
Induced Pluripotent Stem Cell-Derived Cardiomyocytes with SCN5A R1623Q Mutation Associated with Severe Long QT Syndrome in Fetuses and Neonates Recapitulates Pathophysiological Phenotypes

 ,
,  , ,
, , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Generation of iPSCs from Lymphoblastoid Cell Lines (LCLs)
2.3. Characterization of Reprogrammed Cells
2.3.1. Immunocytochemistry
2.3.2. Sequence and Karyotype Analyses
2.3.3. Teratoma Formation
2.4. Differentiation of Cardiomyocytes from iPSCs
2.5. Characterization of iPSC-CMs
2.5.1. Quantitative Real-Time PCR (qRT-PCR)
2.5.2. Field Potential Recording Using the MED64 System and Data Analysis
2.6. Construction of Expression Plasmids
2.7. Cell Culture and Generation of Nav1.5 Stable Expression Cell Lines
2.8. Automated Patch-Clamp Recording
2.9. Statistical Analysis
3. Results
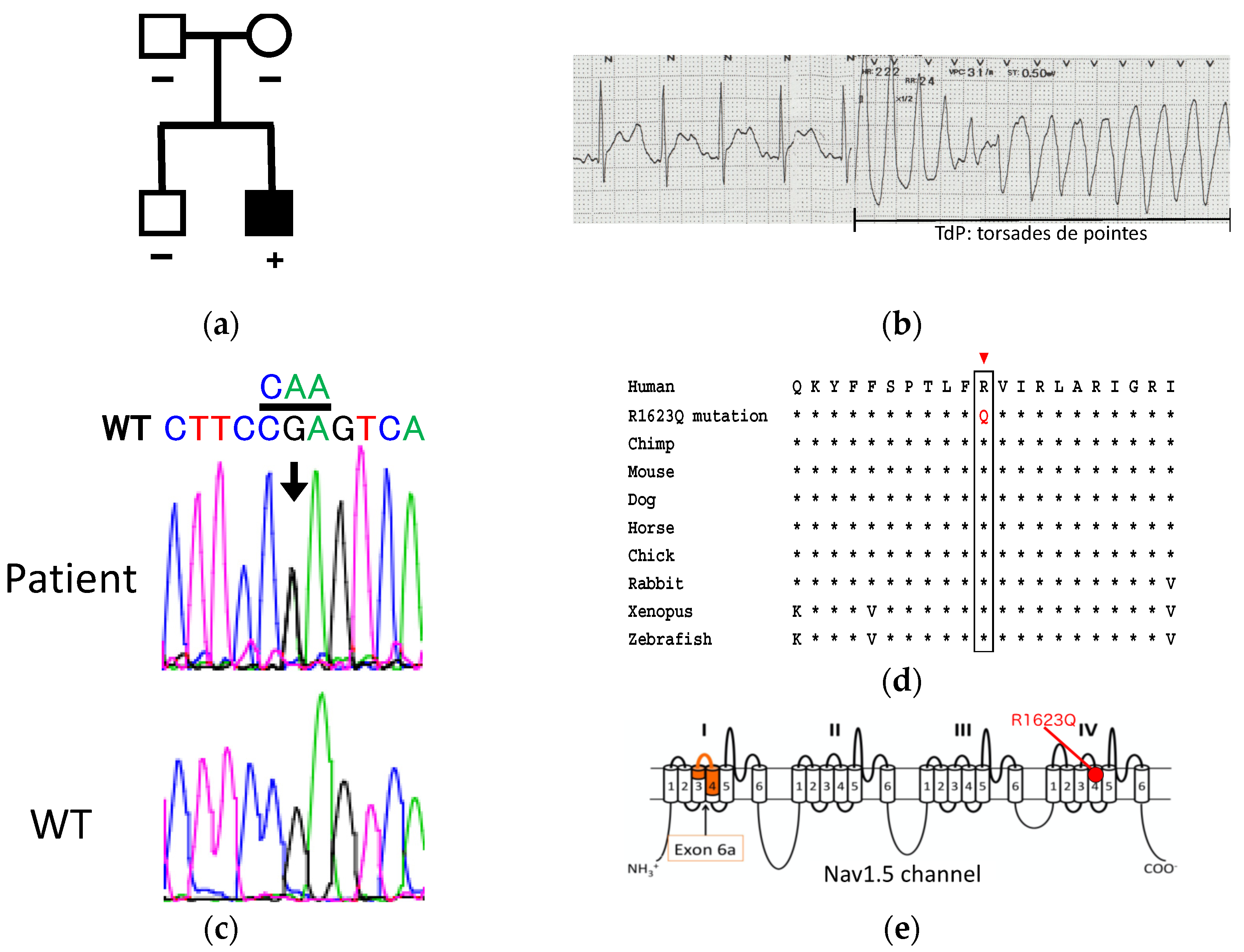
3.1. Patient Information and Genetic Analysis
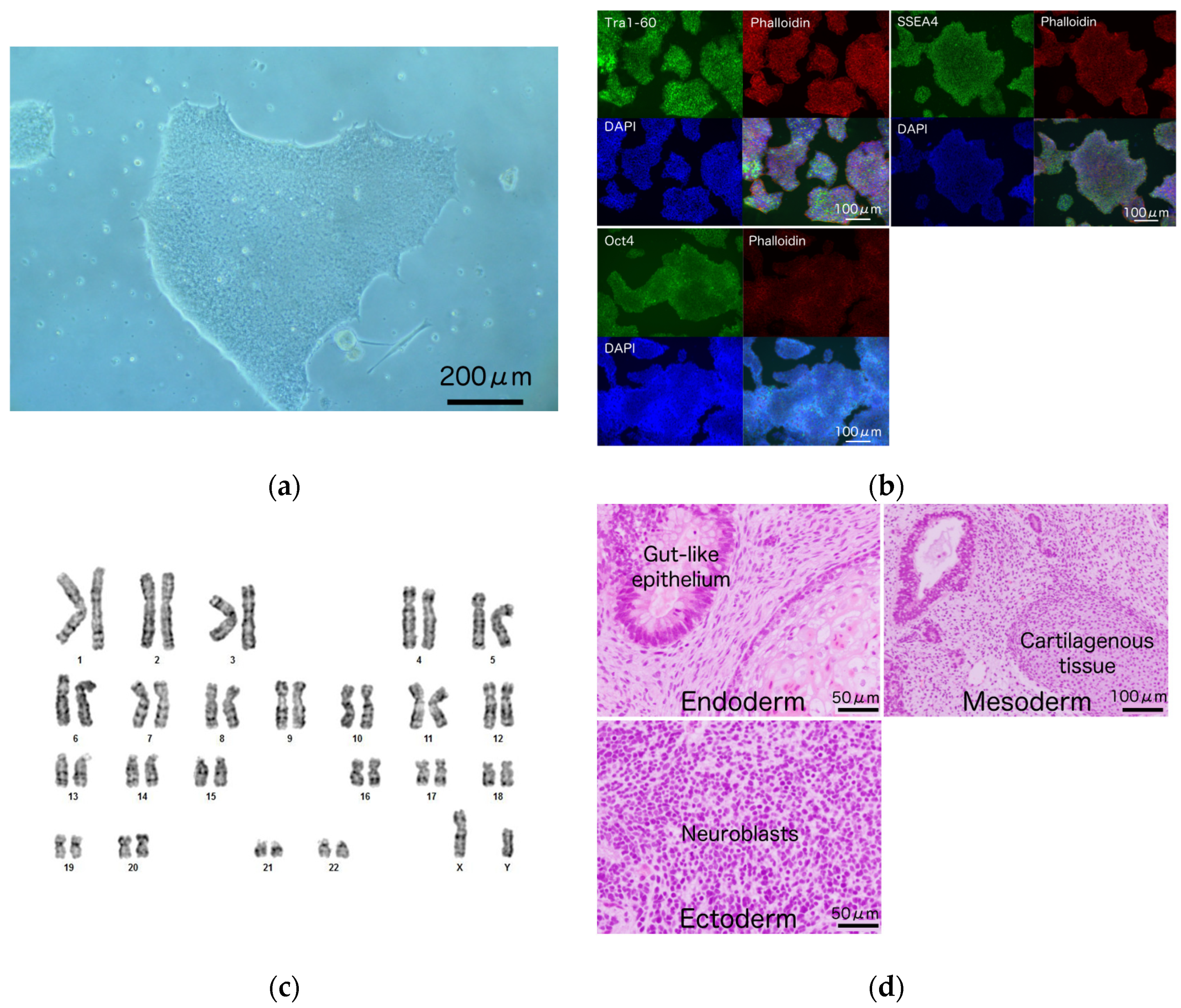
3.2. Generation and Characterization of iPSCs from a Patient Harboring R1623Q Mutation in Nav1.5
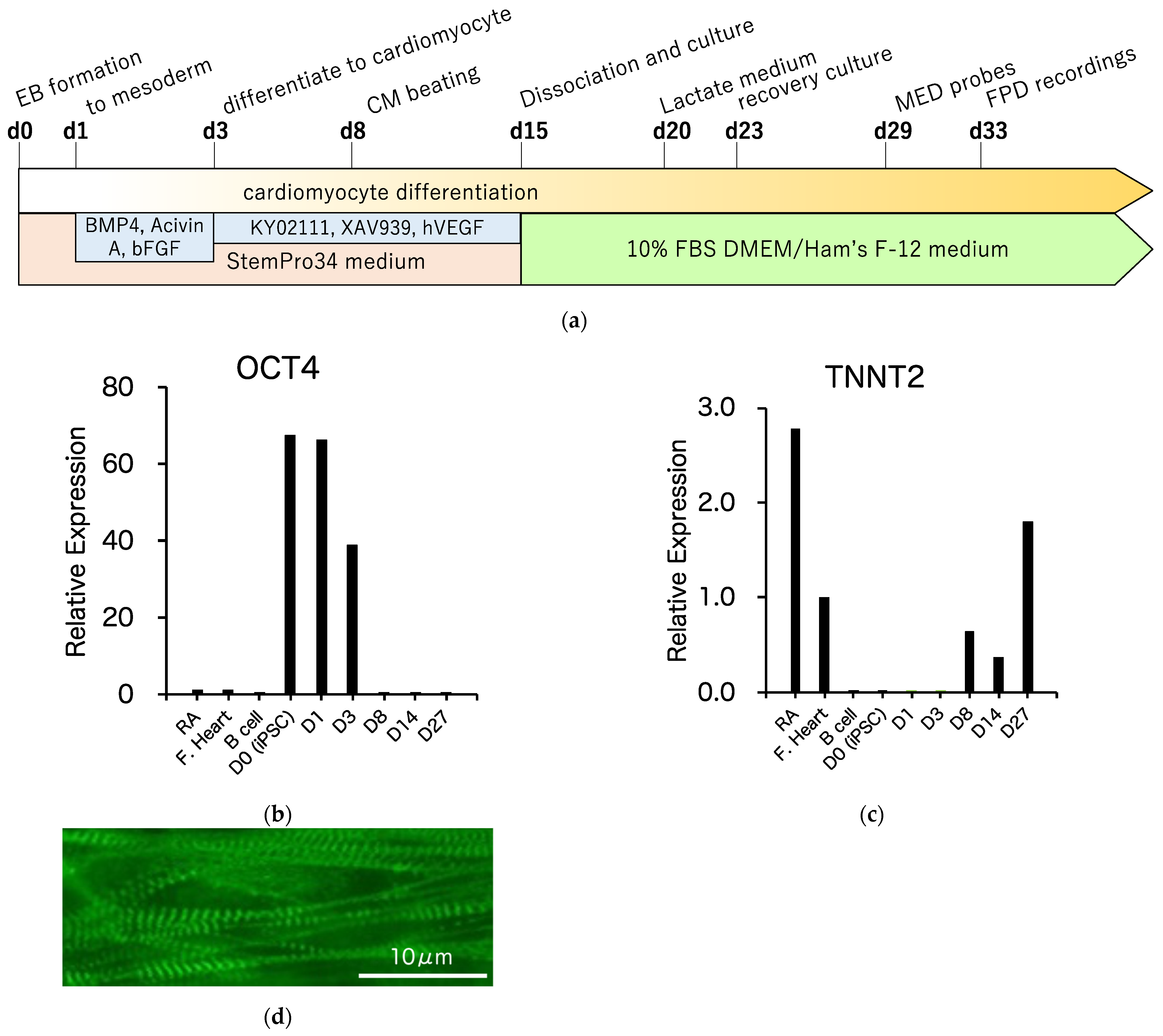
3.3. Differentiation of R1623Q Mutation-Harboring iPSCs into Cardiomyocytes
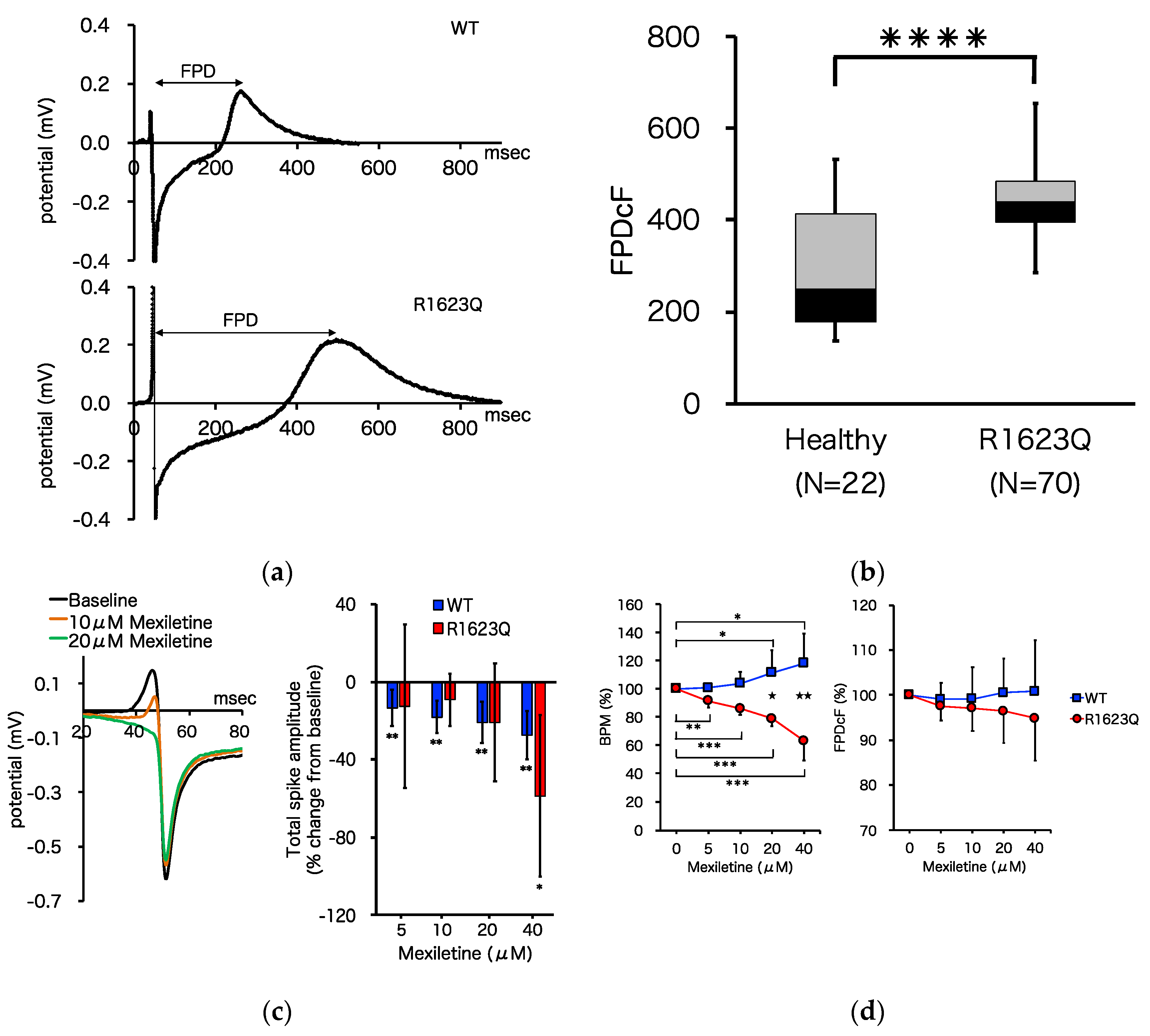
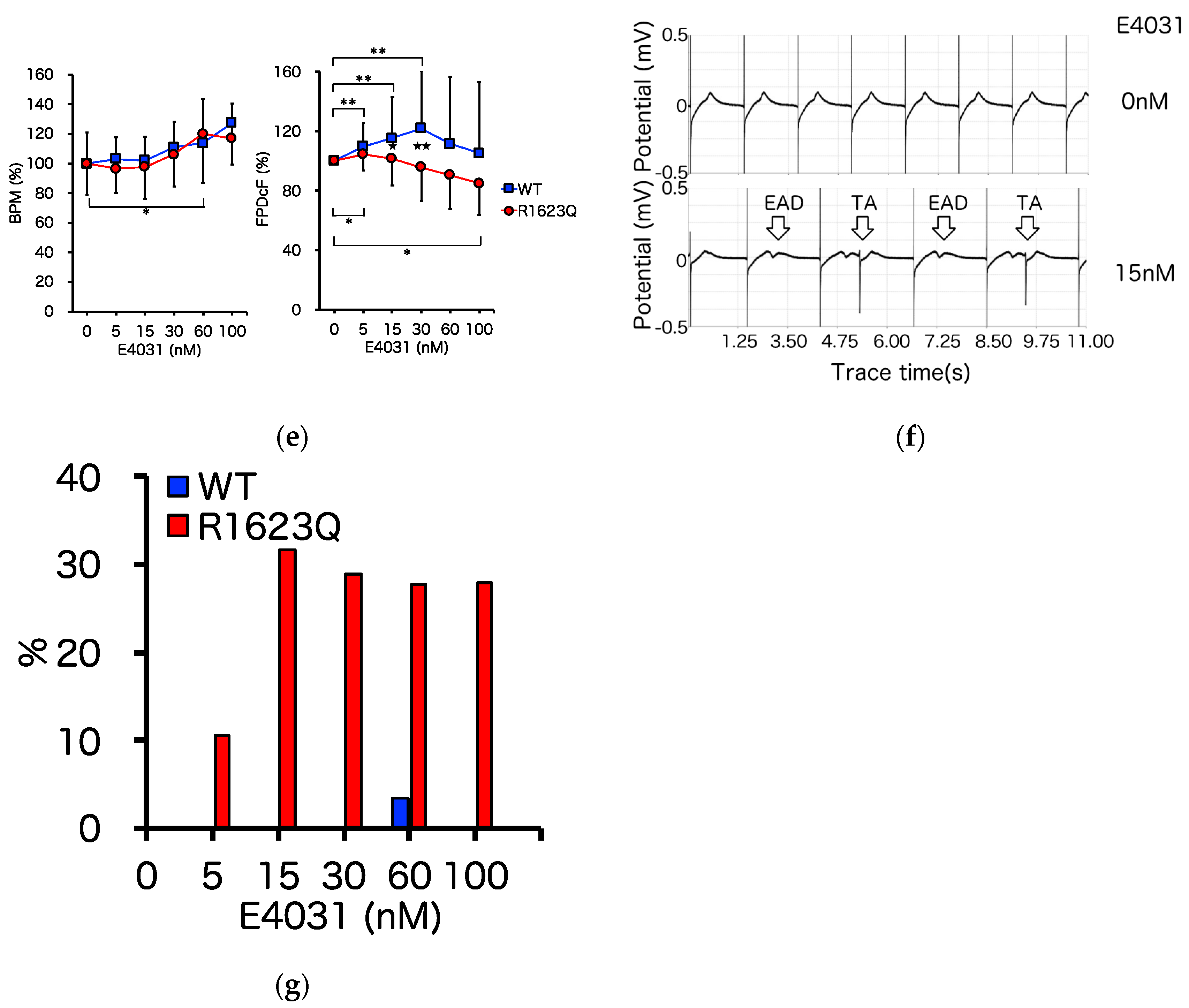
3.4. MED64 Analysis
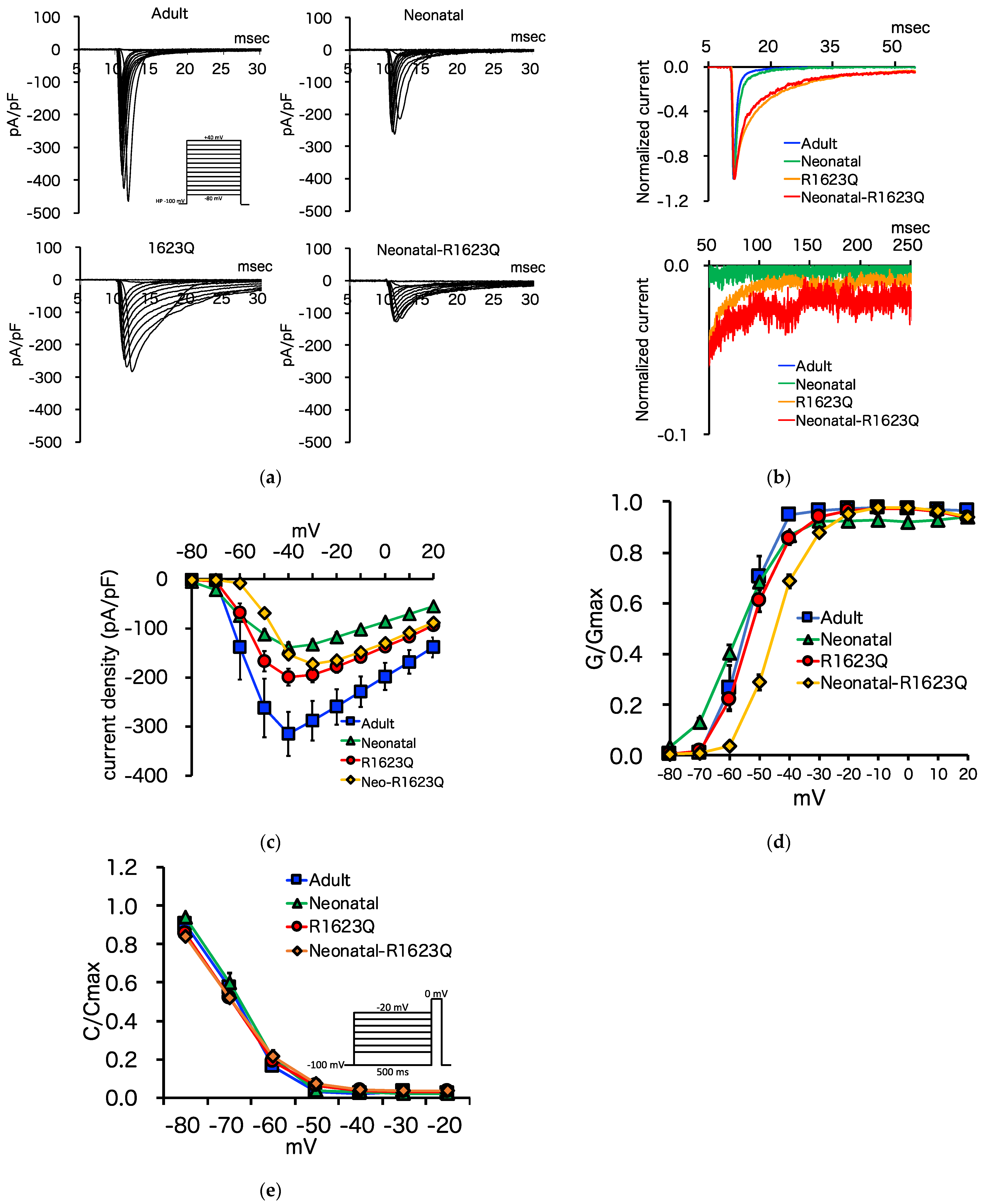
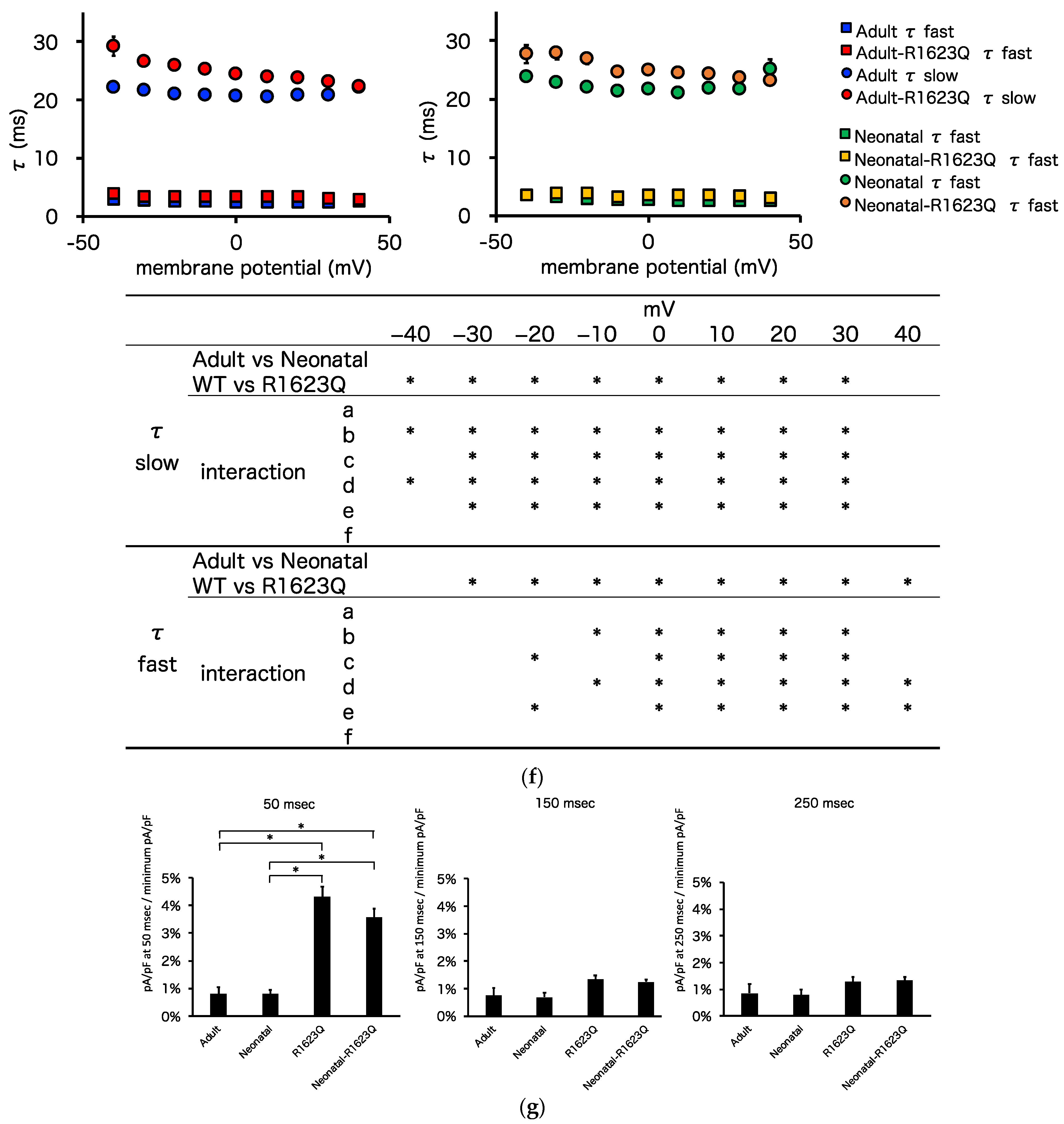
3.5. Automated Patch-Clamp Analysis of Neonatal R1623Q Nav1.5 Variant
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moss, A.J.; McDonald, J. Unilateral cervicothoracic sympathetic ganglionectomy for the treatment of long QT interval syndrome. N. Engl. J. Med. 1971, 285, 903–904. [Google Scholar] [CrossRef]
