3. Results
The present studies explore the physicochemical and biological properties of fluorapatite coatings, which are created by doping hydroxyapatite with fluoride ions in the absence (HApF) or presence of dextran (HApF-Dx).
To assess the stability of the HApF and HApF-Dx suspensions, ultrasound measurements were performed, based on the methodology presented by Povey [
28,
29].
Characterizing the stability of particle suspensions using an ultrasound-based technique offers several advantages compared to the established ζ-potential method. Unlike ζ-potential measurements, which often require dilution, the ultrasound-based technique is non-invasive and does not require the dilution or alteration of the sample, preserving the original suspension properties [
30]. On the other hand, the ultrasound-based technique allows for real-time monitoring of the suspension stability, providing continuous data on the state of the particles over time, making them ideal for dynamic studies [
30]. While the ζ-potential relies on electrophoretic mobility, which can be affected by sample opacity, the ultrasound techniques can penetrate opaque and turbid suspensions, making them more versatile [
31]. Moreover, the ultrasound-based technique can detect early stages of aggregation and sedimentation by measuring changes in the acoustic properties of the suspension, such as attenuation and velocity. In summary, the ultrasound-based technique offers a non-invasive, real-time, and versatile approach to characterizing the stability of particle suspensions, making it an alternative to the established ζ-potential method.
Following the stability evaluation, the suspensions were utilized to produce thin films of HApF and HApF-Dx. A 100 mL quantity of HapF and HApF-Dx suspensions was stirred continuously at room temperature for 15 min at 900 rpm to achieve optimal particle homogeneity. The resulting suspension was transferred to a transparent cubical container, specifically equipped with two coaxial ultrasonic transducers positioned 16 mm apart. The transducers’ axes were located at the mid-height of the container. As soon as the stirring process concluded, the acquisition of 1000 ultrasonic signals commenced. Signals were recorded every 5 s on the digital oscilloscope. To minimize experimental noise, each signal recorded on the oscilloscope represents the average of 32 individual signals.
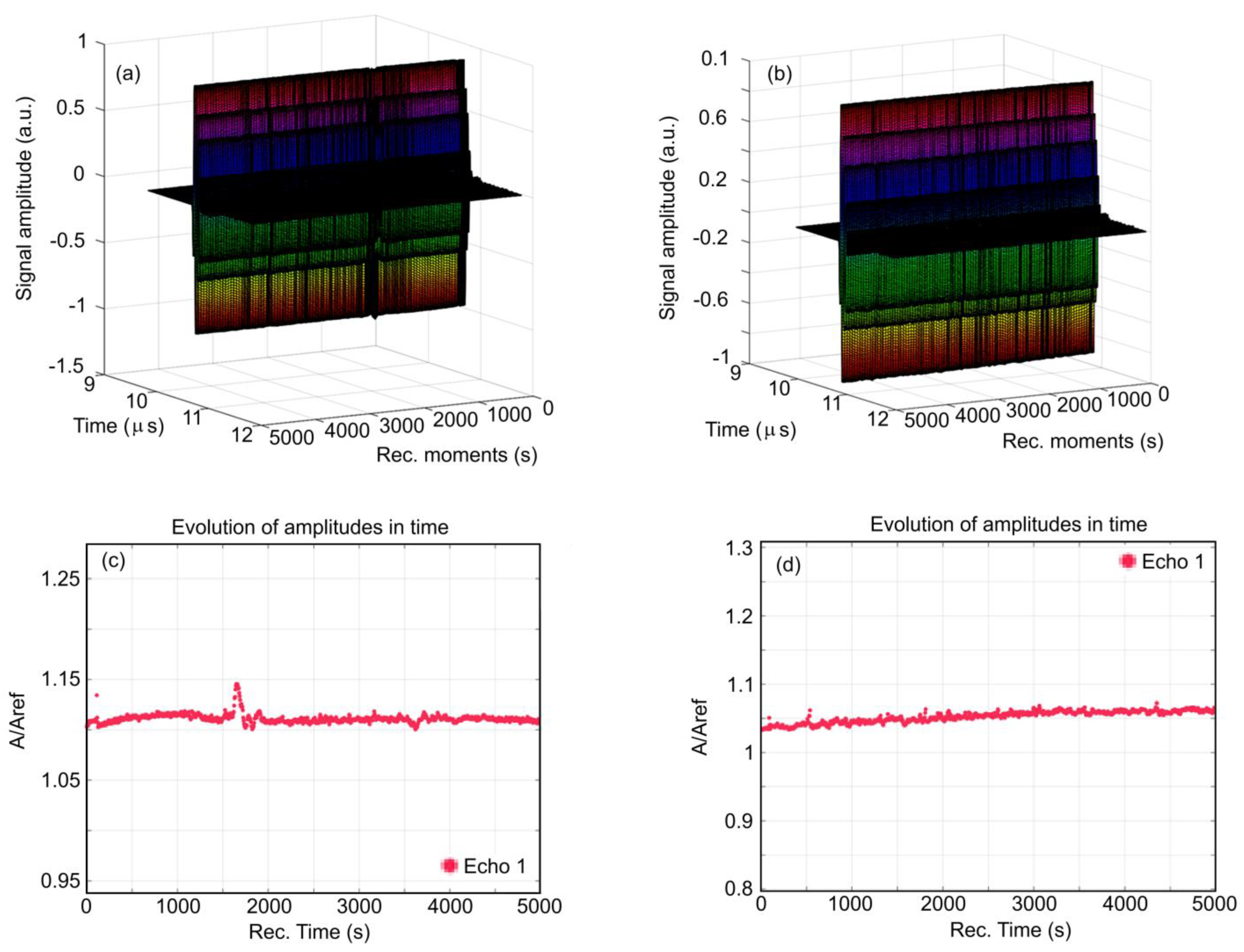
Figure 1 presents a superposition of the 1000 recorded signals for HApF (
Figure 1a) and HApF-Dx (
Figure 1b). These signals, spanning 5000 s of process evolution, are displayed from right to left in a waterfall-style plot. The sedimentation process appears to be extremely slow and is imperceptible in this representation. The evolution of the recorded signal amplitudes for the HApF sample during the experiment is detailed in
Figure 1c, showing an almost constant average amplitude. Exceptions are represented by the rapid passage of a cluster at t = 90 s and a more important slower variation in amplitudes during the 1600–1800 s time interval. For the HApF-Dx sample, this evolution is detailed in
Figure 1d. In the case of the HApF-Dx sample, a slow increase in amplitude was observed, punctuated by small and rapid amplitude variations.
