; van den Brand, M.; Janssen, A.; Diepstra, H.; van den Brandt, F.; van den Heuvel, B.; Hol, F.; Hofste, T.; et al. A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. BMJ Case. Rep. 2009, 2009. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb. Cell 2015, 2, 105–125. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Cízková, A.; Stránecký, V.; Mayr, J.A.; Tesarová, M.; Havlícková, V.; Paul, J.; Ivánek, R.; Kuss, A.W.; Hansíková, H.; Kaplanová, V.; et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008, 40, 1288–1290. [Google Scholar] [CrossRef]
- De Meirleir, L.; Seneca, S.; Lissens, W.; De Clercq, I.; Eyskens, F.; Gerlo, E.; Smet, J.; Van Coster, R. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, R.; Khayat, M.; Shalev, S.A.; Horovitz, Y.; Mandel, H.; Hershkovitz, E.; Barghuti, F.; Shaag, A.; Saada, A.; Korman, S.H.; et al. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: Further delineation of a new syndrome. J. Med. Genet. 2011, 48, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Metodiev, M.D.; Gerber, S.; Hubert, L.; Delahodde, A.; Chretien, D.; Gérard, X.; Amati-Bonneau, P.; Giacomotto, M.C.; Boddaert, N.; Kaminska, A.; et al. Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J. Med. Genet. 2014, 51, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Gerards, M.; Kamps, R.; van Oevelen, J.; Boesten, I.; Jongen, E.; de Koning, B.; Scholte, H.R.; de Angst, I.; Schoonderwoerd, K.; Sefiani, A.; et al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain 2013, 136, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Takeda, A. Mitochondrial Cardiomyopathy. J. Pediatr. Cardiol. Cardiac. Surg. 2020, 4, 53–62. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Gordaliza-Alaguero, I.; Cantó, C.; Zorzano, A. Metabolic implications of organelle-mitochondria communication. EMBO Rep. 2019, 20, e47928. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef]
- Schwartz, P.J. Do animal models have clinical value? Am. J. Cardiol. 1998, 81, 14d–20d. [Google Scholar] [CrossRef]
- Matsa, E.; Ahrens, J.H.; Wu, J.C. Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine. Physiol. Rev. 2016, 96, 1093–1126. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nagata, N.; Kurokawa, H.; Yamanaka, S. iPS cells: A game changer for future medicine. EMBO J. 2014, 33, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Matsa, E.; Wu, J.C. Induced pluripotent stem cells: At the heart of cardiovascular precision medicine. Nat. Rev. Cardiol. 2016, 13, 333–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altomare, C.; Pianezzi, E.; Cervio, E.; Bolis, S.; Biemmi, V.; Benzoni, P.; Camici, G.G.; Moccetti, T.; Barile, L.; Vassalli, G. Human-induced pluripotent stem cell-derived cardiomyocytes from cardiac progenitor cells: Effects of selective ion channel blockade. Europace 2016, 18, iv67–iv76. [Google Scholar] [CrossRef]
- Ruzzenente, B.; Rötig, A.; Metodiev, M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016, 25, R115–R122. [Google Scholar] [CrossRef] [Green Version]
- Uosaki, H.; Taguchi, Y.h. Comparative Gene Expression Analysis of Mouse and Human Cardiac Maturation. Genom. Proteom. Bioinform. 2016, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Anzai, T.; Yamagata, T.; Uosaki, H. Comparative Transcriptome Landscape of Mouse and Human Hearts. Front. Cell. Dev. Biol. 2020, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascenção, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [Green Version]