- Yokoo, N.; Baba, S.; Kaichi, S.; Niwa, A.; Mima, T.; Doi, H.; Yamanaka, S.; Nakahata, T.; Heike, T. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2009, 387, 482–488. [Google Scholar] [CrossRef]
- Aiba, T. Recent understanding of clinical sequencing and gene-based risk stratification in inherited primary arrhythmia syndrome. J. Cardiol. 2019, 73, 335–342. [Google Scholar] [CrossRef]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- Yamagishi, H.; Furutani, M.; Kamisago, M.; Morikawa, Y.; Kojima, Y.; Hino, Y.; Furutani, Y.; Kimura, M.; Imamura, S.; Takao, A.; et al. A de novo missense mutation (R1623Q) of the SCN5A gene in a Japanese girl with sporadic long QT sydrome. Mutations in brief no. 140. Online. Hum. Mutat. 1998, 11, 481. [Google Scholar] [CrossRef]
- Chen, L.Q.; Santarelli, V.; Horn, R.; Kallen, R.G. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J. Gen. Physiol. 1996, 108, 549–556. [Google Scholar] [CrossRef]
- Kambouris, N.G.; Nuss, H.B.; Johns, D.C.; Tomaselli, G.F.; Marban, E.; Balser, J.R. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation 1998, 97, 640–644. [Google Scholar] [CrossRef]
- Kambouris, N.G.; Nuss, H.B.; Johns, D.C.; Marban, E.; Tomaselli, G.F.; Balser, J.R. A revised view of cardiac sodium channel “blockade” in the long-QT syndrome. J. Clin. Investig. 2000, 105, 1133–1140. [Google Scholar] [CrossRef][Green Version]
