
In Situ Hybridization of Pulp Fibers Using Mg-Al Layered Double Hydroxides

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Methods
2.2.1. LDH Synthesis and Fiber Mineralization
2.2.2. X-Ray Diffractometry
2.2.3. ATR-FTIR
2.2.4. Thermogravimetry
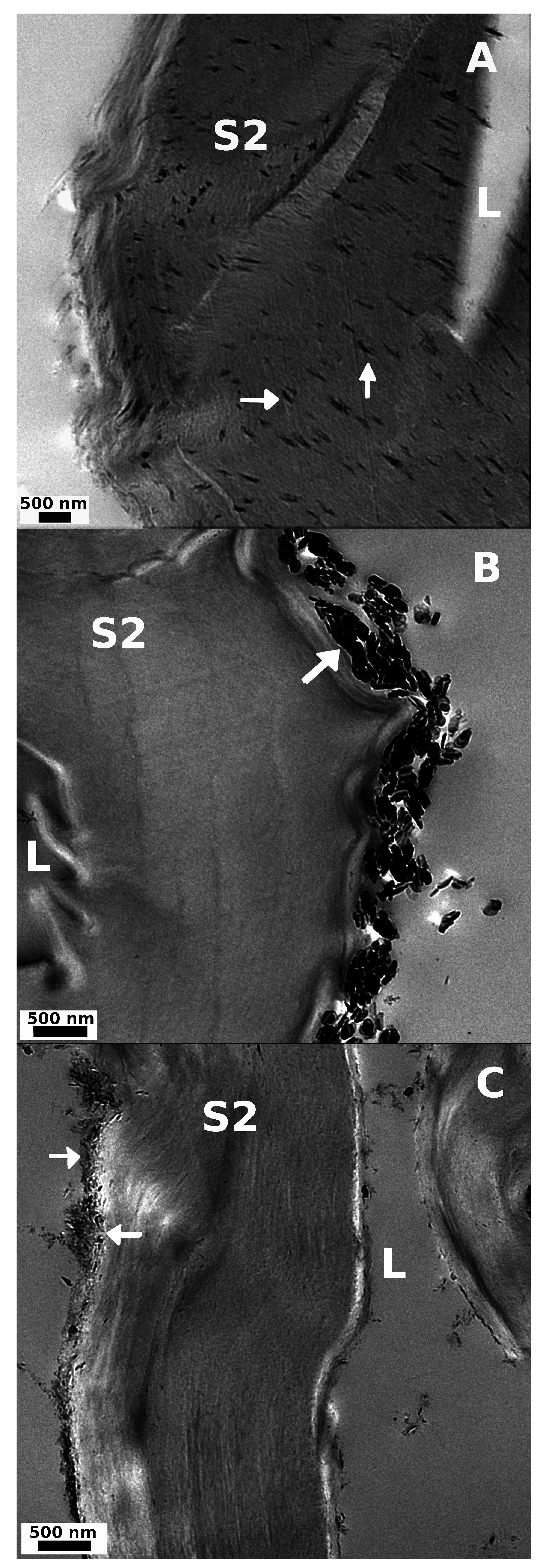
2.2.5. Microscopy
2.2.6. Microrobotic Platform

2.2.7. Capillary Viscometry
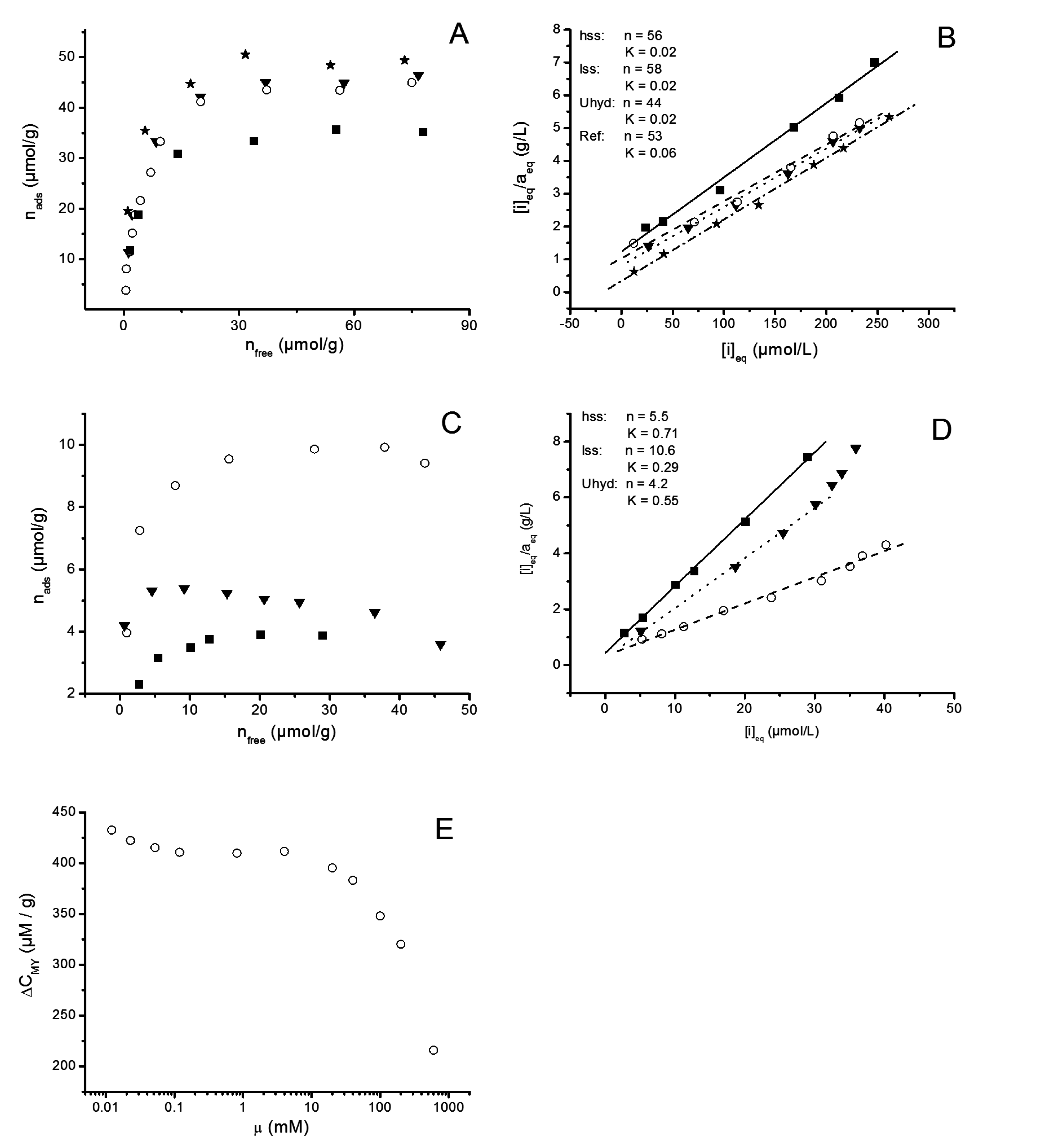
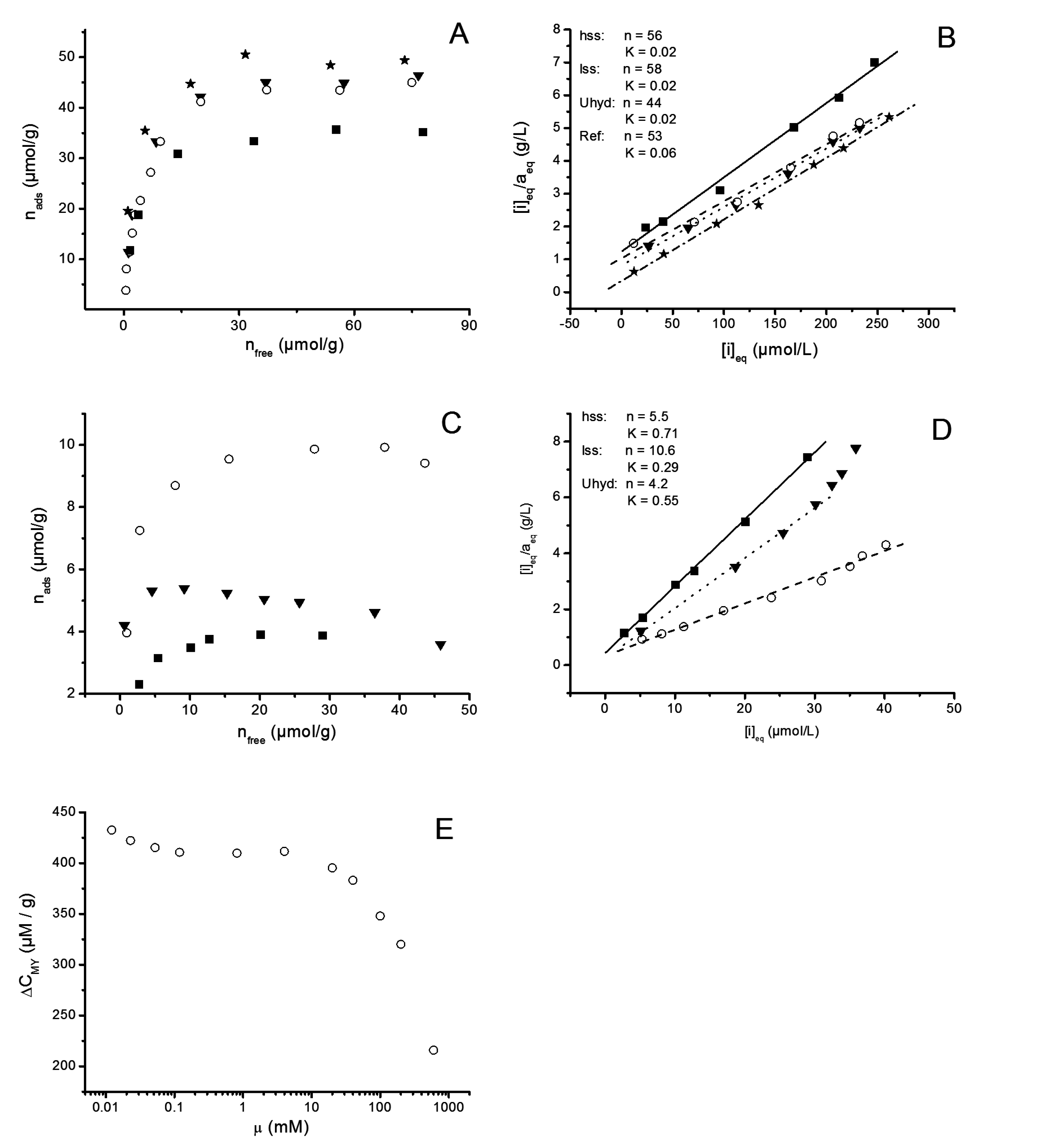
2.2.8. Adsorption Isotherms
3. Results and Discussion
3.1. LDH Synthesis
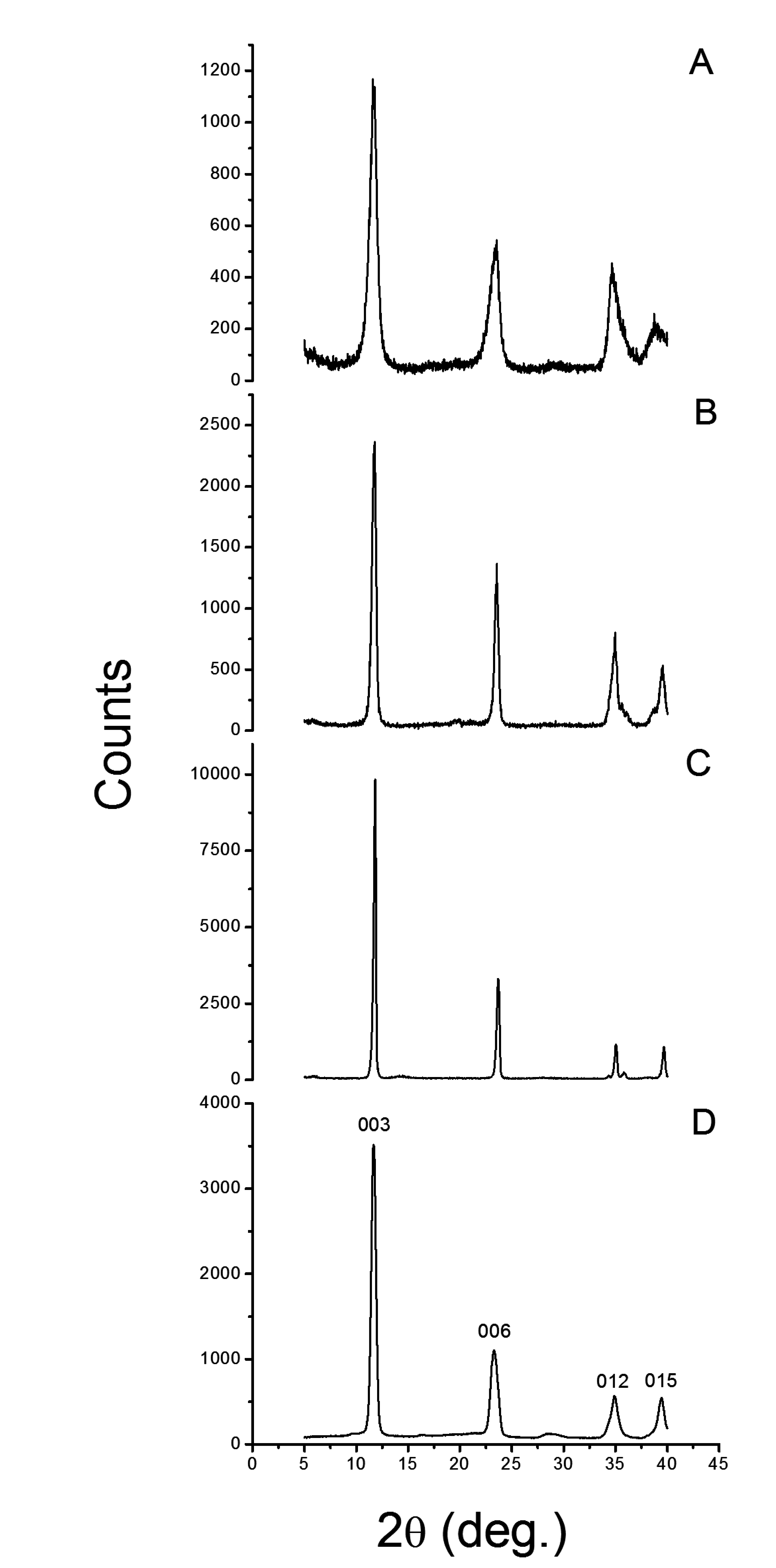
3.1.1. X-Ray Diffractometry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 2θ | a = b | c | l | FWHM | RPH | S/N |
|---|---|---|---|---|---|---|---|
| deg. | Å | Å | Å | deg. | % | ||
| HT | 11.68 | 3.10 | 22.73 | 7.58 | 0.522 | 100 | 360 |
| 23.28 | 0.781 | 29 | 105 | ||||
| 34.90 | 0.763 | 14 | 50 | ||||
| 39.44 | 0.613 | 14 | 50 | ||||
| LDH-U | 11.85 | 3.04 | 22.45 | 7.48 | 0.266 | 100 | 280 |
| 23.71 | 0.309 | 33 | 93 | ||||
| 35.04 | 0.257 | 11 | 31 | ||||
| 39.70 | 0.297 | 11 | 30 | ||||
| LDH-C | 11.75 | 3.08 | 22.61 | 7.54 | 0.459 | 100 | 47 |
| 23.53 | 0.426 | 55 | 26 | ||||
| 34.60 | 1.000 | 32 | 15 | ||||
| 39.52 | 0.613 | 21 | 10 | ||||
| LDH-OH | 11.71 | 3.09 | 22.77 | 7.59 | 0.893 | 100 | 37 |
| 23.44 | 1.125 | 43 | 16 | ||||
| 34.63 | 1.302 | 35 | 13 | ||||
| 38.68 | 1.830 | 14 | 5 |

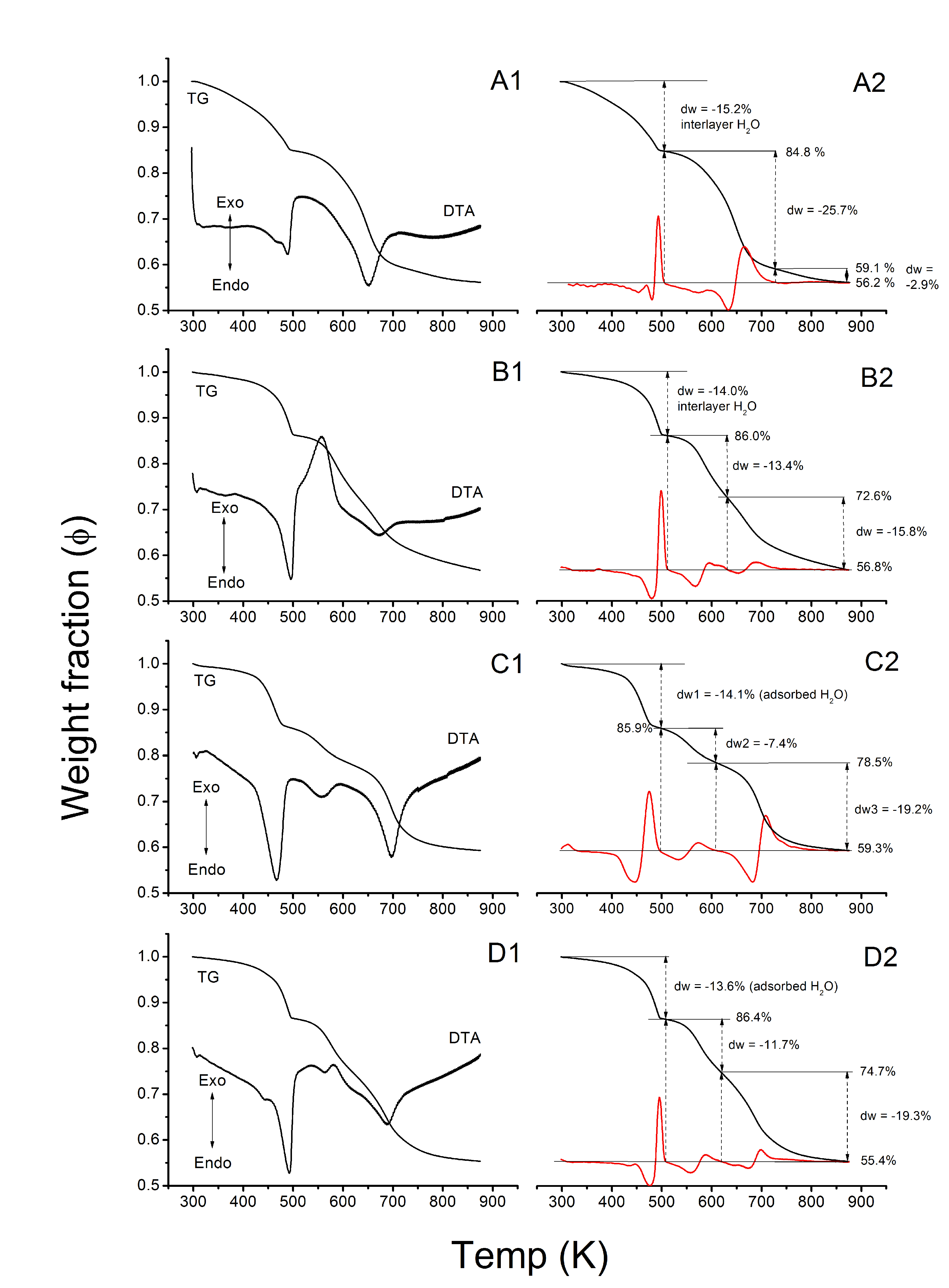
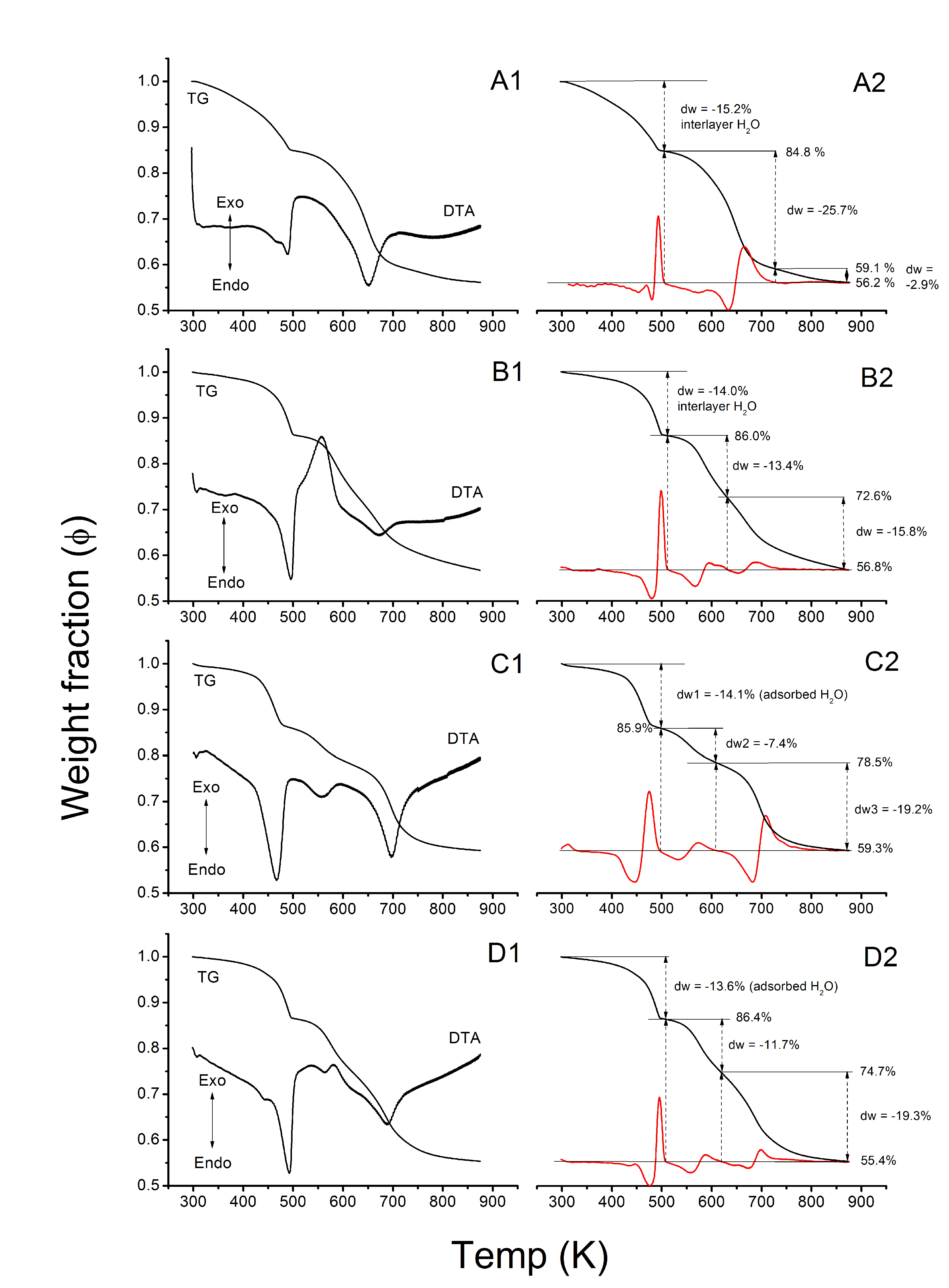
3.1.2. Thermogravimetry