- Kauppila, J.H.K.; Baines, H.L.; Bratic, A.; Simard, M.-L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.-G.; Greaves, L.C.; et al. A Phenotype-Driven Approach to Generate Mouse Models with Pathogenic mtDNA Mutations Causing Mitochondrial Disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef] [Green Version]
- Dunn, K.K.; Palecek, S.P. Engineering Scalable Manufacturing of High-Quality Stem Cell-Derived Cardiomyocytes for Cardiac Tissue Repair. Front. Med. 2018, 5, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Soonpaa, M.H.; Adler, E.D.; Roepke, T.K.; Kattman, S.J.; Kennedy, M.; Henckaerts, E.; Bonham, K.; Abbott, G.W.; Linden, R.M.; et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature 2008, 453, 524–528. [Google Scholar] [CrossRef]
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS ONE 2011, 6, e23657. [Google Scholar] [CrossRef]
- Elliott, D.A.; Braam, S.R.; Koutsis, K.; Ng, E.S.; Jenny, R.; Lagerqvist, E.L.; Biben, C.; Hatzistavrou, T.; Hirst, C.E.; Yu, Q.C.; et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods 2011, 8, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef]
- Pesl, M.; Acimovic, I.; Pribyl, J.; Hezova, R.; Vilotic, A.; Fauconnier, J.; Vrbsky, J.; Kruzliak, P.; Skladal, P.; Kara, T.; et al. Forced aggregation and defined factors allow highly uniform-sized embryoid bodies and functional cardiomyocytes from human embryonic and induced pluripotent stem cells. Heart Vessels 2014, 29, 834–846. [Google Scholar] [CrossRef]
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A Universal and Robust Integrated Platform for the Scalable Production of Human Cardiomyocytes From Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 1482–1494. [Google Scholar] [CrossRef]
- Kim, M.S.; Horst, A.; Blinka, S.; Stamm, K.; Mahnke, D.; Schuman, J.; Gundry, R.; Tomita-Mitchell, A.; Lough, J. Activin-A and Bmp4 levels modulate cell type specification during CHIR-induced cardiomyogenesis. PLoS ONE 2015, 10, e0118670. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct Metabolic Flow Enables Large-Scale Purification of Mouse and Human Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, N.; Tohyama, S.; Nakajima, K.; Kanazawa, H.; Suzuki, T.; Hattori, F.; Seki, T.; Kishino, Y.; Hirano, A.; Okada, M.; et al. A massive suspension culture system with metabolic purification for human pluripotent stem cell-derived cardiomyocytes. Stem. Cells Transl. Med. 2014, 3, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rafatian, N.; Feric, N.T.; Cox, B.J.; Aschar-Sobbi, R.; Wang, E.Y.; Aggarwal, P.; Zhang, B.; Conant, G.; Ronaldson-Bouchard, K.; et al. A Platform for Generation of Chamber-Specific Cardiac Tissues and Disease Modeling. Cell 2019, 176, 913–927.e18. [Google Scholar] [CrossRef] [Green Version]
- Denning, C.; Borgdorff, V.; Crutchley, J.; Firth, K.S.A.; George, V.; Kalra, S.; Kondrashov, A.; Hoang, M.D.; Mosqueira, D.; Patel, A.; et al. Cardiomyocytes from human pluripotent stem cells: From laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta (BBA.)-Mol. Cell Res. 2016, 1863, 1728–1748. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Gnecchi, M.; Schwartz, P.J. Long QT Syndrome Modelling with Cardiomyocytes Derived from Human-induced Pluripotent Stem Cells. Arrhythm. Electrophysiol. Rev. 2019, 8, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Vermglinchan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Heliö, T.; Kallio, P.; et al. Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 2019, 8, 594. [Google Scholar] [CrossRef] [Green Version]