- Makita, N.; Shirai, N.; Nagashima, M.; Matsuoka, R.; Yamada, Y.; Tohse, N.; Kitabatake, A. A de novo missense mutation of human cardiac Na+ channel exhibiting novel molecular mechanisms of long QT syndrome. FEBS Lett. 1998, 423, 5–9. [Google Scholar] [CrossRef]
- Miller, T.E.; Estrella, E.; Myerburg, R.J.; Garcia de Viera, J.; Moreno, N.; Rusconi, P.; Ahearn, M.E.; Baumbach, L.; Kurlansky, P.; Wolff, G.; et al. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation 2004, 109, 3029–3034. [Google Scholar] [CrossRef]
- Ten Harkel, A.D.; Witsenburg, M.; de Jong, P.L.; Jordaens, L.; Wijman, M.; Wilde, A.A. Efficacy of an implantable cardioverter-defibrillator in a neonate with LQT3 associated arrhythmias. Europace 2005, 7, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Hernandez, M.; Edwards, C.; Edkins, E.; Jansen, F.E.; Scheffer, I.E.; Berkovic, S.F.; Mulley, J.C. Neonatal seizures and long QT syndrome: A cardiocerebral channelopathy? Epilepsia 2010, 51, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wehrens, X.H. Enhanced impact of SCN5A mutation associated with long QT syndrome in fetal splice isoform. Heart Rhythm 2012, 9, 598–599. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cuneo, B.F.; Strasburger, J.F.; Yu, S.; Horigome, H.; Hosono, T.; Kandori, A.; Wakai, R.T. In utero diagnosis of long QT syndrome by magnetocardiography. Circulation 2013, 128, 2183–2191. [Google Scholar] [CrossRef]
- Cuneo, B.F.; Etheridge, S.P.; Horigome, H.; Sallee, D.; Moon-Grady, A.; Weng, H.Y.; Ackerman, M.J.; Benson, D.W. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: Risk stratification of perinatal long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2013, 6, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, B.F.; Ambrose, S.E.; Tworetzky, W. Detection and successful treatment of emergent anti-SSA mediated fetal atrioventricular block. Am. J. Obstet. Gynecol. 2016. [Google Scholar] [CrossRef]