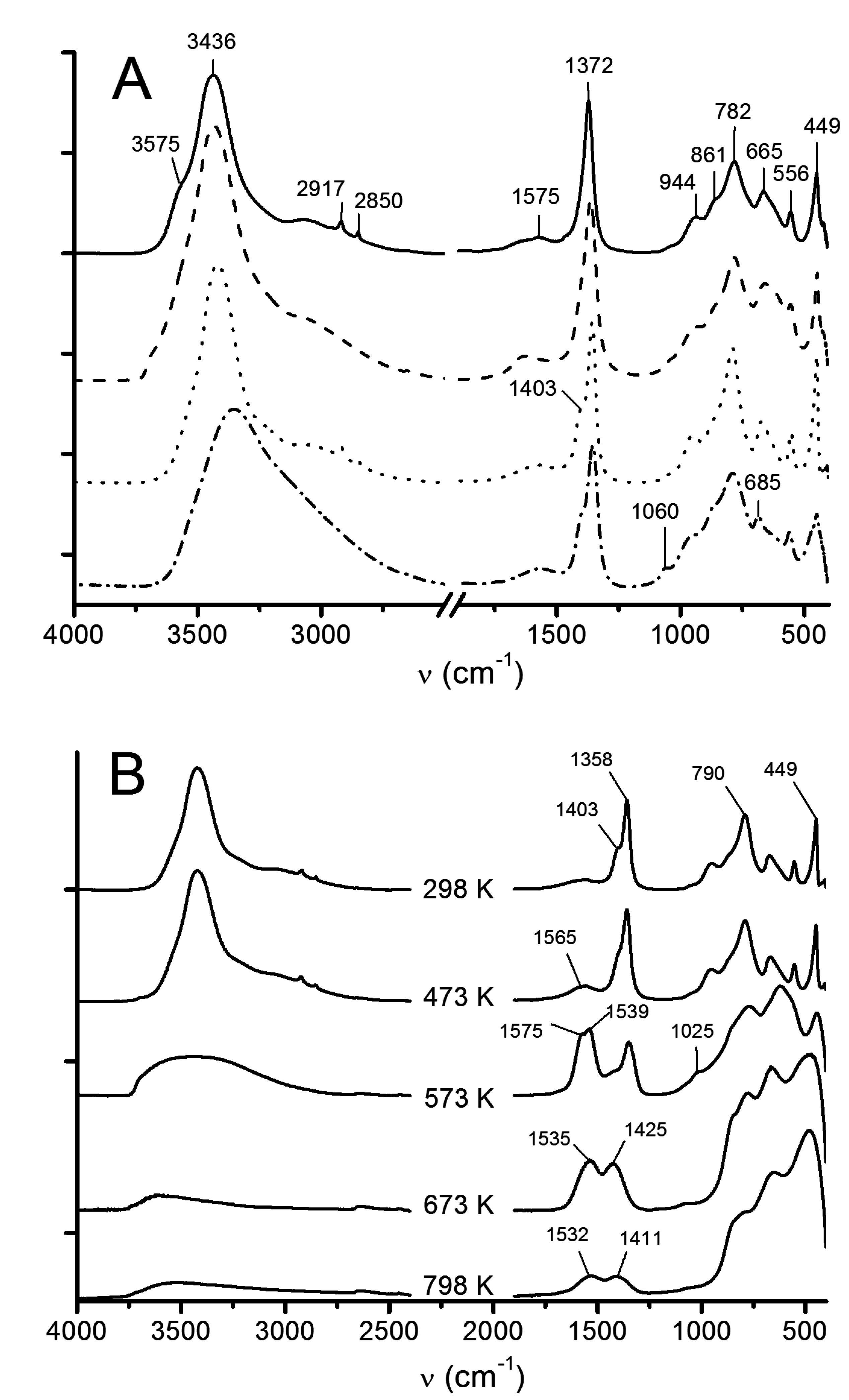
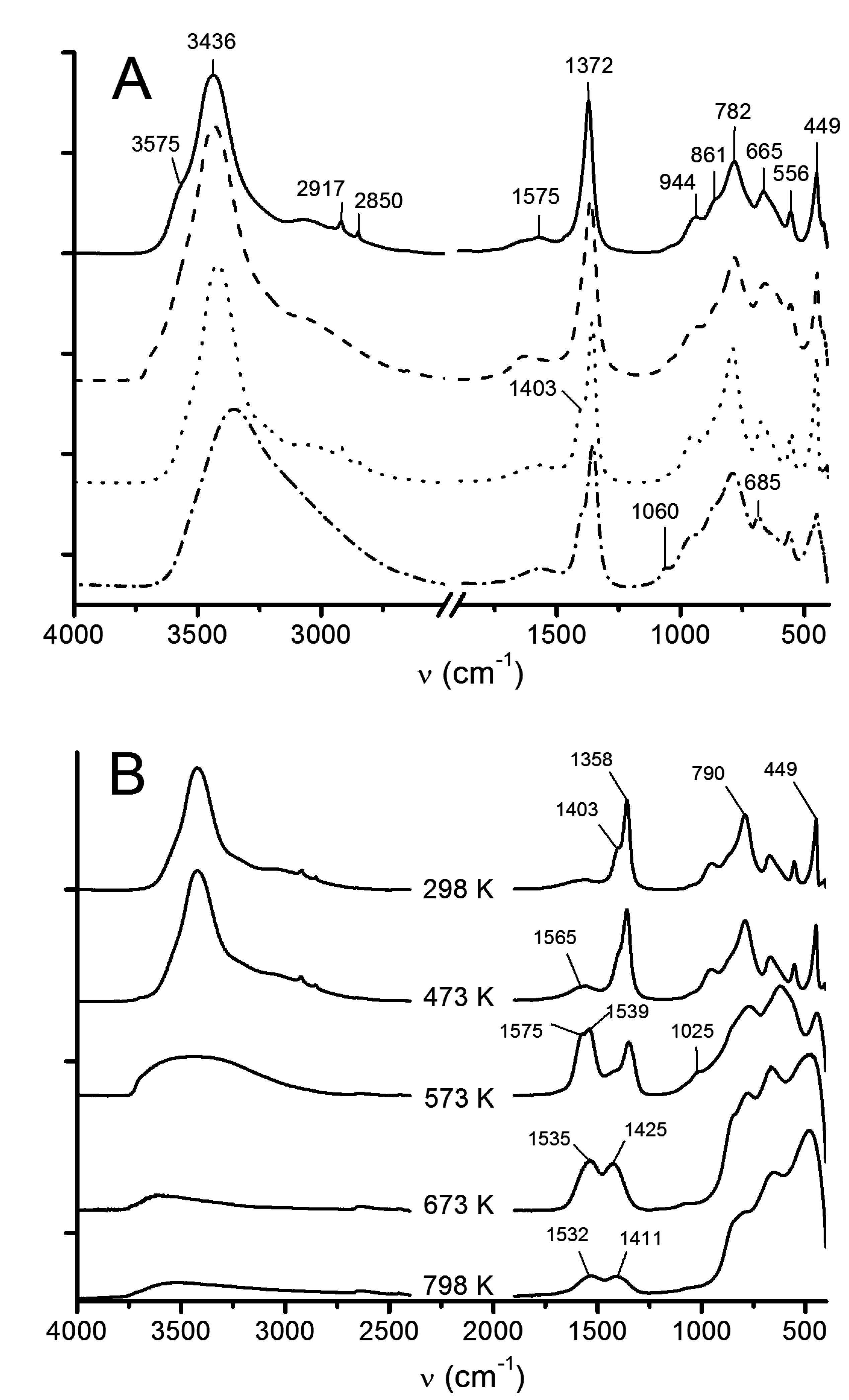
3.1.3. ATR-FTIR
3.2. Mineralization and Coating of Pulp Fibers
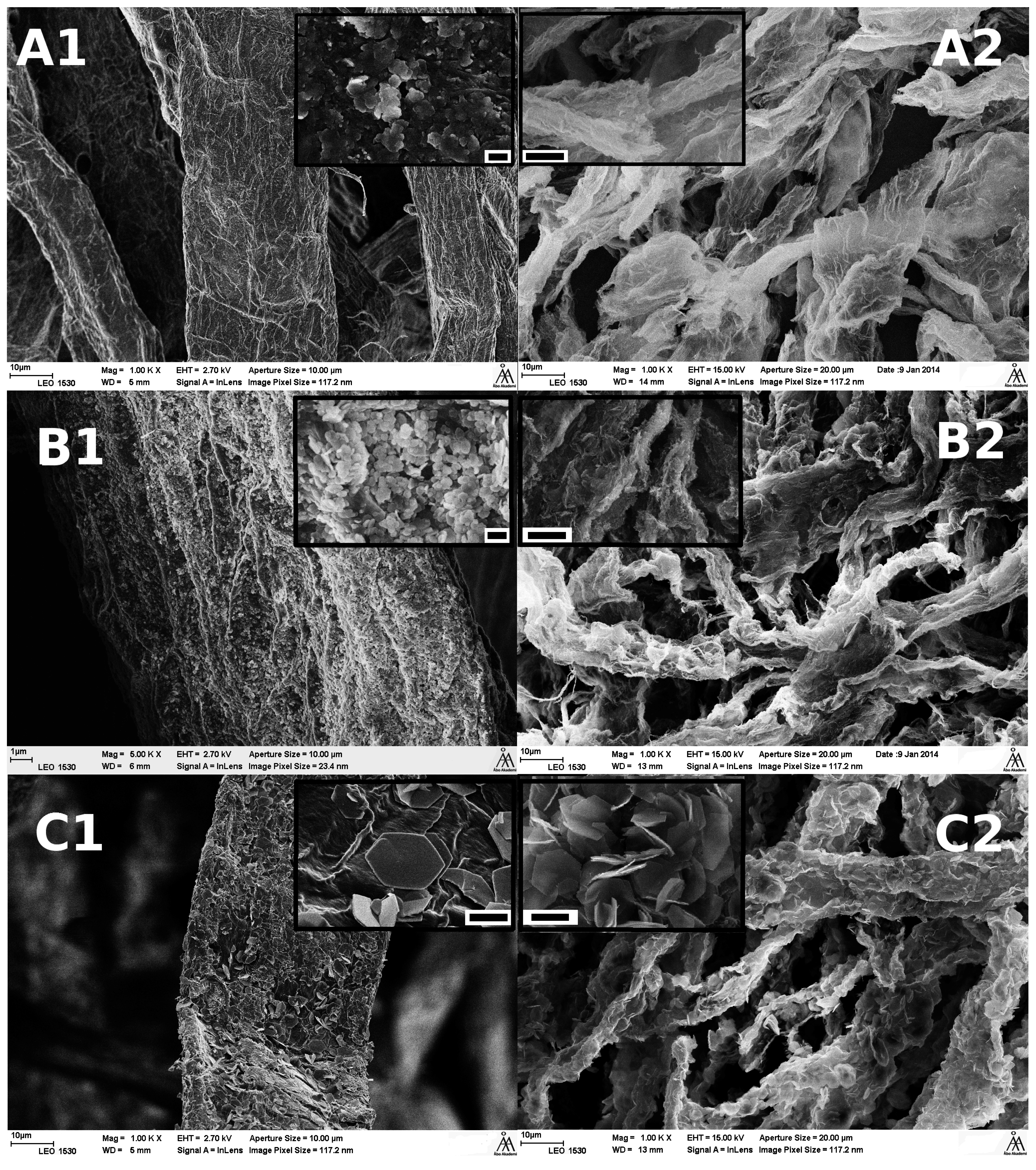
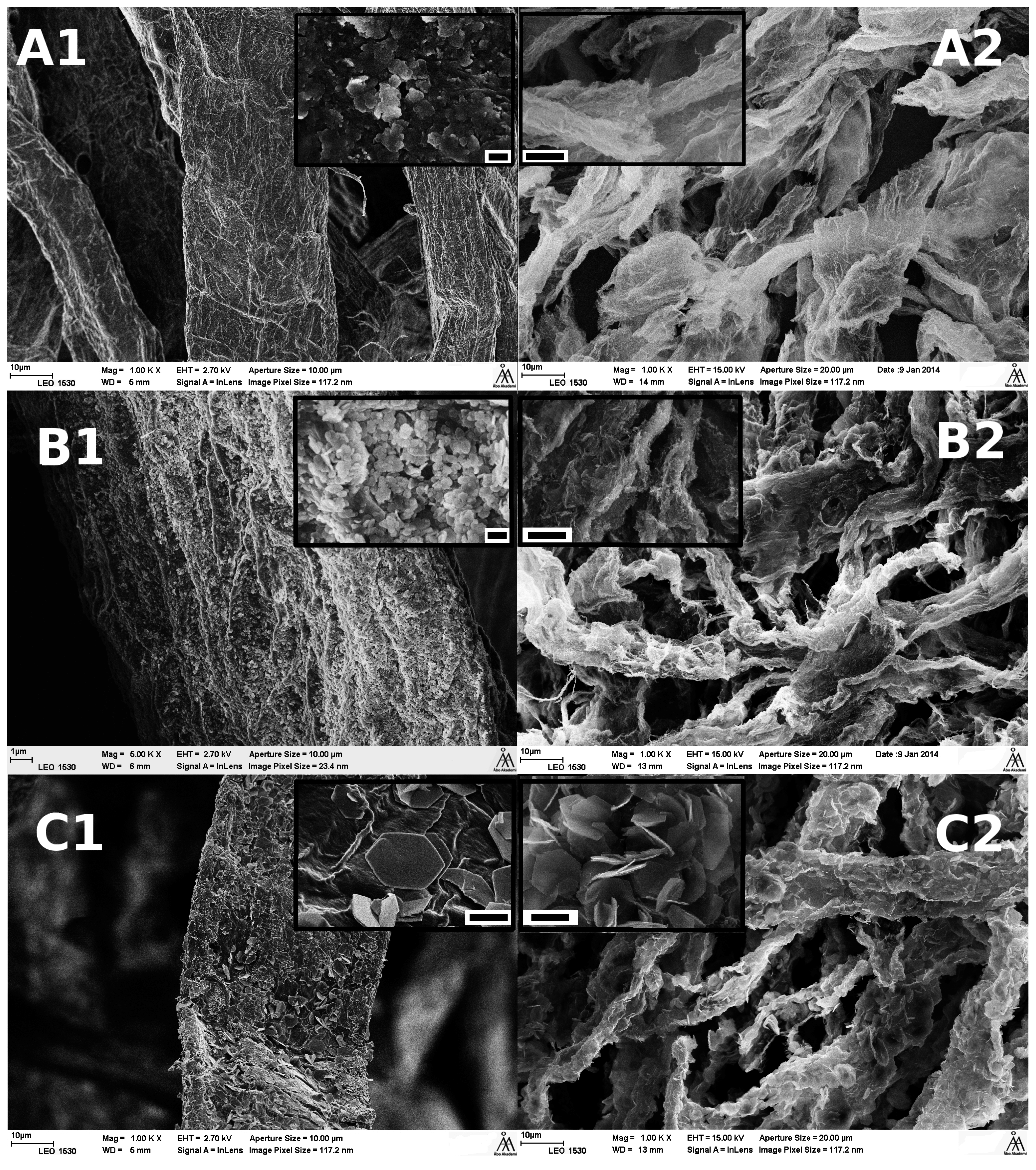
3.2.1. Microscopy
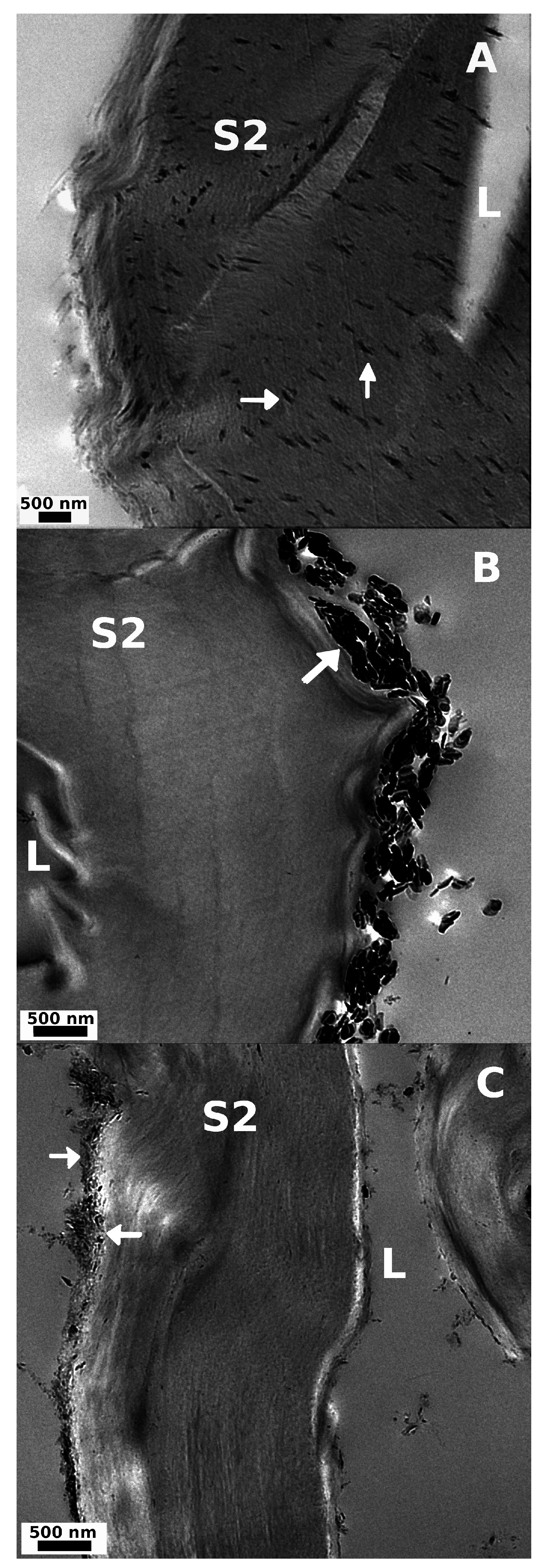
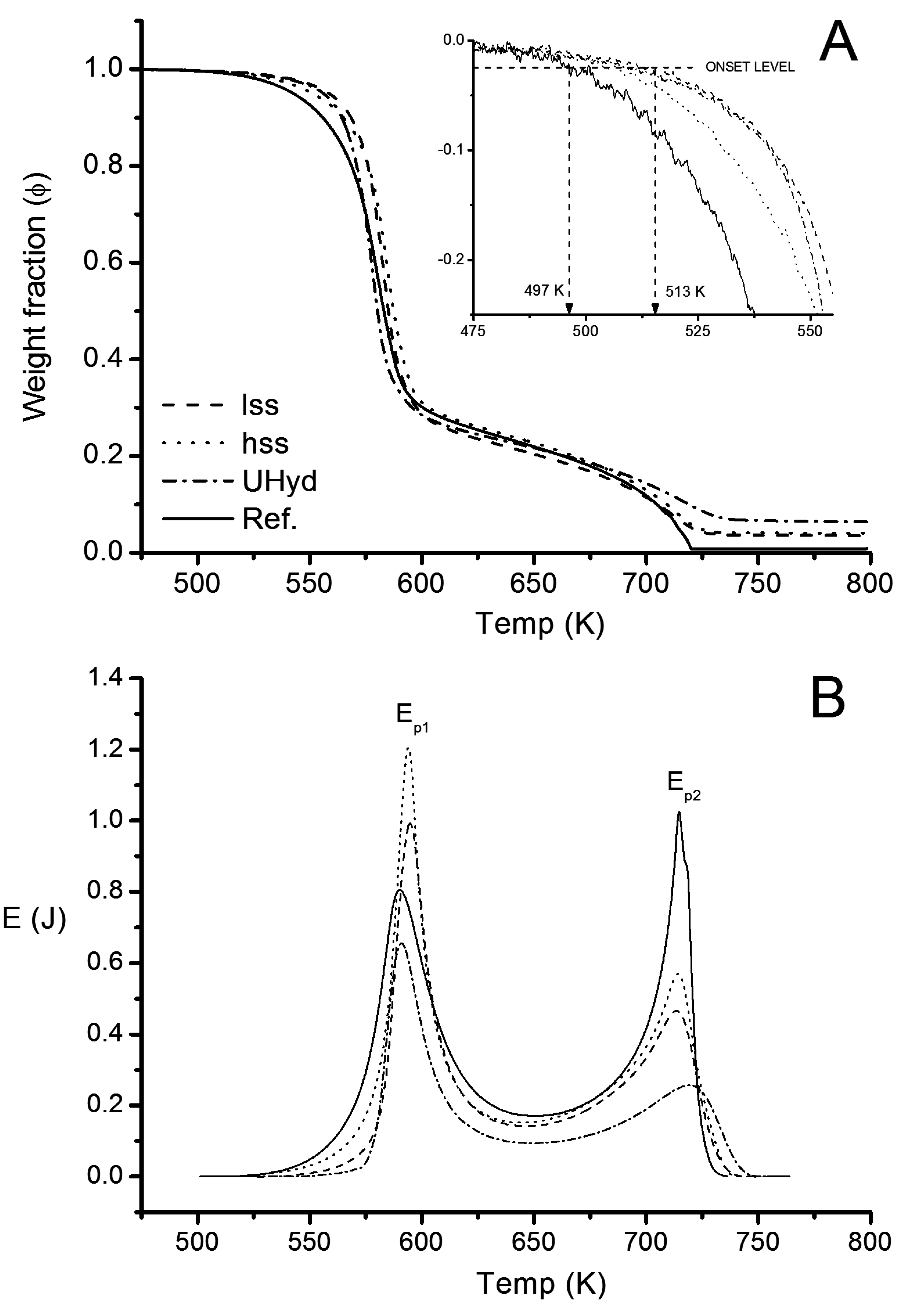
3.2.2. Thermogravimetry
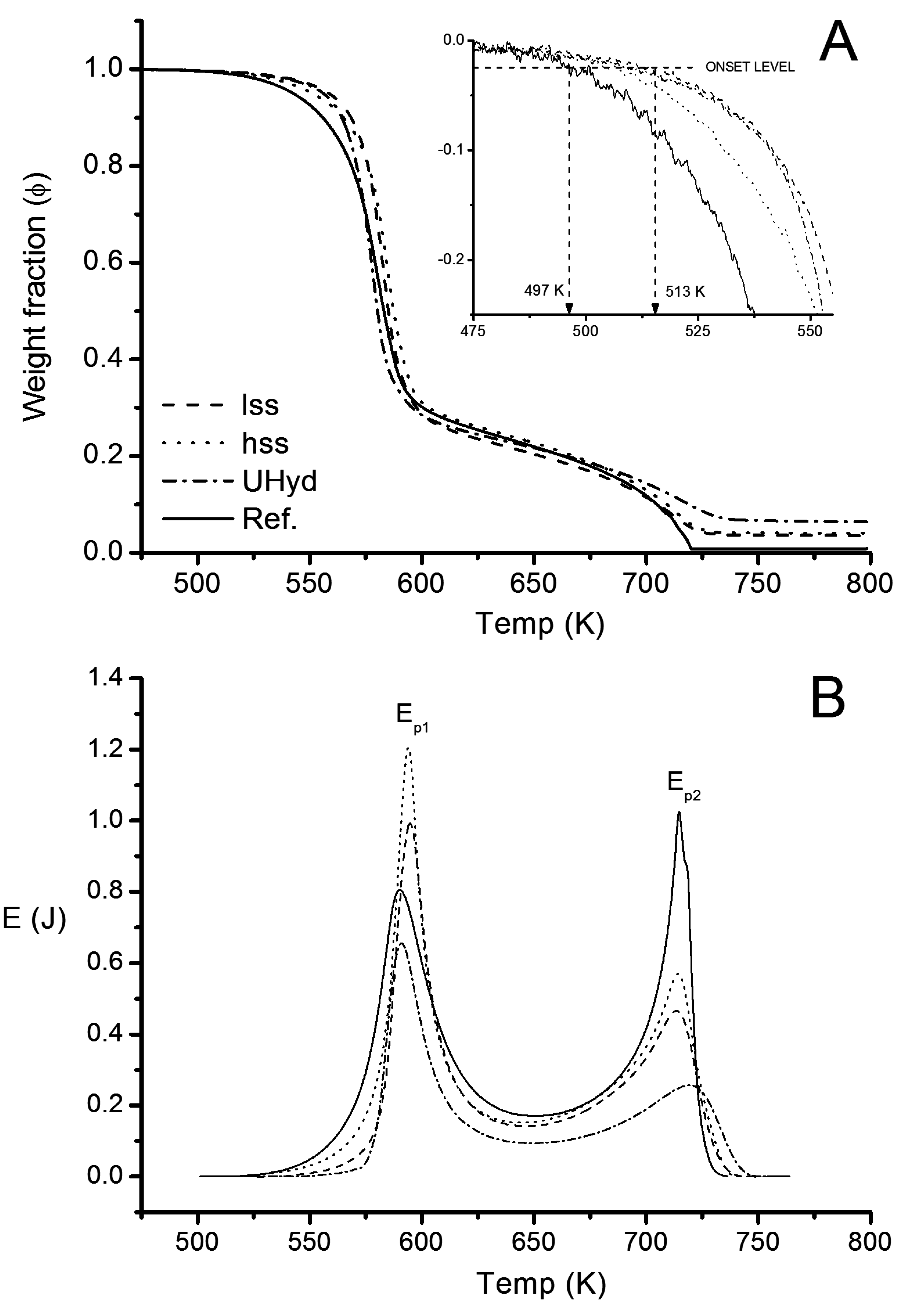
| Sample | ∆Ψ | ∆ΨLDH | Ep1 | Ep2 | ∑E | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | kJ·g−1 | |||||||||||
| Reference | 100 | − | 100 | − | 100 | − | 78 | − | 3.9 | 3.9 | 11.7 | 15.6 |
| lss | 95.0 | 5.0 | 95.8 | 4.2 | 84.5 | 15.5 | 78 | 43.8 | 2.6 | 3.1 | 7.3 | 9.9 |
| hss | 94.2 | 5.8 | 95.0 | 5.0 | 81.8 | 18.2 | 78 | 43.4 | 3.0 | 3.7 | 7.6 | 10.6 |
| Uhyd | 90.4 | 9.7 | 91.7 | 8.3 | 67.0 | 33.0 | 78 | 40.8 | 1.7 | 2.5 | 4.3 | 5.0 |
| Standard | M | ∆fusHm | mp. (Litterature Value) | n | Etot | ∆cUunit |
|---|---|---|---|---|---|---|
| g·mol−1 | J·mol−1 | K | μmol | J | J·K−2 | |
| Pb | 207.20 | 4765 ± 11 | 600.13 (600.61) | 243 | 1.158 | 0.2699 |
| Zn | 65.39 | 7103 ± 31 | 692.25 (692.68) | 275 | 1.951 | 0.3054 |
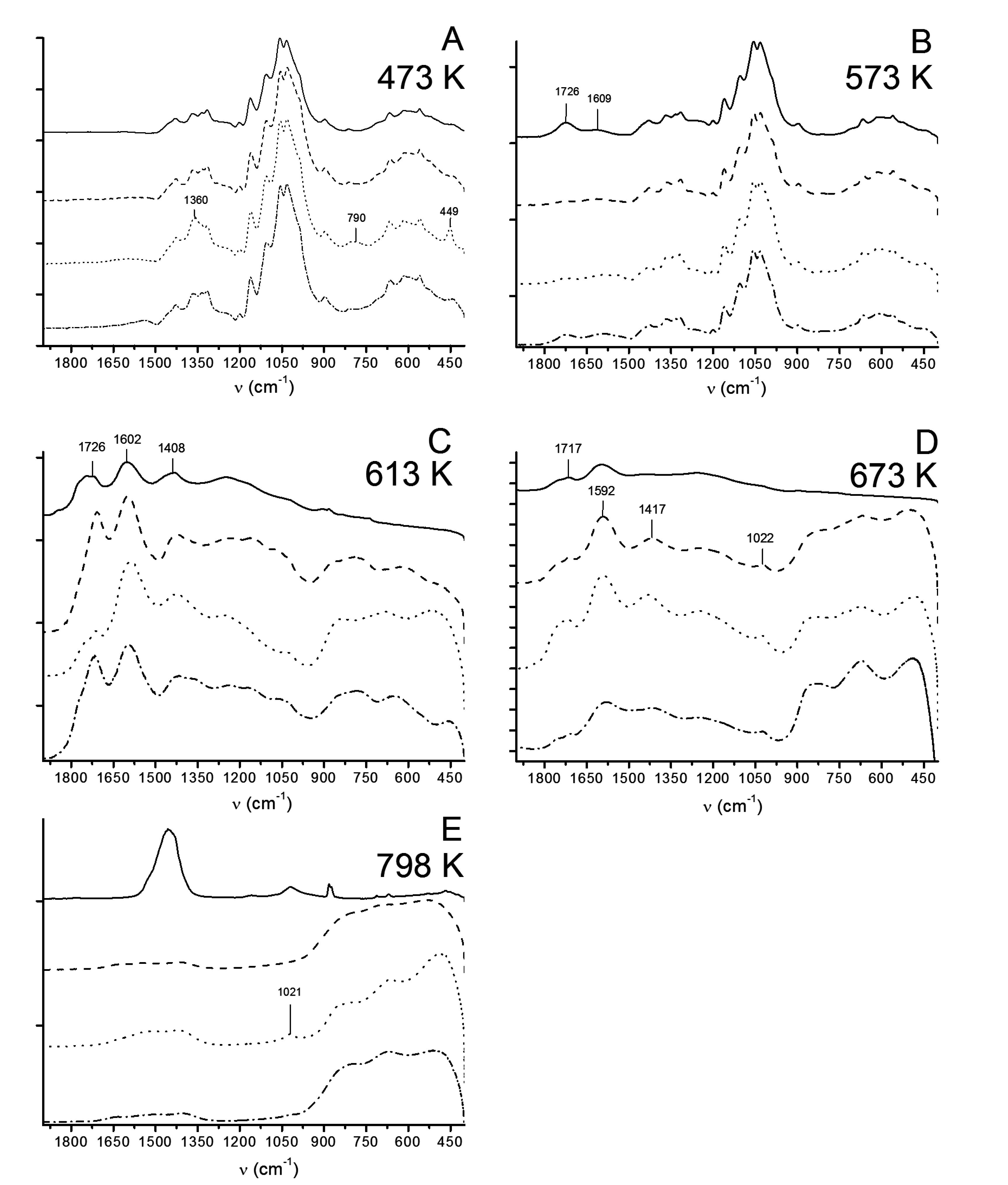
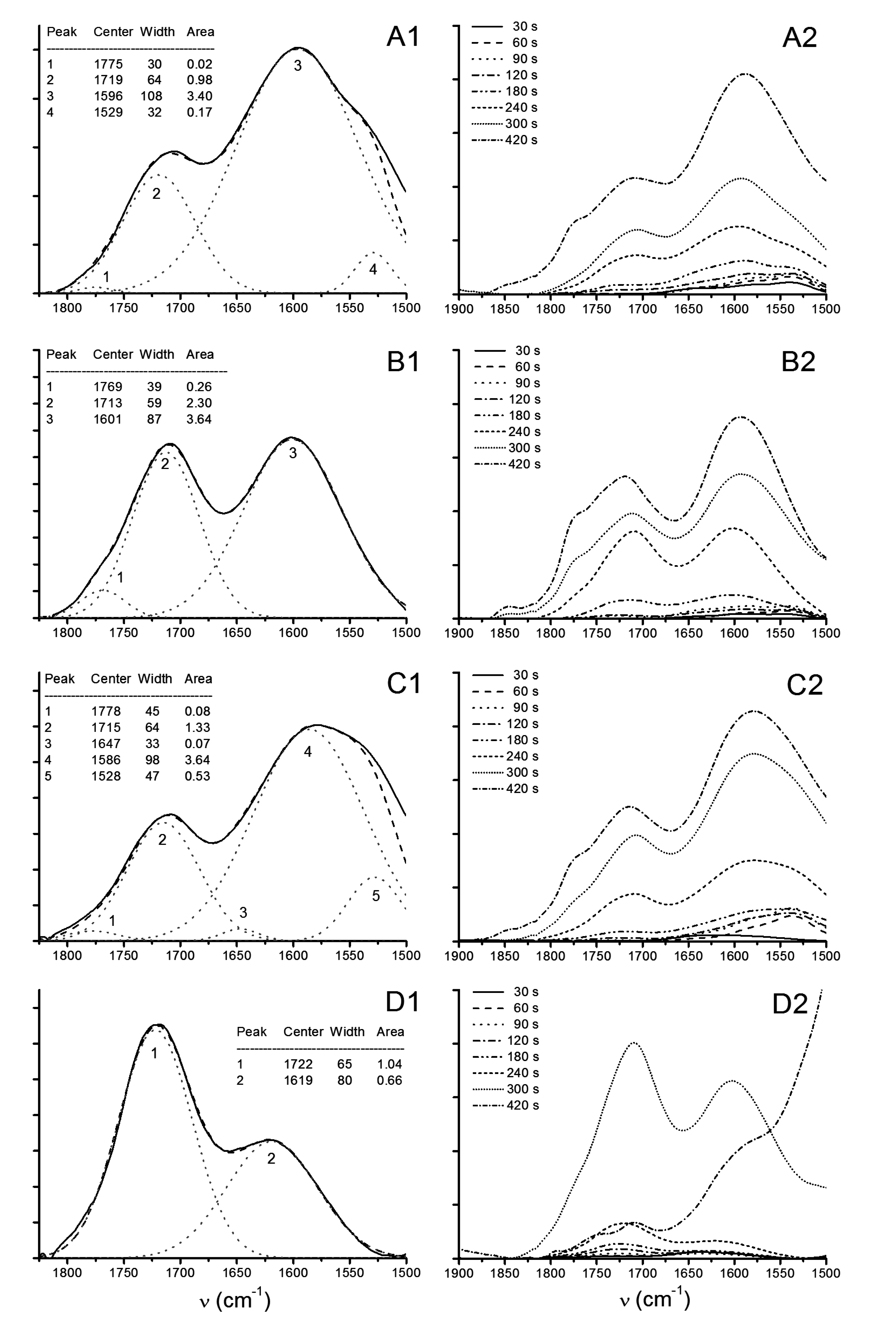
3.2.3. ATR-FTIR
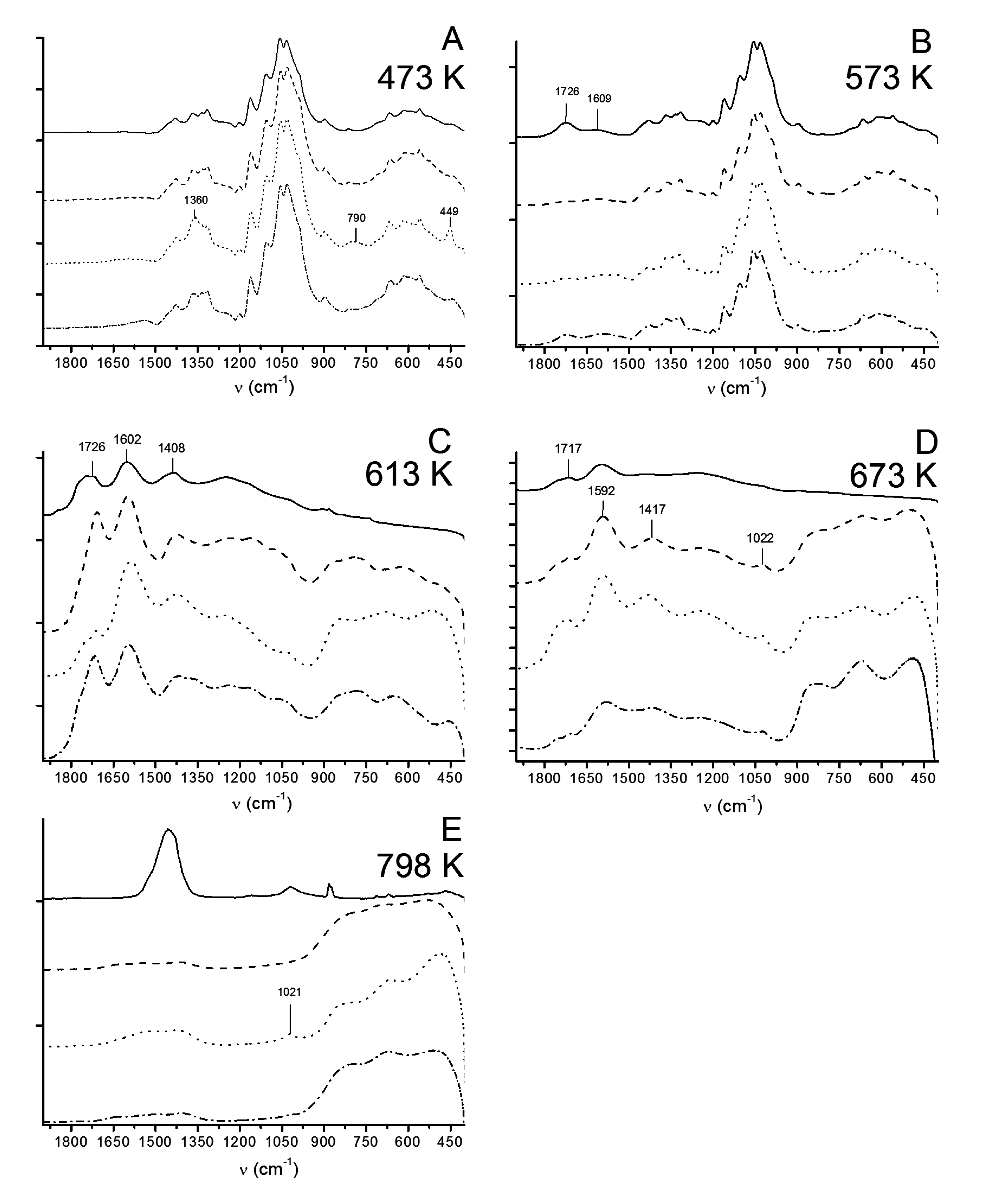
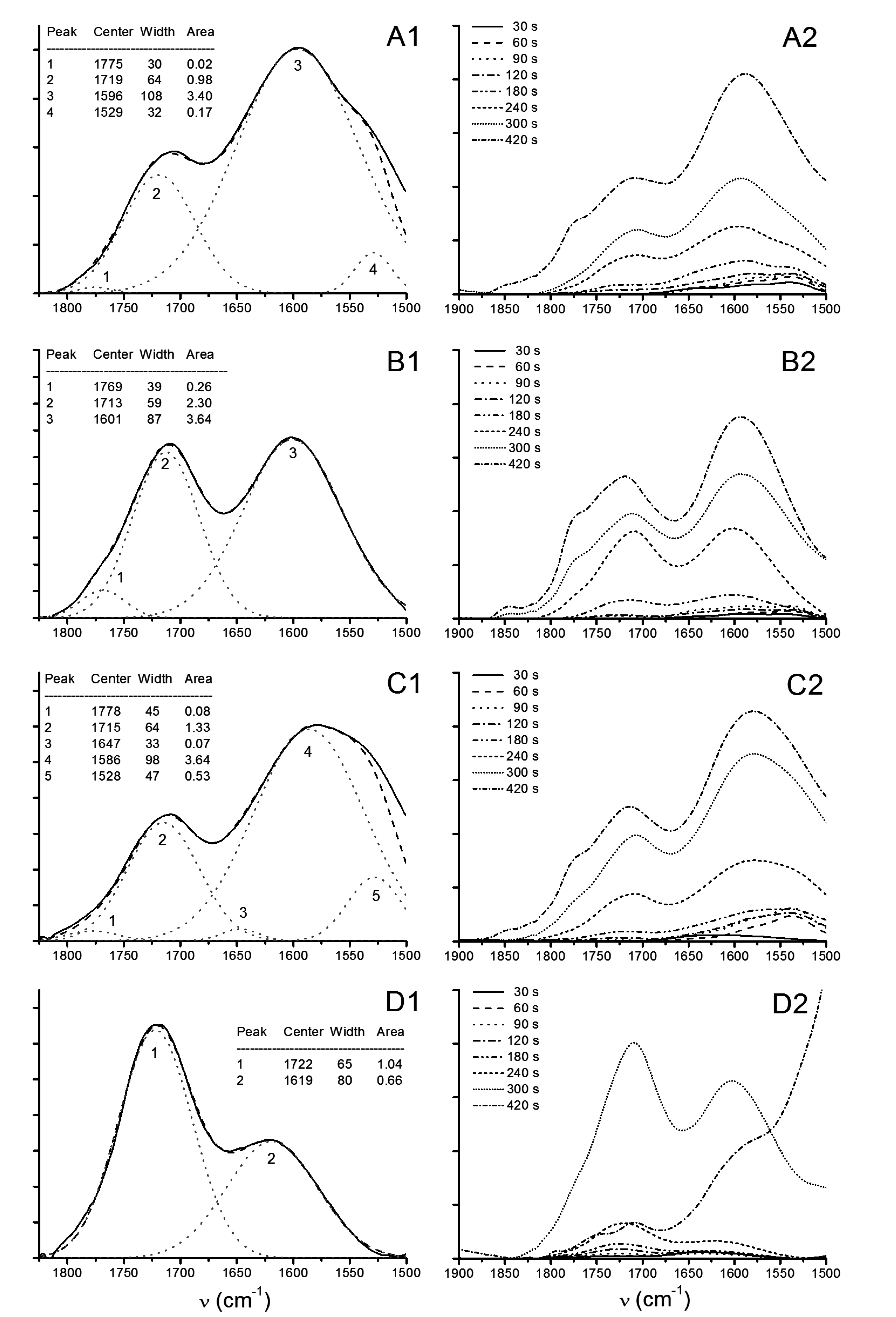
3.2.4. Capillary Viscometry
3.2.5. Microrobotic Instrumentation
| Sample | k−1 | η |
|---|---|---|
| 109 N−1 · m−2 | mL · g−1 | |
| Reference | 7.63(6.93) | 923 |