- Kodo, K.; Ong, S.G.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; InanlooRahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-β signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell. Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W.; Wang, X.; Li, D.; Dai, Y.; et al. Mitochondrial Dysfunctions Contribute to Hypertrophic Cardiomyopathy in Patient iPSC-Derived Cardiomyocytes with MT-RNR2 Mutation. Stem. Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, Y.; Yuasa, S.; Watanabe, Y.; Ito, S.; Egashira, T.; Seki, T.; Hattori, T.; Ohno, S.; Kodaira, M.; Suzuki, T.; et al. Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem. Biophys. Rep. 2017, 9, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Aich, A.; Jain, G.; Wozny, K.; Lüchtenborg, C.; Hartmann, M.; Bernhard, O.; Balleiniger, M.; Alfar, E.A.; Zieseniss, A.; et al. Defective Mitochondrial Cardiolipin Remodeling Dampens HIF-1α Expression in Hypoxia. Cell Rep. 2018, 25, 561–570.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, S.; Guo, X.; Li, Y.; Ogurlu, R.; Lu, F.; Prondzynski, M.; Buzon, S.d.l.S.; Ma, Q.; Zhang, D.; et al. Increased Reactive Oxygen Species–Mediated Ca2+/Calmodulin-Dependent Protein Kinase II Activation Contributes to Calcium Handling Abnormalities and Impaired Contraction in Barth Syndrome. Circulation 2021, 143, 1894–1911. [Google Scholar] [CrossRef]
- Fatica, E.M.; DeLeonibus, G.A.; House, A.; Kodger, J.V.; Pearce, R.W.; Shah, R.R.; Levi, L.; Sandlers, Y. Barth Syndrome: Exploring Cardiac Metabolism with Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Metabolites 2019, 9, 306. [Google Scholar] [CrossRef] [Green Version]
- Rohani, L.; Meng, G.; Machiraju, P.; Liu, S.; Wu, J.; Kovalchuk, I.; Lewis, I.; Shutt, T.; Khan, A.; Rancourt, D.; et al. Modeling the dilated cardiomyopathy with ataxia syndrome (dcma), a pediatric mitochondrial cardiomyopathy, using cardiomyocytes derived from induced pluripotent stem cells. Can. J. Cardiol. 2017, 33, S163–S164. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Ho, P.W.-L.; Schick, R.; Lau, Y.-M.; Lai, W.-H.; Zhou, T.; Li, Y.; Ng, K.-M.; Ho, S.-L.; Esteban, M.A.; et al. Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflügers Arch. Eur. J. Physiol. 2014, 466, 1831–1844. [Google Scholar] [CrossRef]
- Hick, A.; Wattenhofer-Donzé, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; André, C.; Reutenauer, L.; et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Models Mech. 2013, 6, 608–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Turkenburg, M.; Poll-The, B.T.; Karall, D.; Pérez-Cerdá, C.; Morrone, A.; Malvagia, S.; Wanders, R.J.; Kulik, W.; Vaz, F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Schulz, J.B.; Boesch, S.; Bürk, K.; Dürr, A.; Giunti, P.; Mariotti, C.; Pousset, F.; Schöls, L.; Vankan, P.; Pandolfo, M. Diagnosis and treatment of Friedreich ataxia: A European perspective. Nat. Rev. Neurol. 2009, 5, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, Y.; Li, D.; He, X.; Li, S.; Wu, B.; Wang, W.; Gu, S.; Zhu, X.; Wang, X.; et al. The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 2014, 51, 176–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic Instability of iPSCs: Challenges Towards Their Clinical Applications. Stem. Cell Rev. Rep. 2017, 13, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiraju, P.; Greenway, S.C. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J. Stem Cells 2019, 11, 33–43. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering Adolescence. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Peters, N.S.; Severs, N.J.; Rothery, S.M.; Lincoln, C.; Yacoub, M.H.; Green, C.R. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation 1994, 90, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Vreeker, A.; Van Stuijvenberg, L.; Hund, T.J.; Mohler, P.J.; Nikkels, P.G.J.; Van Veen, T.A.B. Assembly of the Cardiac Intercalated Disk during Pre- and Postnatal Development of the Human Heart. PLoS ONE 2014, 9, e94722. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, T.; Makiyama, T.; Sasaki, K.; Yoshida, Y.; Wuriyanghai, Y.; Chen, J.; Hattori, T.; Ohno, S.; Kita, T.; Horie, M.; et al. Ultrastructural Maturation of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes in a Long-Term Culture. Circ. J. 2013, 77, 1307–1314. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef] [PubMed]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.-H.; Gepstein, L. Cardiomyocyte Differentiation of Human Induced Pluripotent Stem Cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm. 2005, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C. Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circ. Res. 2013, 112, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Mummery, C.; Wilde, A.; Bezzina, C.; Verkerk, A. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front. Physiol. 2012, 3, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagi, K.; Takano, M.; Narazaki, G.; Uosaki, H.; Hoshino, T.; Ishii, T.; Misaki, T.; Yamashita, J.K. Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels and T-Type Calcium Channels Confer Automaticity of Embryonic Stem Cell-Derived Cardiomyocytes. Stem Cells 2007, 25, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Mengarelli, I.; Lodder, E.M.; Kosmidis, G.; Bellin, M.; Zhang, M.; Dittmann, S.; Guan, K.; Wilde, A.A.M.; Schulze-Bahr, E.; et al. Switch From Fetal to Adult SCN5A Isoform in Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Unmasks the Cellular Phenotype of a Conduction Disease–Causing Mutation. J. Am. Heart Assoc. 2017, 6, e005135. [Google Scholar] [CrossRef] [PubMed]
- Pesl, M.; Pribyl, J.; Caluori, G.; Cmiel, V.; Acimovic, I.; Jelinkova, S.; Dvorak, P.; Starek, Z.; Skladal, P.; Rotrekl, V. Phenotypic assays for analyses of pluripotent stem cell-derived cardiomyocytes. J. Mol. Recognit. 2017, 30, e2602. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Spafford, M.A.; Marsh, D.R. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol.-Heart Circ. Physiol. 1991, 261, H1698–H1705. [Google Scholar] [CrossRef]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef]
- Kikuchi, C.; Bienengraeber, M.; Canfield, S.; Koopmeiner, A.; Schäfer, R.; Bosnjak, Z.J.; Bai, X. Comparison of Cardiomyocyte Differentiation Potential between Type 1 Diabetic Donor- and Nondiabetic Donor-Derived Induced Pluripotent Stem Cells. Cell Transplant. 2015, 24, 2491–2504. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Anson, B.; Engle, S.; Will, Y. Characterization of Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes: Bioenergetics and Utilization in Safety Screening. Toxicol. Sci. 2012, 130, 117–131. [Google Scholar] [CrossRef] [Green Version]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions in cardiac muscle: A journey to heart failure. Biochim. Biophysica. Acta (BBA) Mol. Basis Dis. 2015, 1852, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Berg, C.W.; Okawa, S.; Chuva De Sousa Lopes, S.M.; Van Iperen, L.; Passier, R.; Braam, S.R.; Tertoolen, L.G.; Del Sol, A.; Davis, R.P.; Mummery, C.L. Transcriptome of human foetal heart compared with cardiomyocytes from pluripotent stem cells. Development 2015, 142, 3231–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedada, F.B.; Chan, S.S.