- Strand, S.; Strasburger, J.F.; Cuneo, B.F.; Wakai, R.T. Complex and Novel Arrhythmias Precede Stillbirth in Fetuses with De Novo Long QT Syndrome. Circ. Arrhythm. Electrophysiol. 2020, 13, e008082. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Gallotti, R.G.; Shannon, K.M.; Bos, J.M.; Sadeghi, E.; Strasburger, J.F.; Wakai, R.T.; Horigome, H.; Clur, S.A.; Hill, A.C.; et al. Genotype Predicts Outcomes in Fetuses and Neonates With Severe Congenital Long QT Syndrome. JACC Clin. Electrophysiol. 2020, 6, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, N.; Hayama, E.; Furutani, Y.; Nakanishi, T. Prospective in vitro models of channelopathies and cardiomyopathies. Stem Cells Int. 2012, 2012, 439219. [Google Scholar] [CrossRef] [PubMed]
- Malan, D.; Friedrichs, S.; Fleischmann, B.K.; Sasse, P. Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ. Res. 2011, 109, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Kaifeng, S.; Dittmann, S.; Xu, G.; Gupta, M.K.; Linke, M.; Zechner, U.; Nguemo, F.; Milting, H.; Farr, M.; et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS ONE 2013, 8, e83005. [Google Scholar] [CrossRef]