| lss | 7.93(4.04) | 813 |
| hss | 8.70(4.21) | 868 |
| Uhyd | 4.30(1.71) | 754 |
| Pair | t | Null (%) |
|---|---|---|
| Uhyd to lss | 2.6182 | 98.2 |
| Uhyd to hss | 3.0581 | 99.3 |
| lss to hss | 0.4163 | 31.8 |
3.2.6. Adsorption Experiments
4. Conclusions
Acknowledgments
Author Contributions
Supplementary Information
Conflicts of Interest
References
- Leroux, F.; Besse, J.P. Polymer Interleaved Layered Double Hydroxide: A New Emerging Class of Nanocomposites. Chem. Mater. 2001, 13, 3507–3515. [Google Scholar] [CrossRef]
- Basu, D.; Das, A.; Stoeckelhuber, K.W.; Wagenknecht, U.; Heinrich, G. Advances in layered double hydroxide (LDH)-based elastomer composites. Progress Polym. Sci. 2014, 39, 594–626. [Google Scholar] [CrossRef]
- Yu, X.Y.; Luo, T.; Jia, Y.; Xu, R.X.; Gao, C.; Zhang, Y.X.; Liu, J.H.; Huang, X.J. Three-dimensional hierarchical flower-like Mg-Al-layered double hydroxides: Highly efficient adsorbents for As(v) and Cr(vi) removal. Nanoscale 2012, 4, 3466–3474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Z.; Wu, Y.Y.; Liu, C.; Orpe, A.; Liu, Q.; Xu, Z.P.; Qian, G.R.; Qiao, S.Z. Effective Self-Purification of Polynary Metal Electroplating Wastewaters through Formation of Layered Double Hydroxides. Environ. Sci. Technol. 2010, 44, 8884–8890. [Google Scholar] [CrossRef] [PubMed]
- Mousty, C.; Walcarius, A. Electrochemically assisted deposition by local pH tuning: A versatile tool to generate ordered mesoporous silica thin films and layered double hydroxide materials. J. Solid State Electrochem. 2014, 1–27. [Google Scholar] [CrossRef]