-K.; Metzger, S.K.; Zhang, L.; Zhang, J.; Garry, D.J.; Kamp, T.J.; Kyba, M.; Metzger, J.M. Acquisition of a Quantitative, Stoichiometrically Conserved Ratiometric Marker of Maturation Status in Stem Cell-Derived Cardiac Myocytes. Stem Cell Rep. 2014, 3, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Uosaki, H.; Cahan, P.; Lee, D.I.; Wang, S.; Miyamoto, M.; Fernandez, L.; Kass, D.A.; Kwon, C. Transcriptional Landscape of Cardiomyocyte Maturation. Cell Rep. 2015, 13, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; He, J.; Wang, Y.; Guo, Y.; Zhang, J.; Peng, L.; Wang, D.; Lin, Q.; Zhang, J.; Guo, Z.; et al. Qualitative transcriptional signatures for evaluating the maturity degree of pluripotent stem cell-derived cardiomyocytes. Stem. Cell Res. Ther. 2019, 10, 113. [Google Scholar] [CrossRef]
- Klein, I.; Ojamaa, K. Thyroid Hormone and the Cardiovascular System. N. Engl. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Funakoshi, S.; Fernandes, I.; Mastikhina, O.; Wilkinson, D.; Tran, T.; Dhahri, W.; Mazine, A.; Yang, D.; Burnett, B.; Lee, J.; et al. Generation of mature compact ventricular cardiomyocytes from human pluripotent stem cells. Nat. Commun. 2021, 12, 3155. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Horikoshi, Y.; Yan, Y.; Terashvili, M.; Wells, C.; Horikoshi, H.; Fujita, S.; Bosnjak, Z.J.; Bai, X. Fatty Acid-Treated Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes Exhibit Adult Cardiomyocyte-Like Energy Metabolism Phenotypes. Cells 2019, 8, 1095. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Minami, I.; Braas, D.; Pappoe, H.; Wu, X.; Sagadevan, A.; Vergnes, L.; Fu, K.; Morselli, M.; Dunham, C.; et al. Glucose inhibits cardiac muscle maturation through nucleotide biosynthesis. eLife 2017, 6, e29330. [Google Scholar] [CrossRef]
- Yoshida, S.; Miyagawa, S.; Fukushima, S.; Kawamura, T.; Kashiyama, N.; Ohashi, F.; Toyofuku, T.; Toda, K.; Sawa, Y. Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes by Soluble Factors from Human Mesenchymal Stem Cells. Mol. Ther. 2018, 26, 2681–2695. [Google Scholar] [CrossRef] [Green Version]
- Talman, V.; Kivelä, R. Cardiomyocyte—Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front. Cardiovasc. Med. 2018, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abecasis, B.; Gomes-Alves, P.; Rosa, S.; Gouveia, P.J.; Ferreira, L.; Serra, M.; Alves, P.M. Unveiling the molecular crosstalk in a human induced pluripotent stem cell-derived cardiac model. Biotechnol. Bioeng. 2019, 116, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, T.; Okano, S.; Ichimura, H.; Kadota, S.; Tanaka, Y.; Minami, I.; Uesugi, M.; Wada, Y.; Saito, N.; Okada, K.; et al. Impact of extracellular matrix on engraftment and maturation of pluripotent stem cell-derived cardiomyocytes in a rat myocardial infarct model. Sci. Rep. 2017, 7, 8630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herron, T.J.; Rocha, A.M.D.; Campbell, K.F.; Ponce-Balbuena, D.; Willis, B.C.; Guerrero-Serna, G.; Liu, Q.; Klos, M.; Musa, H.; Zarzoso, M.; et al. Extracellular Matrix–Mediated Maturation of Human Pluripotent Stem Cell–Derived Cardiac Monolayer Structure and Electrophysiological Function. Circ. Arrhythmia Electrophysiol. 2016, 9, e003638. [Google Scholar] [CrossRef] [Green Version]