- Malan, D.; Zhang, M.; Stallmeyer, B.; Muller, J.; Fleischmann, B.K.; Schulze-Bahr, E.; Sasse, P.; Greber, B. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res. Cardiol. 2016, 111, 14. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Makiyama, T.; Melgari, D.; Yamamoto, Y.; Wuriyanghai, Y.; Yokoi, F.; Nishiuchi, S.; Harita, T.; Hayano, M.; Kohjitani, H.; et al. Propranolol Attenuates Late Sodium Current in a Long QT Syndrome Type 3-Human Induced Pluripotent Stem Cell Model. Front. Cell Dev. Biol. 2020, 8, 761. [Google Scholar] [CrossRef] [PubMed]
- Onkal, R.; Mattis, J.H.; Fraser, S.P.; Diss, J.K.; Shao, D.; Okuse, K.; Djamgoz, M.B. Alternative splicing of Nav1.5: An electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms and critical involvement of a lysine residue. J. Cell Physiol. 2008, 216, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Sarao, R.; Gupta, S.K.; Auld, V.J.; Dunn, R.J. Developmentally regulated alternative RNA splicing of rat brain sodium channel mRNAs. Nucleic Acids Res. 1991, 19, 5673–5679. [Google Scholar] [CrossRef]
- Gustafson, T.A.; Clevinger, E.C.; O’Neill, T.J.; Yarowsky, P.J.; Krueger, B.K. Mutually exclusive exon splicing of type III brain sodium channel alpha subunit RNA generates developmentally regulated isoforms in rat brain. J. Biol. Chem. 1993, 268, 18648–18653. [Google Scholar] [CrossRef]
- Chioni, A.M.; Fraser, S.P.; Pani, F.; Foran, P.; Wilkin, G.P.; Diss, J.K.; Djamgoz, M.B. A novel polyclonal antibody specific for the Na(v)1.5 voltage-gated Na(+) channel ‘neonatal’ splice form. J. Neurosci. Methods 2005, 147, 88–98. [Google Scholar] [CrossRef]
- Murphy, L.L.; Moon-Grady, A.J.; Cuneo, B.F.; Wakai, R.T.; Yu, S.; Kunic, J.D.; Benson, D.W.; George, A.L., Jr. Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm 2012, 9, 590–597. [Google Scholar] [CrossRef]