- Ishizaki, T.; Chiba, S.; Watanabe, K.; Suzuki, H. Corrosion resistance of Mg-Al layered double hydroxide container-containing magnesium hydroxide films formed directly on magnesium alloy by chemical-free steam coating. J. Mater. Chem. A 2013, 1, 8968–8977. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, L.; Chen, H.; Xu, S.; Evans, D.; Duan, X. Corrosion Resistance of Superhydrophobic Layered Double Hydroxide Films on Aluminum. Angew. Chem. Int. Ed. 2008, 47, 2466–2469. [Google Scholar] [CrossRef] [PubMed]
- He, S.; An, Z.; Wei, M.; Evans, D.G.; Duan, X. Layered double hydroxide-based catalysts: Nanostructure design and catalytic performance. Chem. Commun. 2013, 49, 5912–5920. [Google Scholar] [CrossRef] [PubMed]
- Parida, K.; Mohapatra, L. Recent progress in the development of carbonate-intercalated Zn/Cr LDH as a novel photocatalyst for hydrogen evolution aimed at the utilization of solar light. Dalton Trans. 2012, 41, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Huang, X.; Li, G.; Li, Z. Layered Double Hydroxide Nano- and Microstructures Grown Directly on Metal Substrates and Their Calcined Products for Application as Li-Ion Battery Electrodes. Adv. Funct. Mater. 2008, 18, 1448–1458. [Google Scholar] [CrossRef]
- Faraji, S.; Ani, F.N. Microwave-assisted synthesis of metal oxide/hydroxide composite electrodes for high power supercapacitors-A review. J. Power Sour. 2014, 263, 338–360. [Google Scholar] [CrossRef]