- Chun, Y.W.; Balikov, D.A.; Feaster, T.K.; Williams, C.H.; Sheng, C.C.; Lee, J.-B.; Boire, T.C.; Neely, M.D.; Bellan, L.M.; Ess, K.C.; et al. Combinatorial polymer matrices enhance in vitro maturation of human induced pluripotent stem cell-derived cardiomyocytes. Biomaterials 2015, 67, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.-C.; Ting, S.; Lee, Y.-K.; Ng, K.-M.; Zhang, J.; Chen, Z.; Siu, C.-W.; Oh, S.K.W.; Tse, H.-F. Electrical Stimulation Promotes Maturation of Cardiomyocytes Derived from Human Embryonic Stem Cells. J. Cardiovasc. Transl. Res. 2013, 6, 989–999. [Google Scholar] [CrossRef]
- Mirbagheri, M.; Adibnia, V.; Hughes, B.R.; Waldman, S.D.; Banquy, X.; Hwang, D.K. Advanced cell culture platforms: A growing quest for emulating natural tissues. Mater. Horiz. 2019, 6, 45–71. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Cho, G.-S.; Lee, D.I.; Tampakakis, E.; Murphy, S.; Andersen, P.; Uosaki, H.; Chelko, S.; Chakir, K.; Hong, I.; Seo, K.; et al. Neonatal Transplantation Confers Maturation of PSC-Derived Cardiomyocytes Conducive to Modeling Cardiomyopathy. Cell Rep. 2017, 18, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hua, Y.; Miyagawa, S.; Zhang, J.; Li, L.; Liu, L.; Sawa, Y. hiPSC-Derived Cardiac Tissue for Disease Modeling and Drug Discovery. Int. J. Mol. Sci. 2020, 21, 8893. [Google Scholar] [CrossRef] [PubMed]
- Cavero, I.; Holzgrefe, H. CiPA: Ongoing testing, future qualification procedures, and pending issues. J. Pharmacol. Toxicol. Methods 2015, 76, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Blinova, K.; Dang, Q.; Millard, D.; Smith, G.; Pierson, J.; Guo, L.; Brock, M.; Lu, H.R.; Kraushaar, U.; Zeng, H.; et al. International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep. 2018, 24, 3582–3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, Y.; Yamazaki, D.; Osada, T.; Yoshinaga, T.; Sawada, K. Development of torsadogenic risk assessment using human induced pluripotent stem cell-derived cardiomyocytes: Japan iPS Cardiac Safety Assessment (JiCSA) update. J. Pharmacol. Sci. 2018, 138, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Yamazaki, D.; Kurokawa, J.; Inutsuka, T.; Sekino, Y. Points to consider for a validation study of iPS cell-derived cardiomyocytes using a multi-electrode array system. J. Pharmacol. Toxicol. Methods. 2016, 81, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Ando, H.; Yoshinaga, T.; Yamamoto, W.; Asakura, K.; Uda, T.; Taniguchi, T.; Ojima, A.; Shinkyo, R.; Kikuchi, K.; Osada, T.; et al. A new paradigm for drug-induced torsadogenic risk assessment using human iPS cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods. 2017, 84, 111–127. [Google Scholar] [CrossRef]
- Yamazaki, D.; Kitaguchi, T.; Ishimura, M.; Taniguchi, T.; Yamanishi, A.; Saji, D.; Takahashi, E.; Oguchi, M.; Moriyama, Y.; Maeda, S.; et al. Proarrhythmia risk prediction using human induced pluripotent stem cell-derived cardiomyocytes. J. Pharmacol. Sci. 2018, 136, 249–256. [Google Scholar] [CrossRef]
- Weissig, V. Drug Development for the Therapy of Mitochondrial Diseases. Trends Mol. Med. 2020, 26, 40–57. [Google Scholar] [CrossRef] [PubMed]