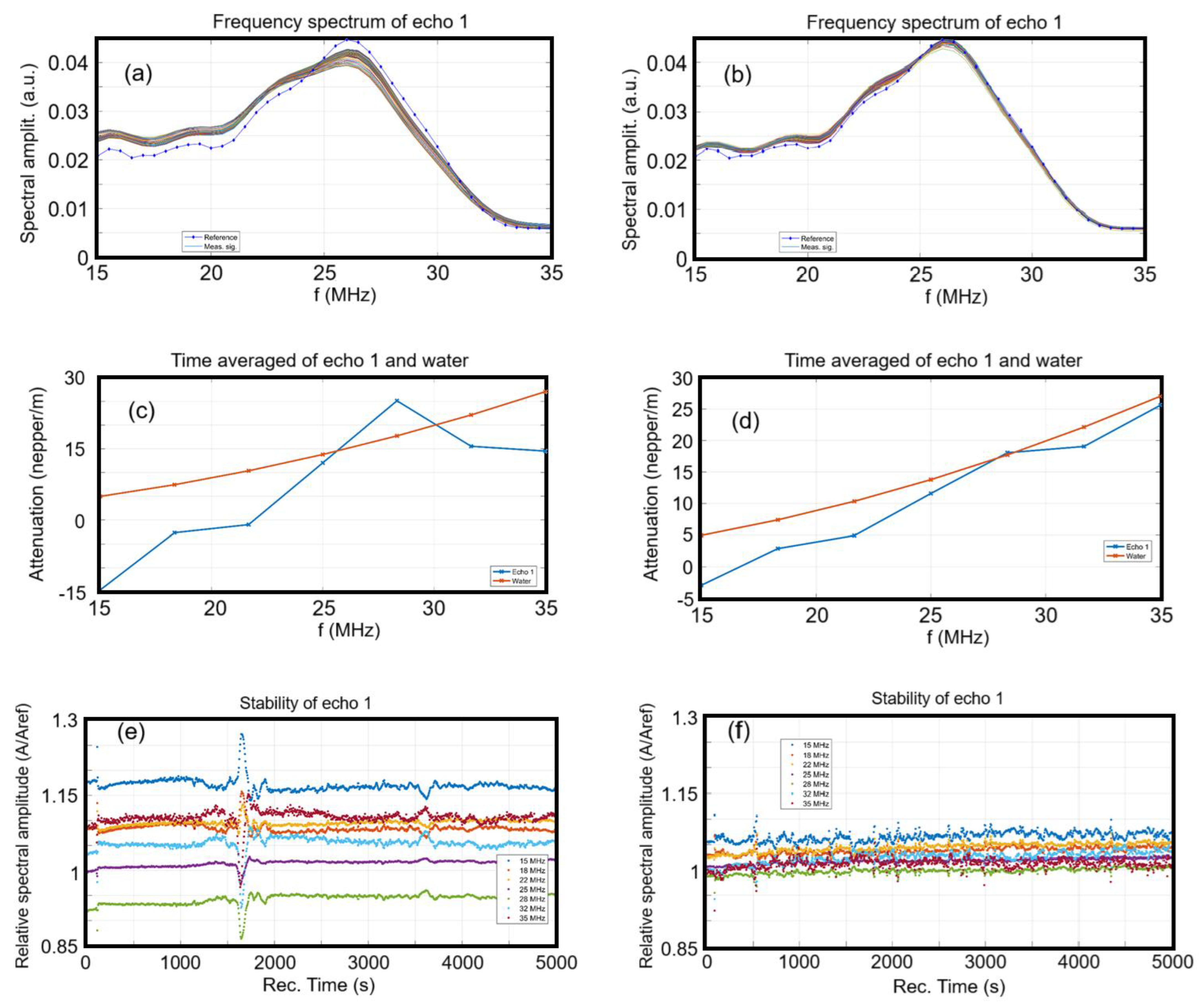
Figure 2 illustrates the evolution of the frequency spectra for each of the 1000 recorded signals. For comparison, the spectrum of the reference liquid (double-distilled water) is also displayed as a dotted blue line. For both samples, the spectra are observed to cluster closely around the spectrum of the reference liquid, demonstrating that the nanoparticles exhibit elastic properties nearly identical to those of the reference liquid, as well as a high degree of suspension stability. However, for the HApF sample (
Figure 2a), the grouped plots of the spectra indicate higher amplitudes values for lower frequencies of the spectrum and lower amplitudes towards the higher frequencies, all relative to the central frequency of the ultrasonic transducers, which is 26 MHz. From the point of view of ultrasonic particle characterization, the HApF-Dx has a more stable behavior, since the 1000 curves, determined at 5 s intervals, are more closely packed together than in the case of the HApF. Also, the grouped 1000 spectra of the HApF are distributing the ultrasonic incident energy more towards the lower frequencies of the spectrum (15–25 MHz) and less on the higher frequencies (25–30 MHz), representing an acoustic signature of the HApF. On the other hand, the HApF-Dx sample has an average behavior closer to that of the reference liquid, by its mean density and compressibility.