- Walzik, S.; Schroeter, A.; Benndorf, K.; Zimmer, T. Alternative splicing of the cardiac sodium channel creates multiple variants of mutant T1620K channels. PLoS ONE 2011, 6, e19188. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, S.; Fujita, J.; Hishiki, T.; Matsuura, T.; Hattori, F.; Ohno, R.; Kanazawa, H.; Seki, T.; Nakajima, K.; Kishino, Y.; et al. Glutamine Oxidation Is Indispensable for Survival of Human Pluripotent Stem Cells. Cell Metab. 2016, 23, 663–674. [Google Scholar] [CrossRef]
- Puisieux, F.L.; Adamantidis, M.M.; Dumotier, B.M.; Dupuis, B.A. Cisapride-induced prolongation of cardiac action potential and early afterdepolarizations in rabbit Purkinje fibres. Br. J. Pharmacol. 1996, 117, 1377–1379. [Google Scholar] [CrossRef]
- Gintant, G.A.; Limberis, J.T.; McDermott, J.S.; Wegner, C.D.; Cox, B.F. The canine Purkinje fiber: An in vitro model system for acquired long QT syndrome and drug-induced arrhythmogenesis. J. Cardiovasc. Pharmacol. 2001, 37, 607–618. [Google Scholar] [CrossRef]
- Barandi, L.; Virag, L.; Jost, N.; Horvath, Z.; Koncz, I.; Papp, R.; Harmati, G.; Horvath, B.; Szentandrassy, N.; Banyasz, T.; et al. Reverse rate-dependent changes are determined by baseline action potential duration in mammalian and human ventricular preparations. Basic Res. Cardiol. 2010, 105, 315–323. [Google Scholar] [CrossRef]
- Sutanto, H.; Heijman, J. Beta-Adrenergic Receptor Stimulation Modulates the Cellular Proarrhythmic Effects of Chloroquine and Azithromycin. Front. Physiol. 2020, 11, 587709. [Google Scholar] [CrossRef]