- Kitani, T.; Ong, S.G.; Lam, C.K.; Rhee, J.W.; Zhang, J.Z.; Oikonomopoulos, A.; Ma, N.; Tian, L.; Lee, J.; Telli, M.L.; et al. Human-Induced Pluripotent Stem Cell Model of Trastuzumab-Induced Cardiac Dysfunction in Patients With Breast Cancer. Circulation 2019, 139, 2451–2465. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, L.R.; Chapman, K.; Xie, M.; Maifoshie, E.; Jenkins, M.; Golforoush, P.A.; Bellahcene, M.; Noseda, M.; Faust, D.; Jarvis, A.; et al. MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Cell Stem Cell 2019, 24, 579–591.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef]
- Morén, C.; Juárez-Flores, D.L.; Cardellach, F.; Garrabou, G. The Role of Therapeutic Drugs on Acquired Mitochondrial Toxicity. Curr. Drug Metab. 2016, 17, 648–662. [Google Scholar] [CrossRef]
- Finsterer, J.; Segall, L. Drugs interfering with mitochondrial disorders. Drug Chem. Toxicol. 2010, 33, 138–151. [Google Scholar] [CrossRef]
- Song, M.; Dorn, G.W., II. Mitoconfusion: Noncanonical Functioning of Dynamism Factors in Static Mitochondria of the Heart. Cell Metab. 2015, 21, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Qi, Y.; Ye, Y.; Yue, P.; Zhang, D.; Li, Y. Mechanotranduction Pathways in the Regulation of Mitochondrial Homeostasis in Cardiomyocytes. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Filosto, M.; Simoncini, C.; Siciliano, G. Drugs and mitochondrial diseases: 40 queries and answers. Expert Opin. Pharmacother. 2012, 13, 527–543. [Google Scholar] [CrossRef]
- Coutinho, E.; Batista, C.; Sousa, F.; Queiroz, J.; Costa, D. Mitochondrial Gene Therapy: Advances in Mitochondrial Gene Cloning, Plasmid Production, and Nanosystems Targeted to Mitochondria. Mol. Pharm. 2017, 14, 626–638. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| a. Genes in Mitochondrial DNA to Disease Relationship for Mitochondrial Disorders | |||
|---|---|---|---|
| Gene | OMIM ID | Cardiac Phenotype | Other Phenotypes/Mitochondrial Diseases |
| Subunits of respiratory chain complex | |||
| MT-ND1 | 516000 | HCM, LVNC | LHON (Leber’s hereditary optic neuropathy) |
| MT-ND4 | 516003 | HCM | LHON, progressive dystonia |
| MT-ND5 | 516005 | HCM. WPW | Leigh syndrome |
| MT-ATP6/8 | 516060 | HCM | |
| MT-ATP6 | 516060 | HCM | NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa.), Leigh disease |
| MT-ND6 | 516006 | DCM, HCM | LHON, MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) |
| MT-CYB | 516020 | HCM | Septo-optic dysplasia |
| Mitochondrial protein synthesis | |||
| MT-TL1 | 590050 | HCM, DCM, RCM, LVNC | MELAS, Leigh syndrome, CPEO (chronic progressive external ophthalmoplegia), mitochondrial myopathy |
| MT-TI | 590045 | HCM, DCM | |
| MT-TK | 590060 | HCM, DCM | MERRF (myoclonus epilepsy associated with ragged red fibers), Leigh syndrome |
| MT-TV | 590105 | HCM | Leigh syndrome |
| MT-RNR1 | 561000 | RCM | Maternally inherited deafness |
| b. Genes in Nuclear DNA to Disease Relationship for Mitochondrial Disorders | |||
| Gene | OMIM ID | Cardiac Phenotype | Other phenotypes/mitochondrial diseases |
| Subunits of the respiratory chain complex | |||
| NDUFS2 | 252010 | HCM | Mitochondrial complex I deficiency |
| NDUFV2 | 252010 | HCM | Mitochondrial complex I deficiency |
| NDUFA11 | 252010 | HCM | Mitochondrial complex I deficiency |
| NDUFB11 | 300403 | LVNC, WPW | Mitochondrial complex I deficiency |
| SDHA | 252011 | DCM, LVNC | Mitochondrial complex II deficiency |
| Assembly factor | |||
| NDUFAF1 | 252010 | HCM | Mitochondrial complex I deficiency |
| ACAD9 | 611126 | HCM | Mitochondrial complex I deficiency |
| SCO2 | 604377 | HCM | Cytochrome c oxidase deficiency |
| COX10 | 220110 | HCM | Mitochondrial complex IV deficiency |
| COX15 | 615119 | HCM | Cytochrome c oxidase deficiency |
| COA6 | 614772 | HCM | |