The amplitudes in the frequency spectra of the HApF and HApF-Dx samples were averaged across the 1000 spectra displayed in
Figure 2c,d. Subsequently, the natural logarithm of the amplitude ratios relative to the reference liquid was calculated and is also presented in
Figure 2c,d. The HApF suspension (
Figure 2c) exhibits lower attenuations compared to the reference liquid in the lower higher frequency range of the ultrasonic transducer. However, at a frequency of 28 MHz, the attenuation of the HApF sample surpasses that of the reference liquid by 7 neper/m. Conversely, the HApF-Dx suspension (
Figure 2d) closely resembles the reference liquid. Notably, in the lower frequency range, the attenuation is slightly reduced compared to the reference liquid, which is typical for the presence of solid nanoparticles immersed in the suspension. At a frequency of 28 MHz there is a good superposition of amplitudes of the HApF-Dx sample and reference liquid. Lower attenuations than in the reference liquid indicate the better ultrasonic wave propagation through the suspended solid nanoparticles, which in this case are less attenuating than the surrounding liquid.
We are equally interested in the spectral stability of the HApF and HApF-Dx samples, representing the amplitude of the frequency component of each spectrum, as a function of time (
Figure 2e,f). Some remarkable features can be noted for the HApF sample (
Figure 2e). After the first 90 s when there occurs a first rapid passage of a first cluster in front of the transducers, there is a relative slow and continuous variation in the spectral amplitudes. During the time interval 1600–1800 s, the spectral amplitudes suffer increases in amplitudes for the lower frequencies (15–25 MHz) and a reduction in amplitudes for the higher frequencies of the investigated spectrum (28–35 MHz). These features are attributed to the slow passage of the sedimentation surface in front of the transducers. In the HApF-Dx sample (
Figure 2f), after the initial 90 s, a rapid passage of the first cluster in front of the transducers is observed. Subsequently, numerous small yet rapid variations in the ultrasonic signal occur. While these variations are less evident in the average amplitude (
Figure 2f), they are clearly visible in this figure. These phenomena are attributed to the formation of clusters, which move more quickly between the opposing transducers, with the most significant event occurring approximately 550 s into the experiment [
32].
We determine a quantitative stability parameter for the HApF and HApF-Dx sample
, the amplitudes
A being shown in
Figure 1c,d and the resulting values are shown in
Table 1.
This S parameter indicates the long-term evolution of the signal amplitudes by an indication of their average slope in the plots shown in
Figure 1c,d.
A best linear fit over a selected interval of recorded amplitudes provides the slope dA/dt, which is then divided by the average amplitude over the selected time interval. The selected time interval covers 50% of the recorded signals as a sliding window, in search of the best long-term evolution of the amplitudes. Possible initial large jumps in amplitudes occurring for some samples with rapid sedimentation are thus avoided. Obviously, the double-distilled water has constant amplitude of ultrasonic signals for any time lapse, so that S = 0 (1/s) in this case. Samples with S = 1 × 10−7 … 1 × 10−4 (1/s) are considered to categorized from very stable to stable, and larger than S = 1 × 10−4 (1/s) are considered to be unstable, based on a large number of samples. The estimation error is also indicated for each sample, being related to the choice of the analyzed time interval. The HApF sample is considered to be stable and HApF-Dx is very stable.
The stability parameter calculated from the ultrasonic measurements reflects how the amplitude of the ultrasonic echoes changes over time, providing insights into the stability of hydroxyapatite suspensions doped with different ions. Studies on the evaluation of the stability of hydroxyapatite doped with different ions by ultrasonic measurements have shown that different parameters such as synthesis conditions (temperature and mixing time) play a very important role. For suspensions based on hydroxyapatite doped with silver ions (Ag:HAp), the stability parameter is influenced by factors such as stirring time and temperature during preparation [
29]. Gels treated at higher temperatures tend to show greater stability, indicated by a slower variation in the amplitude of the ultrasonic signal [
29]. The previous study [
29] showed that suspensions stirred at room temperature showed a linear increase in amplitude during the initial period, while those stirred at higher temperatures showed a decrease followed by a slow increase. The phenomenon is explained by the concentration of particles in front of the separation surface and the variation in the acoustic wave speed between the suspension and the solvent. Following the obtained results, it can be concluded that ultrasonic stability measurements are particularly useful for characterizing opaque and concentrated suspensions, where traditional methods, such as light scattering, may not be effective.
The XRD patterns of the HApF and HApF-Dx thin films (illustrated in
Figure 3) were compared to the HAp reference model (JCPDS 09-432). Both films, obtained by the spin-coating method, exhibited similar diffraction patterns corresponding to the characteristic peaks of hexagonal HAp crystals. The absence of non-HAp peaks indicates the successful formation of pure-phase apatite. The broadness of the XRD peaks suggests that the particles are nanometric in size. The (0 0 2) reflection at 25.9°, being distinct, was selected to calculate particle size. In the case of HApF-Dx, the peaks appear slightly broader compared to HApF. This behavior is in agreement with the calculated size for the two samples. The crystallite sizes derived from the XRD data, revealing a reduction in crystallite size for the HApF-Dx sample (21.48 ± 2 nm) compared to HApF sample (26.56 ± 2 nm).