- Shryock, J.C.; Song, Y.; Rajamani, S.; Antzelevitch, C.; Belardinelli, L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc. Res. 2013, 99, 600–611. [Google Scholar] [CrossRef]
- Veerman, C.C.; Mengarelli, I.; Lodder, E.M.; Kosmidis, G.; Bellin, M.; Zhang, M.; Dittmann, S.; Guan, K.; Wilde, A.A.M.; Schulze-Bahr, E.; et al. Switch From Fetal to Adult SCN5A Isoform in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Unmasks the Cellular Phenotype of a Conduction Disease-Causing Mutation. J. Am. Heart. Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Abdelsayed, M.; Ruprai, M.; Ruben, P.C. The efficacy of Ranolazine on E1784K is altered by temperature and calcium. Sci. Rep. 2018, 8, 3643. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | ‘Adult’ Nav1.5 | ‘Neonatal’ Nav1.5 | ‘R1623Q’ Nav1.5 | ‘Neonatal-R1623Q’ Nav1.5 | Two-Way ANOVA (p-Value) | Interaction (p-Value) | |
|---|---|---|---|---|---|---|---|
| Adult vs. Neonatal | WT vs. R1623Q | ||||||
| Peak sodium current density (pA/pF) | −315.0 ± 44.9 | −139.5 ± 11.0 | −199.3 ± 17.3 | −172.6 ± 9.9 | * | * | * a, * b, * c |
| Steady-state activation (V1/2, mV) | 55.4 ± 1.3 | −57.9 ± 1.0 | −52.6 ± 1.2 | −45.0 ± 0.8 | * | * | * c, * d, * e, * f |
| Steady-state activation (k) | 4.4 ± 0.4 | 4.9 ± 0.3 | 4.7 ± 0.3 | 4.6 ± 0.2 | - | ||
| Steady-state inactivation (V1/2, mV) | −70.1 ± 1.0 | −68.0 ± 1.1 | −70.7 ± 0.8 | −70.3 ± 0.6 | - | ||