- Fan, X.; Yang, Z.; Xie, X.; Long, W.; Wang, R.; Hou, Z. The electrochemical behaviors of Zn-Al-La-hydrotalcite in Zn-Ni secondary cells. J. Power Sour. 2013, 241, 404–409. [Google Scholar] [CrossRef]
- Goncalves, N.A.; Caio, T.R.N.; Boaventura de Moraes, S.; Lona, L.M.F. Synthesis and characterization of biodegradable poly(L-lactide)/layered double hydroxide nanocomposites. Polym. Bull. 2014, 71, 2235–2245. [Google Scholar] [CrossRef]
- Dou, Y.; Xu, S.; Liu, X.; Han, J.; Yan, H.; Wei, M.; Evans, D.G.; Duan, X. Transparent, Flexible Films Based on Layered Double Hydroxide/Cellulose Acetate wiht Excellent Oxygen Barrier Property. Adv. Funct. Mater. 2014, 24, 514–521. [Google Scholar] [CrossRef]
- Schmidt, B.; Katiyar, V.; Plackett, D.; Larsen, E.H.; Gerds, N.; Koch, C.; Bender; Petersen, J.H. Migration of nanosized layered double hydroxide platelets from polylactide nanocomposite films. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Von Haartman, S.; Heikkilä, E.; Lange, C.; Fardim, P. Potential Applications of Hybrid Layered Double Hydroxide (LDH) Particles in Pulp and Paper Production. BioResources 2014, 9, 2274–2288. [Google Scholar] [CrossRef]
- Winters, R.; Schomaker, E.; de Vos, S.C. Polymer-Containing Composition, Its Preparation and Use. Netherlands WO2009112441 A1, 17 September 2009. [Google Scholar]
- Beckham, G.T.; Biddy, M.J.; Chmely, S.C.; Sturgeon, M. Hydroxide Catalysts for Lignin Depolymerization. USA US 20140107381 A1, 17 April 2014. [Google Scholar]