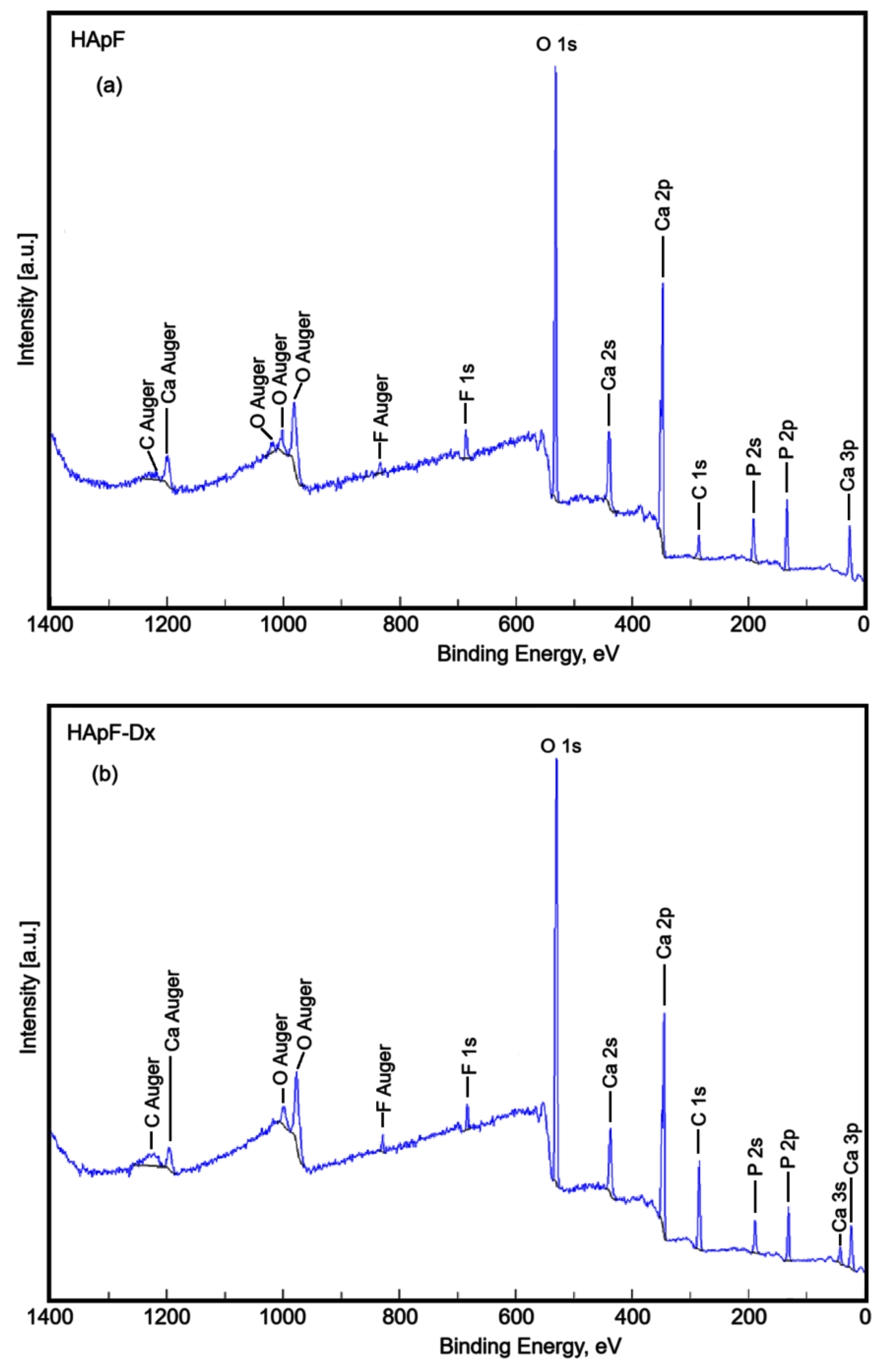
XPS studies were performed to examine the chemical composition of the HApF and HApF-Dx samples. All measured binding energies were referenced to the main C 1s photoelectron line at 284 eV. The overall XPS spectrum of the HApF-Dx sample is shown in
Figure 4b. For comparison, the spectrum of the HApF sample (
Figure 4a) was also shown. It is noted that F, Ca, and P were detected in both samples.
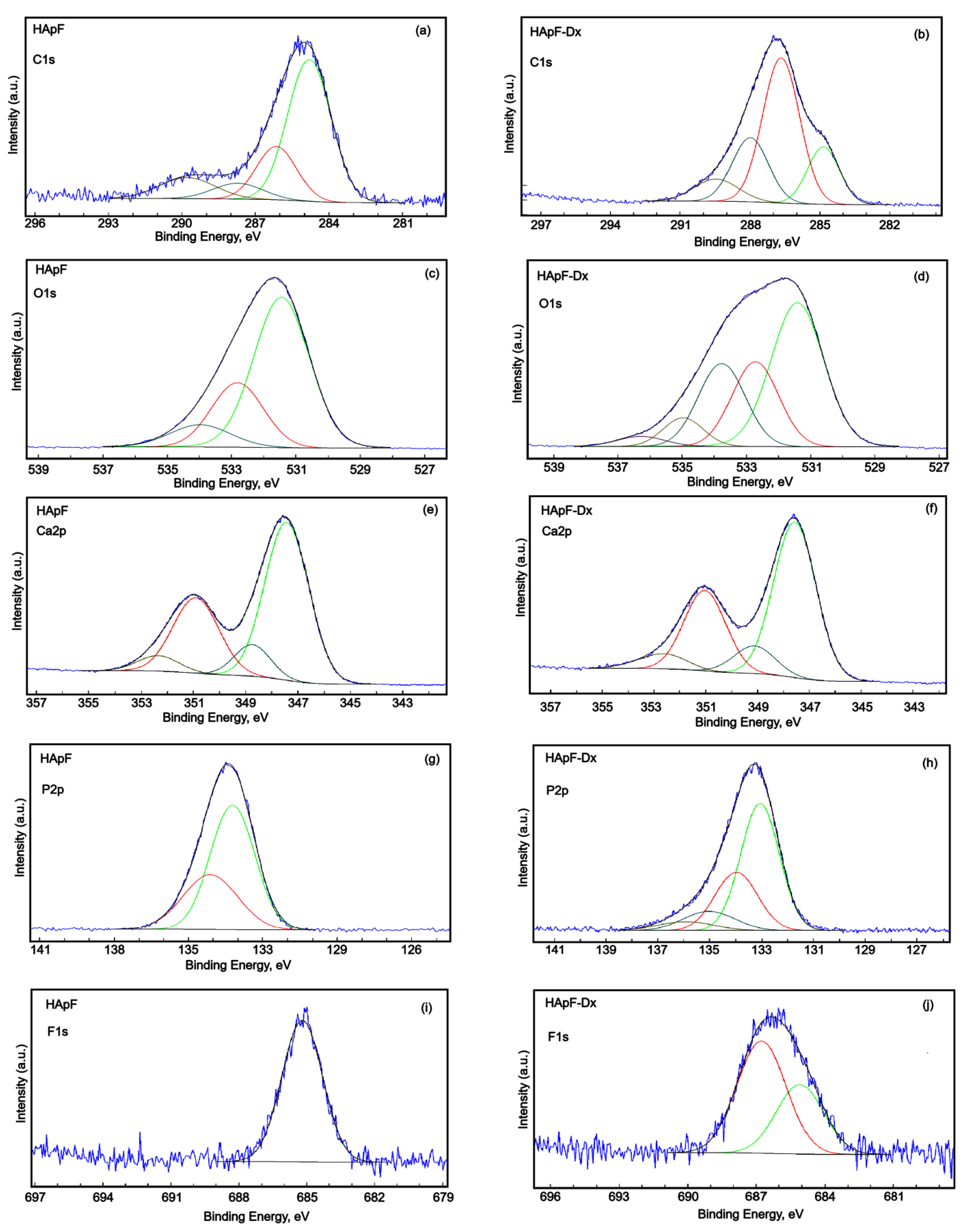
High-energy resolution analysis of C 1s, O 1s, Ca 2p, and F 1s is presented in
Figure 5. The high-resolution XPS spectrum of C 1s was deconvoluted into four components for both the HApF and HApF-Dx samples (
Figure 5a,b). In the HApF sample, the four components appeared at binding energies (BE) of 284.4, 286.14, 287.72, and 289.78 eV (
Figure 5a). Meanwhile, for the HApF-Dx sample, the components were observed at BE values of 284.85, 286.67, 287.99, and 289.46 eV (
Figure 5b). These components correspond to C-C and C-H single bonds, C-O single bonds, C=O, O-C-O double bonds, as well as contaminants containing –COOR groups. For the HApF-Dx sample, the oxygen bonds observed are in the highest proportion due to dextran. Additionally, an increase in the total carbon concentration at the surface was noted. Furthermore, the binding energies were slightly higher. This behavior can be due to the structural characteristics of dextran.
The higher binding energy associated with dextran probably stems from its structural characteristics, particularly its polysaccharide composition and functional groups [
33]. Thus, the higher binding energy associated with dextran may be due to the interaction between the chemical environment and the molecular structure of dextran as revealed by X-ray photoelectron spectroscopy (XPS). The oxygen–carbon bonds (e.g., C-O, C=O) in dextran contribute to higher binding energies for carbon and oxygen atoms compared to their binding energies observed in simpler or alternative chemical environments. Moreover, the different functional groups in dextran, such as hydroxyl (-OH) groups, may also play a role in altering binding energy. These groups can interact with one another and their surroundings, further affecting the binding energy dynamics.
The high-resolution spectrum of O 1s for the HApF sample contains three components (
Figure 5c). The first component appearing at the binding energy of 531.45 eV can be attributed to the O in hydroxyapatite and the PO
43− groups of HAp. The two observable components at the binding energy of 532.82 and 533.98 eV are attributed to simple C-O bonds and adsorbed H
2O and other contaminants with simple C-O bonds.
The high-resolution spectrum of O1 from the HApF-Dx sample contains five components (