| Steady-state inactivation (k) | 4.4 ± 0.1 | 4.8 ± 0.1 | 6.0 ± 0.1 | 6.8 ± 0.2 | * | * | * b, * c, * d, * e, * f |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayama, E.; Furutani, Y.; Kawaguchi, N.; Seki, A.; Nagashima, Y.; Okita, K.; Takeuchi, D.; Matsuoka, R.; Inai, K.; Hagiwara, N.; et al. Induced Pluripotent Stem Cell-Derived Cardiomyocytes with SCN5A R1623Q Mutation Associated with Severe Long QT Syndrome in Fetuses and Neonates Recapitulates Pathophysiological Phenotypes. Biology 2021, 10, 1062. https://doi.org/10.3390/biology10101062
Hayama E, Furutani Y, Kawaguchi N, Seki A, Nagashima Y, Okita K, Takeuchi D, Matsuoka R, Inai K, Hagiwara N, et al. Induced Pluripotent Stem Cell-Derived Cardiomyocytes with SCN5A R1623Q Mutation Associated with Severe Long QT Syndrome in Fetuses and Neonates Recapitulates Pathophysiological Phenotypes. Biology. 2021; 10(10):1062. https://doi.org/10.3390/biology10101062
Chicago/Turabian StyleHayama, Emiko, Yoshiyuki Furutani, Nanako Kawaguchi, Akiko Seki, Yoji Nagashima, Keisuke Okita, Daiji Takeuchi, Rumiko Matsuoka, Kei Inai, Nobuhisa Hagiwara, and et al. 2021. "Induced Pluripotent Stem Cell-Derived Cardiomyocytes with SCN5A R1623Q Mutation Associated with Severe Long QT Syndrome in Fetuses and Neonates Recapitulates Pathophysiological Phenotypes" Biology 10, no. 10: 1062. https://doi.org/10.3390/biology10101062
APA StyleHayama, E., Furutani, Y., Kawaguchi, N., Seki, A., Nagashima, Y., Okita, K., Takeuchi, D., Matsuoka, R., Inai, K., Hagiwara, N., & Nakanishi, T. (2021). Induced Pluripotent Stem Cell-Derived Cardiomyocytes with SCN5A R1623Q Mutation Associated with Severe Long QT Syndrome in Fetuses and Neonates Recapitulates Pathophysiological Phenotypes. Biology, 10(10), 1062. https://doi.org/10.3390/biology10101062