- Swanson, C.; Stimpfling, T.; Troutier-Thulliez, A.L.; Hintze-Bruening, H.; Leroux, F. Layered double hydroxide platelets exfoliation into a water-based polyester. J. Appl. Polym. Sci. 2013, 128, 2954–2960. [Google Scholar] [CrossRef]
- Costantino, U.; Marmottini, F.; Nocchetti, M.; Vivani, R. New Synthetic Routes to Hydrotalcite-Like Compounds: Characterisation and Properties of the Obtained Materials. Eur. J. Inorg. Chem. 1998, 1998, 1439–1446. [Google Scholar] [CrossRef]
- Mostafa Moujahid, E.; Besse, J.P.; Leroux, F. Poly(styrene sulfonate) layered double hydroxide nanocomposites. Stability and subsequent structural transformation with changes in temperature. J. Mater. Chem. 2003, 13, 258–264. [Google Scholar] [CrossRef]
- Abello, S.; Mitchell, S.; Santiago, M.; Stoica, G.; Perez-Ramirez, J. Perturbing the properties of layered double hydroxides by continuous coprecipitation with short residence time. J. Mater. Chem. 2010, 20, 5878–5887. [Google Scholar] [CrossRef]
- Wang, J.; Fan, G.; Li, F. A hybrid nanocomposite precursor route to synthesize dispersion-enhanced Ni catalysts for the selective hydrogenation of o-chloronitrobenzene. Catal. Sci. Technol. 2013, 3, 982–991. [Google Scholar] [CrossRef]
- Abelló, S.; Bolshak, E.; Montané, D. Ni-Fe catalysts derived from hydrotalcite-like precursors for hydrogen production by ethanol steam reforming. Appl. Catal. A Gen. 2013, 450, 261–274. [Google Scholar] [CrossRef]