Figure 5d). The first component appears at the binding energy of 531.41 eV and can be attributed to the O from hydroxyapatite and the PO
43− groups of HAp. The second component observed at the BE of 532.72 eV was assigned to C-O single bonds. The third component observed at the BE of 533.77 eV was attributed to possible traces of water and other contaminants with C-O single bonds. In agreement with previous studies [
34], oxygen bound to organic components can range in binding energy from 530.9 eV to 533.8 eV (corrected here to C 1s (C-C, C-H)). The fourth and fifth components were observed at BE values of 534.96 and 536.28 eV. According to previous studies [
35,
36], the BE values of O 1s observed at 534.96 and 536.28 eV in the XPS spectrum are usually attributed to oxygen species in high oxidation states or environments with strong electronegative interactions. Thus, these binding energies could be attributed to oxygen in fluorinated compounds (O-F bonds) [
35,
36]. These peaks with higher binding energy can be attributed to oxygen in hydroxyl (OH
−) groups or oxygen species adsorbed on the surface. In fluorine-doped hydroxyapatite, some of the hydroxyl groups might be substituted by fluoride ions, which can affect the local electronic environment by moving the binding energy of the remaining oxygen atoms in the hydroxyl groups. The absence of peaks associated with F-O bonds in the high-resolution XPS spectrum of O 1s for fluorine-doped hydroxyapatite (HApF) could be attributed to several factors such as overlap with other peaks and surface effects. Consequently, the binding energy of the F-O bonds may overlap with other oxygen-related peaks, such as those from hydroxyl groups, making it challenging to resolve them distinctly. On the other hand, it is known that XPS mainly analyzes the surface of the material. Therefore, if it is concentrated deeper into the sample, the surface analysis may not capture the F-O bonds.
The high-resolution XPS spectrum of Ca 2p (
Figure 5e,f) shows four components. The two specific components (2p3/2 and 2p1/2) were observed at 347.42 eV and 350.90 eV for the HApF sample and 347.56 eV and 351.13 eV for the HApF-Dx sample [
37,
38]. The two components were separated by ~3.6 eV in binding energy for both analyzed samples. For both samples, the ratio of the areas of the two components is 2:1. The binding energy is characteristic of hydroxyapatite. The third and fourth components were observed at BE values of 348.74 and 352.35 eV (HApF sample) and 349.12 and 352.63 eV (HApF-Dx sample) [
39,
40]. This doublet observed in both samples is at a high bond energy, which may suggest the presence of Ca-F bonds.