| TMEM70 | 614052 | HCM | Mitochondrial complex V (ATP synthase) deficiency |
| Mitochondrial protein synthesis | |||
| AARS2 | 614096 | HCM | COXPD (combined oxidative phosphorylation deficiency) 8 |
| MRPS22 | 611719 | HCM | COXPD8 |
| TSFM | 610505 | HCM | COXPD3 |
| GTPBP3 | 616198 | HCM, DCM | COXPD23 |
| MTO1 | 614702 | HCM | COXPD10 |
| ELAC2 | 615440 | HCM | COXPD17 |
| Maintenance of mitochondrial integrity | |||
| TAZ | 302060 | DCM, LVNC | BTHS (Barth syndrome) |
| AGK | 212350 | HCM | Sengers syndrome |
| SLC22A5 | 212140 | HCM, DCM | Systemic primary carnitine deficiency |
| ACADVL | 201475 | HCM, DCM | Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency |
| HADHA | 609015 | DCM | Mitochondrial trifunctional protein (MTP) deficiency with myopathy and neuropathy |
| ATAD3A-C dup | 612316 | HCM | |
| Mitochondrial DNA stability | |||
| SLC25A4 | 615418 | HCM | Mitochondrial DNA depletion syndrome-12 |
| QRSL1 | 617209 | HCM | COXPD40 |
| KARS | 619147 | HCM | Infantile-onset progressive leukoencephalopathy with or without deafness |
| TOP3A | 601243 | DCM | |
| Iron homeostasis | |||
| FXN | 229300 | HCM | Friedreich ataxia |
| BOLA3 | 614299 | HCM | Multiple mitochondrial dysfunctions syndrome-2 with hyperglycinemia |
| Coenzyme Q10 biosynthesis | |||
| COQ9 | 614654 | HCM | Coenzyme Q10 deficiency 5 |
| COQ4 | 616276 | HCM | Coenzyme Q10 deficiency 7 |
| Mitochondrial protein transport | |||
| DNAJC19 | 610198 | DCM, LVNC | 3-methylglutaconic aciduria type V |
| Gene | Variants | Protein | Disease | Phenotype | Reference |
|---|---|---|---|---|---|
| TAZ | c.517delG | Tafazzin | Barth syndrome | Impaired sarcomere structure and function | [16,128,129,130] |
| c.328T > C | Increased reactive oxygen species | ||||
| DNAJC19 | (rs137854888) | Mitochondrial import inner membrane translocase subunit TIM14 | Dilated cardiomyopathy with ataxia syndrome (DCMA) | Impaired mitochondria | [131] |
| Conduction defects | |||||
| FXN | Expanded GAA repeats | Frataxin | Friedreich ataxia (hypertrophic cardiomyopathy) | Disorganized mitochondria | [132] |
| Impaired Ca2+ handling | |||||
| Increased BNP expression | |||||
| Disrupted iron homeostasis | |||||
| Mitochondrial dysfunction and degeneration | [133] | ||||
| Decreased mitochondrial membrane potential | |||||
| MT-RNR2 | m.2336T > C | Mitochondrial encoded16S rRNA | Hypertrophic cardiomyopathy | Mitochondrial dysfunction | [126] |
| decreased mitochondrial potential | |||||
| Electrophysiological disturbances |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokuyama, T.; Ahmed, R.E.; Chanthra, N.; Anzai, T.; Uosaki, H. Disease Modeling of Mitochondrial Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells. Biology 2021, 10, 981. https://doi.org/10.3390/biology10100981
Tokuyama T, Ahmed RE, Chanthra N, Anzai T, Uosaki H. Disease Modeling of Mitochondrial Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells. Biology. 2021; 10(10):981. https://doi.org/10.3390/biology10100981
Chicago/Turabian StyleTokuyama, Takeshi, Razan Elfadil Ahmed, Nawin Chanthra, Tatsuya Anzai, and Hideki Uosaki. 2021. "Disease Modeling of Mitochondrial Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells" Biology 10, no. 10: 981. https://doi.org/10.3390/biology10100981
APA StyleTokuyama, T., Ahmed, R. E., Chanthra, N., Anzai, T., & Uosaki, H. (2021). Disease Modeling of Mitochondrial Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells. Biology, 10(10), 981. https://doi.org/10.3390/biology10100981