- Lin, J.K.; Uan, J.Y.; Wu, C.P.; Huang, H.H. Direct growth of oriented Mg-Fe layered double hydroxide (LDH) on pure Mg substrates and in vitro corrosion and cell adhesion testing of LDH-coated Mg samples. J. Mater. Chem. 2011, 21, 5011–5020. [Google Scholar] [CrossRef]
- Ma, S.; Wang, J.; Du, L.; Fan, C.; Yahong, S.; Sun, Y.; Sun, G.; Yang, X. Co-Assembly of LDH Nanosheets with Crown Ethers: Structural Transformation and Water-Adsorption Behaviour. Eur. J. Inorg. Chem. 2013, 2013, 1363–1370. [Google Scholar] [CrossRef]
- He, J.; Wei, M.; Li, B.; Kang, Y.; Evans, D.G.; Duan, X. Preparation of Layered Double Hydroxides. In Layered Double Hydroxides; Duan, X., Evans, D.G., Eds.; Springer-Verlag: Berlin, Germany, 2006; Volume 119, pp. 90–103. [Google Scholar]
- Lange, C.; Lundin, T.; Fardim, P. Hydrophobisation of mechanical pulp fibres with sodium dodecyl sulphate functionalised layered double hydroxide particles. Holzforschung 2011, 66, 433–441. [Google Scholar] [CrossRef]
- Lange, C.; Touaiti, F.; Fardim, P. Hybrid clay functionalized biofibres for composite applications. Compos. Part B Eng. 2013, 47, 260–266. [Google Scholar] [CrossRef]
- Saketi, P.; Treimanis, A.; Fardim, P.; Ronkanen, P.; Kallio, P. Microrobotic Platform for Manipulation and Flexibility Measurement of Individual Paper Fibres. In Proceedings of the IEEE/RSJ International Conference on Intelligent Robots and Systems, Taipei, Taiwan, 18–22 October 2010; pp. 5764–5766.