The high-resolution XPS spectra of P 2p were revealed in
Figure 5g,h. The two specific components (2p3/2 and 2p1/2) observed at 133.22 eV and 134.12 eV for the HApF sample and 135.02 eV and 135.92 eV for the HApF-Dx sample are assigned to the P2p doublet are separated by 0.9 eV with an area ratio close to 2:1. The binding energy of this doublet is specific to hydroxyapatite (-PO
4). The result was in agreement with previous studies [
41]. The doublet observed at BE values of 135.02 and 135.92 eV in the HApF-Dx sample (
Figure 5h) could be assigned to the phosphorus in phosphate groups, such as those found in hydroxyapatite (HAp). These binding energies correspond to the chemical state of phosphorus in its oxidized form, specifically within the PO
43− environment. This is consistent with the presence of phosphate in calcium phosphate compounds like hydroxyapatite.
The high-resolution spectrum of F 1s for the HApF sample (
Figure 5i) presents a single component at the BE of 685.19 eV. The peak observed at 685.19 eV is assigned to the F- in the FHAp-Dx structure and confirms that fluoride ions have been successfully incorporated into the HAp structure by substituting hydroxyl groups (-OH). The result aligns with the findings reported in previous studies [
42,
43,
44,
45]. The high-resolution spectrum of F 1s for the HApF-Dx sample (
Figure 5j) shows two components at BE values of 685.09 eV and 686.77 eV due to the distinct chemical environments of fluoride ions within the structure. The binding energies observed at 685.09 eV and 686.77 eV in the XPS high-energy resolution spectra of F 1s are attributed to fluoride ions substituting for hydroxyl groups (-OH) in the hydroxyapatite structure. In the FHAp-Dx sample, two key processes are observed. We have on the one hand the substitution of hydroxyl groups and on the other hand the interaction with the dextran matrix. Fluoride ions replace hydroxyl groups (-OH) in the hydroxyapatite lattice, resulting in the formation of fluorapatite [
42,
43,
44,
45]. This substitution leads to fluoride ions binding to calcium atoms, represented by the component located at the BE of 685.09 eV [
42,
43,
44,
45]. Additionally, the dextran matrix introduces additional interactions between fluoride ions and the polysaccharide environment, which can modify the electronic structure of fluoride ions (the second component observed at the BE of 686.77 eV). Together, these processes underscore the intricate interactions between fluoride ions, the hydroxyapatite lattice, and the dextran matrix.
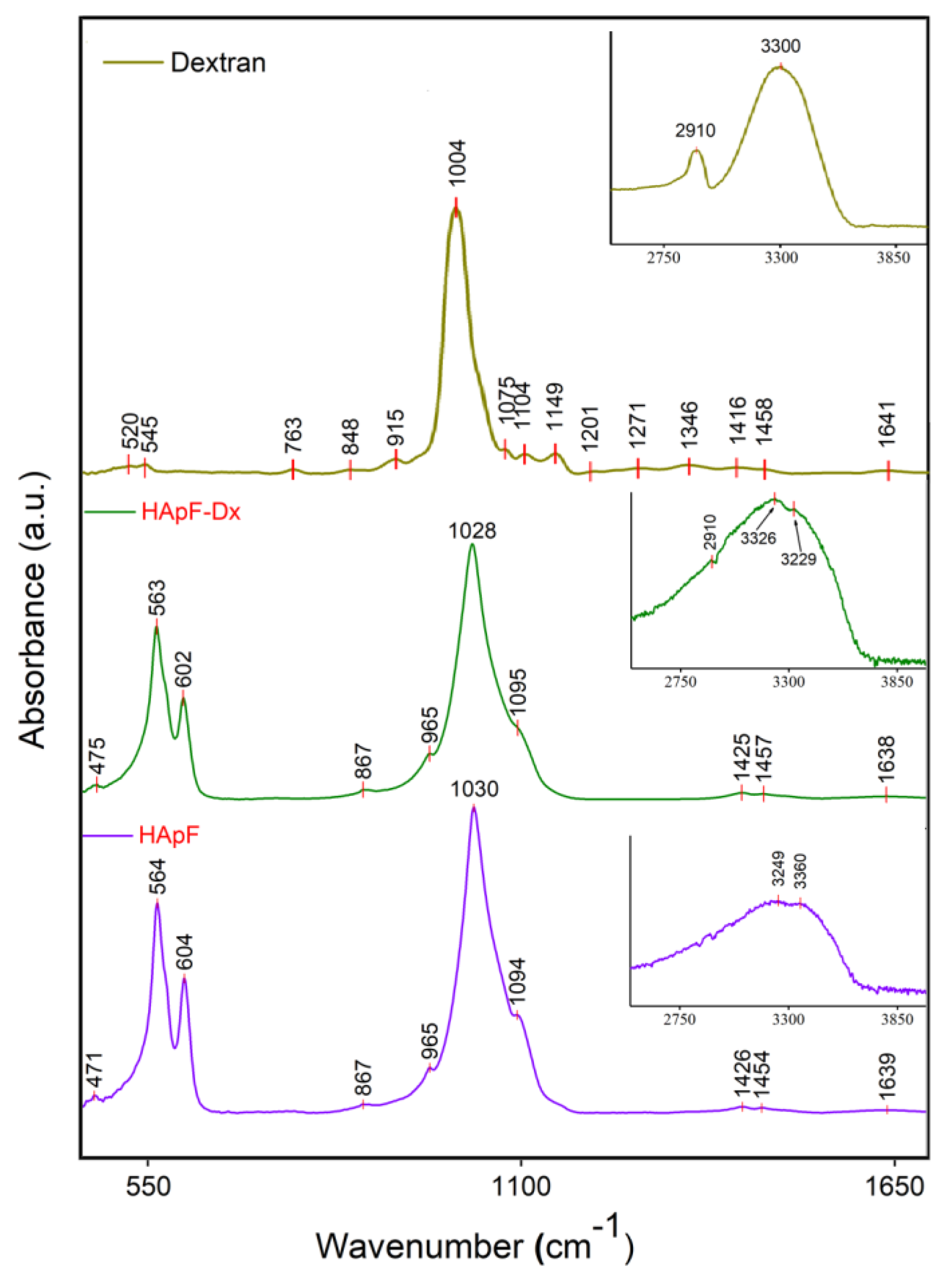
The functional groups present in both the HApF and HApF-Dx thin films were highlighted by the results of the FTIR analysis. Therefore, the findings of the FTIR analysis performed on the HApF and HApF-Dx thin films are presented in the FTIR general spectra shown in
Figure 6. Both spectra were recorded over a range from 450 to 4000 cm
−1. In the case of the HApF thin films, the obtained FTIR spectra reveal the presence of the bands associated with the PO
43− groups that are observed at approximately 471 cm
−1 (for ν
2 of [PO
43−]), 564 cm
−1 and 604 cm
−1 (for ν
4 of [PO
43−]), 965 cm
−1 (for ν
1 of [PO
43−]), 1030 cm
−1 and at about 1094 cm
−1 (for ν
3 of [PO
43−]) [
46,
47,
48,
49,
50,
51].
In addition, the bands specific to the ν
3 vibration of the carbonate [CO
32−] group appear at approximately 1454 cm
−1 and 1426 cm
−1 [
46,
47,
48,
49,
50,
51]. Similar findings were presented by Predoi, D., et al. [
46] in their studies regarding the exploration of the fabrication, properties, and morphology of fluoride -doped hydroxyapatite coatings developed by the vacuum deposition technique.
The FTIR spectra of dextran presented in
Figure 6 exhibit bands in the spectral region between 848 and 1075 cm
−1 that correspond to the stretching vibrations of C–O–C linkages in dextran. Furthermore, bands identified within the 1416–1458 cm
−1 interval could be associated with C–O–H deformation vibrations and may also include contributions from the symmetric stretching vibrations of the carboxylate O–C–O groups [
52,
53]. In the FTIR spectra of dextran could be observed the vibrational bands around 1635–1645 cm
−1 that are assigned to the bending vibrations of the adsorbed water molecules, as well as a strong absorption vibrational band near 3300 cm
−1, characteristic of the stretching vibrations of water molecules [
52,
53]. Moreover, the presence of a peak at around 2900 cm
−1 in the dextran spectra could be associated with the C-H stretching vibration [
52,
53].
The FTIR general spectra of the HApF-Dx thin films (
Figure 6) display a band at approximately 475 cm
−1, attributed to the characteristic ν
2 vibration of the [PO
43−] group from the HAp structure [
51]. The bands observed at around 563 cm
−1 and 602 cm
−1 correspond to the ν
4 vibration of the [PO
43−] group [
51]. The broad band centered at 965 cm
−1 in
Figure 6 indicates the formation of HAp and is associated with the ν
1 of the [PO
43−]) groups [
51]. The intense band centered at 1028 cm
−1 and the one observed at about 1095 cm
−1 appear due to the ν
3 vibration of the [PO
43−] group in HAp. In the FTIR spectra of the HApF-Dx thin film, the bands observed in the range of 1400–1426 cm
−1 can be attributed to the dextran molecule’s vibration such as ν(C–H) and δ(C–H) [
54]. Also, according to recent studies [
51,
54], the presence of bands at 867 cm
−1 and in the 1418–1554 cm
−1 spectral domain of the FTIR spectra indicates the presence of carbonate groups (to ν
2 and ν
3, respectively). The bands observed in the range of 1415–1470 cm
−1 could be associated with C–O–H deformation vibrations and contributions from the O–C–O symmetric stretching vibrations of the carboxylate group [
52,
53,
55,
56], from the dextran structure. The band centered at 2910 cm
−1 could be associated with the ν (C–H) from the dextran structure [
52,
53]. The band observed at 1639 cm
−1 (in the FTIR spectra of the HApF and HApF-Dx thin films) may be attributed to the bending vibration of the O–H bond. Furthermore, the intense bands observed in the spectral range of 3100–3500 cm
−1 could be associated with adsorbed water suggesting that the thin films are well hydrated (
Figure 6). In addition, the FTIR analysis shows that the presence of dextran in the HApF-Dx thin film causes broadening and minor shifts in the peak position related to the functional groups. Another effect induced by the presence of the dextran in the HApF-Dx sample consists of the widening of the vibration bands (as can be seen in
Figure 6). These results are in concordance with the one previously reported by Predoi, D. and collaborators in their work entitled “New Physico-Chemical Analysis of Magnesium-Doped Hydroxyapatite in Dextran Matrix Nanocomposites” [
54].