- Martin, A. Toward a referee viscosity method for cellulose. Tappi J. 1951, 34, 363–366. [Google Scholar]
- Fardim, P.; Holmbom, B. Fast determination of anionic groups in different pulp fibres by methylene blue sorption. TAPPI J. 2003, 2, 28–31. [Google Scholar]
- Evans, D.G.; Slade, R.C. Structure and Bonding. In Layered Double Hydroxides; Duan, X., Evans, D.G., Eds.; Springer-Verlag: Berlin, Germany, 2006; Volume 119, pp. 1–87. [Google Scholar]
- Miyata, S. Physico-chemical properties of synthetic hydrotalcites in relation to composition. Clays Clay Miner. 1980, 28, 50–56. [Google Scholar] [CrossRef]
- Theiss, F.L.; Ayoko, G.A.; Frost, R.L. Thermogravimetric analysis of selected layered double hydroxides. J. Therm. Anal. Calorim. 2013, 112, 649–657. [Google Scholar] [CrossRef]
- Hibino, T.; Yamashita, Y.; Kosuge, K.; Tsunashima, A. Decarbonation behavior of Ma-Al-CO3 Hydrotalcite-like compounds during heat treatment. Clays Clay Miner. 1995, 43, 427–432. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies, 3rd ed.; John Wiley & Sons, LTD: Chichester, England, 2004. [Google Scholar]
- Stanimirova, T.; Hibino, T.; Balek, V. Thermal behhavior of Mg-Al-CO3 layered double hydroxide characterized by emanation thermal analysis. J. Therm. Anal. Calorim. 2006, 84, 473–478. [Google Scholar] [CrossRef]
- Adler, H.H.; Kerr, P.F. Infrared spectra, symmetry and structure rrelation of some carcarbon minerals. Am. Mineral. 1963, 48, 839–853. [Google Scholar]
- Rey, F.; Fornés, V.; Rojo, J.M. Thermal decomposition of hydrotalcites. J. Chem. Soc. Faraday Trans. 1992, 88, 2233–2238. [Google Scholar] [CrossRef]
- Sixta, H. Pulp Properties and Applications. In Handbook of Pulp; Sixta, H., Ed.; WILEY-VCH Verlag GmbH&Co.: Weinheim, Germany, 2006; Volume 1, pp. 1009–1067. [Google Scholar]
- Fan, G.; Xiang, X.; Fan, J.; Li, F. Template-assisted fabrication of macroporous NiFe2O4 films with tunable microstructural, magnetic and interfacial properties. J. Mater. Chem. 2010, 20, 7378–7385. [Google Scholar] [CrossRef]
- Stanimirova, T.; Balek, V. Characterization of layered double hydroxide Mg-Al-CO3 prepared by re-hydration of Mg-Al mixed oxide. J. Therm. Anal. Calorim. 2008, 94, 477–481. [Google Scholar] [CrossRef]
- Stone, J.; Scallan, A. The Effect of Component Removal upon the Porous Structure of the Cell Wall of Wood. Pulp Pap. Mag. Can. 1968, T288–T293. [Google Scholar] [CrossRef]
- Stone, J.; Scallan, A. A structural Model for the Cell Wall of Water-swollen Wood Pulp Fibres Based on Their Accessibility to Macromolecules. Cell. Chem. Technol. 1968, 2, 343–358. [Google Scholar]
- Alince, B. Porosity of swollen pulp fibers revisited. Nord. Pulp Pap. Res. J. 2002, 17, 71–73. [Google Scholar] [CrossRef]
- Scallan, A. On Non-Solvent Water in Cellulosic Fibres as Determined by Salt Exclusion. Cell. Chem. Technol. 1987, 21, 215–223. [Google Scholar]
- Yuichiro, S.; Takeshi, K. Swelling of pulp fibers in hot alkaline solution. Sen’i Gakkaishi 1962, 18, 595–599. [Google Scholar]
- Gellerstedt, F.; Wå gberg, L.; Gatenholm, P. Swelling behaviour of succinylated fibers. Cellulose 2000, 7, 67–86. [Google Scholar] [CrossRef]
- Teleman, A.; Harjunpää, V.; Tenkanen, M.; Buchert, J.; Hausalo, T.; Drakenberg, T.; Vuorinen, T. Characterisation of 4-deoxy-β-L-threo-hex-4-enopyranosyluronic acid attached to xylan in pine kraft pulp and pulpulp liquor by 1H and 13CNMR spectroscopy. Carbohydr. Res. 1995, 272, 55–71. [Google Scholar] [CrossRef]
- Susilo, R.; Chandraghatgi, R.; Li, X.S.; Englezos, P. Iron, manganese and copper equiligria with wood fibres in single salt aqueous suspensions. Can. J. Chem. Eng. 2005, 83, 537–547. [Google Scholar] [CrossRef]
- Eriksson, G.; Grén, U. Pulp washing: Sorption equilibria of metal ions on kraft pulps. Nord. Pulp Pap. Res. J. 1996, 11, 164–170. [Google Scholar] [CrossRef]
- Su, P.; Granholm, K.; Harju, L.; Ivaska, A. Determination of equilibrium constants for sorption of metal ions to pulp by a batch method. Nord. Pulp Pap. Res. J. 2013, 28, 521–528. [Google Scholar]
- Andersson, R.; Liden, J.; Öhman, L.O. The Donnan theory applied to pulp washing—Experimental studies on the removal of anionic substances from an assumed fiber lumen volume and from the fiber wall. Nord. Pulp Pap. Res. J. 2003, 18, 405–412. [Google Scholar] [CrossRef]
- Towers, M.; Scallan, A. Predicting the ion-exchange of kraft pulps using Donnan theory. J. Pulp Pap. Sci. 1996, 22, J332–J337. [Google Scholar]
- Koukkari, P.; Pajarre, R.; Pakarinen, H. Modeling of the ion exchange in pulp suspensions by Gibbs energy Minimization. J. Solut. Chem. 2002, 31, 627–638. [Google Scholar] [CrossRef]
- Li, X.S.; Englezos, P. Application of the NICA-Donnan approach to calculate equilibrium between proton and metal ions with lignocellulosic materials. J. Colloid Interface Sci. 2005, 281, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Bygrave, G.; Englezos, P. A thermodynamics-based model and data for Ca, Mg and Na ion partitioning in kraft pulp fibre suspensions. Nord. Pulp Pap. Res. J. 2000, 15, 155–159. [Google Scholar] [CrossRef]
- Athley, K.; Ulmgren, P. Interaction between divalent metal ions and oxygen-delignified kraft pulps. Nord. Pulp Pap. Res. J. 2001, 16, 204–214. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Benítez-Guerrero, M.; López-Beceiro, J.; Sánchez-Jiménez, P.E.; Pascual-Cosp, J. Comparison of thermal behavior of natural and hot-washed sisal fibers based on their main components: Cellulose, xylan and lignin. TG-FTIR analysis of volatile products. Thermochim. Acta 2014, 581, 70–86. [Google Scholar] [CrossRef]
- Stølen, S.; Grønvold, F. Critical assessment of the enthalpy of fusion of metals used as enthalpy standards at moderate to high temperatures. Thermochim. Acta 1999, 327, 1–32. [Google Scholar] [CrossRef]
- Amigó, J.; Chanh, N. DTA study of thermal combustion of textile fibers. J. Therm. Anal. 1975, 7, 183–185. [Google Scholar] [CrossRef]
- Navirin Vhathvarothai, J.N.; Yu, J. An investigation of thermal behaviour of biomass and coal during co-combustion using thermogravimetric analysis (TGA). Int. J. Energy Res. 2013, 38, 804–812. [Google Scholar]
- Ditmars, D.A. Calibration standards for differential scanning calorimetry I. Zinc: Absolute calorimetric measurement of Tfus and ∆fusHm. J. Chem. Thermodyn. 1990, 22, 639–651. [Google Scholar] [CrossRef]
- Zahra, C.; Zahra, A.M. The Perkin-Elmer 1020 series thermal analysis system. Thermochim. Acta 1996, 276, 161–174. [Google Scholar] [CrossRef]
- Lojewska, J.; Misśkowiec, P.; Lojewski, T.; Proniewicz, L.M. Cellulose oxidative and hydrolytic degradation: In situ FTIR approach. Polym. Degrad. Stab. 2005, 88, 512–520. [Google Scholar] [CrossRef]
- Dahlman, O.; Jacobs, A.; Liljenberg, A.; Olsson, A.I. Analysis of carbohydrates in wood and pulps employing enzymatic hydrolysis and subsequent capillary zone electrophoresis. J. Chromatogr. A 2000, 891, 157–174. [Google Scholar] [CrossRef]
- Chirat, C.; Hostachy, J.C.; Paloniemi, J.; Pelin, K.; Pohjanvesi, S.; Nordén, S.; Vesala, R.; Wennerström, M. Bleaching. In Papermaking Science and Technology, Chemical Pulping Part 1: Fibre Chemistry and Technology; Fardim, P., Ed.; Paper Engineers Association/Paperi ja Puu Oy: Porvoo, Finland, 2011; Volume 6, p. 461. [Google Scholar]
- Sundman, O.; Öhman, L.O. Acid/base and metal adsorption properties of CMC-type softwood Kraft ppulp of different charge. Nord. Pulp Pap. Res. J. 2006, 21, 372–381. [Google Scholar] [CrossRef]
- Barzyk, D.; Page, D.; Ragauskas, A. Acid Group Topochemistry and Fibre-to-Fibre Specific Bond Strength. J. Pulp Pap. Sci. 1997, 23, J59–J61. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lange, C.-E.; Lastusaari, M.; Reza, M.; Latifi, S.K.; Kallio, P.; Fardim, P. In Situ Hybridization of Pulp Fibers Using Mg-Al Layered Double Hydroxides. Fibers 2015, 3, 103-133. https://doi.org/10.3390/fib3020103
Lange C-E, Lastusaari M, Reza M, Latifi SK, Kallio P, Fardim P. In Situ Hybridization of Pulp Fibers Using Mg-Al Layered Double Hydroxides. Fibers. 2015; 3(2):103-133. https://doi.org/10.3390/fib3020103
Chicago/Turabian StyleLange, Carl-Erik, Mika Lastusaari, Mehedi Reza, Seyed Kourosh Latifi, Pasi Kallio, and Pedro Fardim. 2015. "In Situ Hybridization of Pulp Fibers Using Mg-Al Layered Double Hydroxides" Fibers 3, no. 2: 103-133. https://doi.org/10.3390/fib3020103
APA StyleLange, C.-E., Lastusaari, M., Reza, M., Latifi, S. K., Kallio, P., & Fardim, P. (2015). In Situ Hybridization of Pulp Fibers Using Mg-Al Layered Double Hydroxides. Fibers, 3(2), 103-133. https://doi.org/10.3390/fib3020103