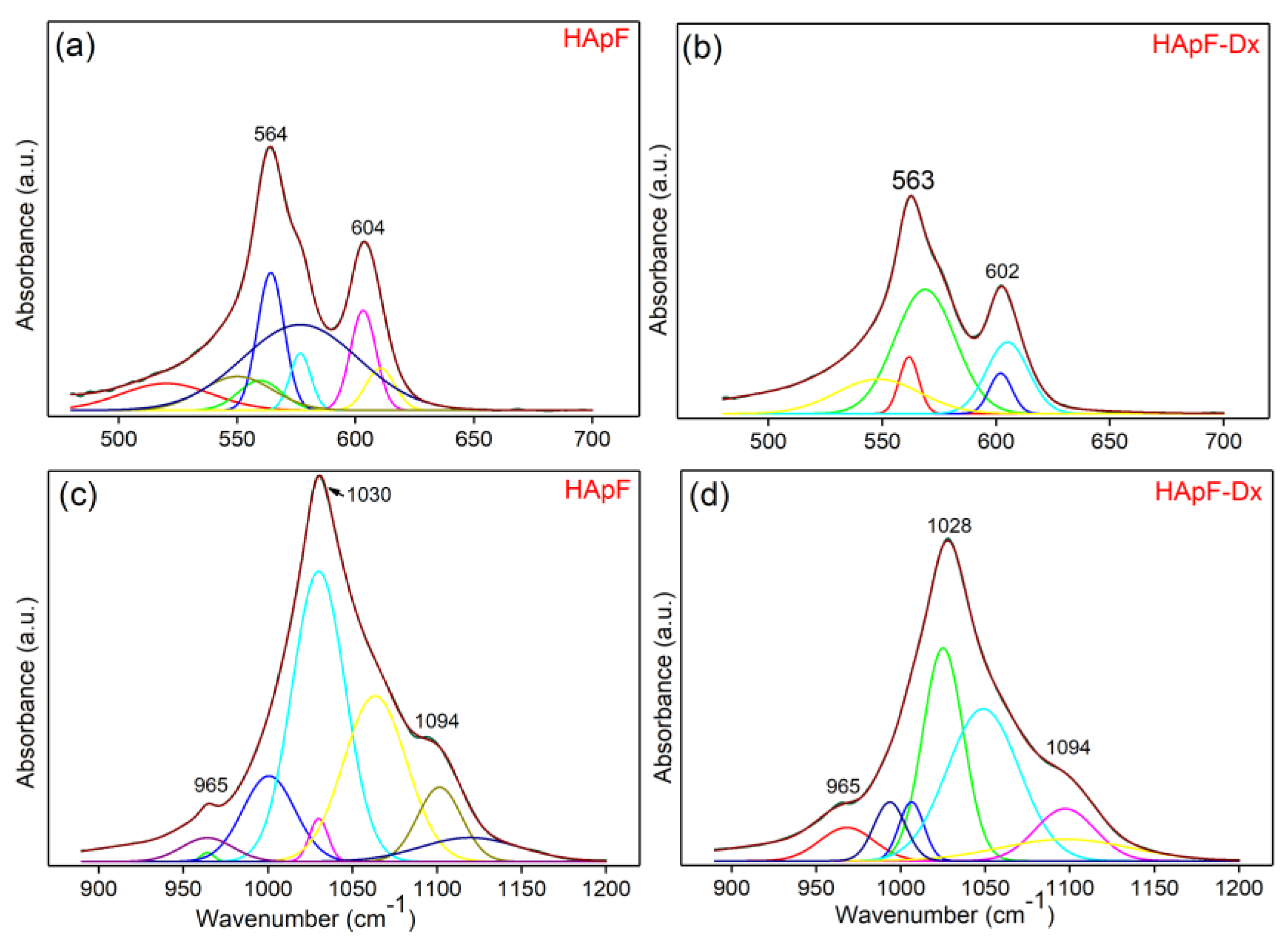
The obtained FTIR data were analyzed with the curve fitting analysis in the 450–700 cm
−1 and 850–1200 cm
−1 spectral ranges. These two spectral ranges were chosen due to the fact that the bands found here belongs to the ν
1, ν
2, ν
3, and ν
4 vibrations of the [PO
43−] groups from HAp. The results of the curve fitting analysis are depicted in
Figure 7.
In
Figure 7a, it can be observed that for the deconvolution of the FTIR spectra of the HApF thin films (in the 450–700 cm
−1 spectral domain) eight components are used for the achievement of a satisfactory fit. In the same spectral region but for the HApF-Dx thin films (
Figure 7b), five sub-bands are needed for obtaining a satisfactory fit of the FTIR spectra. Furthermore, a good fit of the FTIR spectra in the 850–1200 cm
−1 spectral domain (for the HApF thin films) was obtained with the aid of eight components (
Figure 7c). For the FTIR spectra (850–1200 cm
−1 spectral domain) of the HApF-Dx thin films, a satisfactory fit was obtained with the aid of seven sub-bands (
Figure 7d).
The differences in chemical components and binding energy shifts observed in the high-resolution XPS spectra and fitted FTIR spectra of HApF and HApF-Dx can be attributed to several factors such as fluoride substitution effects, dextran influence on surface chemistry, changes in local atomic environment, surface effects, etc. Fluoride ions partially replace the hydroxyl (-OH) groups in the hydroxyapatite structure in the two analyzed samples, and the degree of substitution and the binding environment influence the observed changes. The partial replacement of hydroxyl (-OH) groups in the hydroxyapatite structure leads to changes in the binding energy in the XPS spectra. On the other hand, the presence of dextran in the HApF-Dx sample introduces additional functional groups containing oxygen which leads to the modification of the chemical bonds. As a result, larger changes in the binding energy were observed in the XPS spectra of the HApF-Dx sample compared to the HApF sample. The dextran matrix affects the surface chemistry and adsorption properties which may lead to the appearance of additional spectral components in the XPS and FTIR spectra of the HApF-Dx sample. Furthermore, the incorporation of dextran affects the coordination of calcium and phosphate groups, leading to variations in the central levels of Ca 2p, P 2p, and O 1s in high-resolution XPS spectra. The additional organic components in HApF-Dx contribute to higher oxidation states by changing the binding energies. Changes in vibrational modes due to the presence of dextran were also observed in the FTIR spectra of the HApF-Dx sample. Thus, the C-O and C-H stretching vibrations of dextran overlap with the phosphate and hydroxyl bands, leading to a broadening of the peaks and lower intensities compared to the HApF sample. The presence of dextran introduces additional functional groups (C-O, C=O and -OH) that contribute to the FTIR spectrum by shifting the existing peaks. The differences in chemical composition and binding energy shifts observed in the high-resolution XPS spectra and deconvoluted FTIR spectra of HApF and HApF-Dx can be attributed to the unique chemical environments, electron density distributions, and intermolecular interactions introduced by the presence of dextran. These factors collectively influence the binding energies and vibrational frequencies of the constituent atoms, resulting in the differences observed in the XPS and FTIR spectra.
In order to obtain valuable information regarding the surface features of the HApF and HApF-Dx thin films, the scanning electron microscopy technique was employed. The results of the SEM studies are revealed in
Figure 8a,b. In the case of the HApF thin films, it could be observed that their surface is nanostructured and mainly consists of agglomerated nanoparticles that form nanoagglomerates. Furthermore, the SEM studies revealed that the nanostructured surface of the HApF-Dx thin films consists of agglomerated nanoparticles covered with dextran. Also, it could be observed that both studied thin films are covered with a continuous and homogenous layer. Furthermore, the results of the SEM studies confirmed the absence of cracks on the HApF and HApF-Dx thin films’ surface.
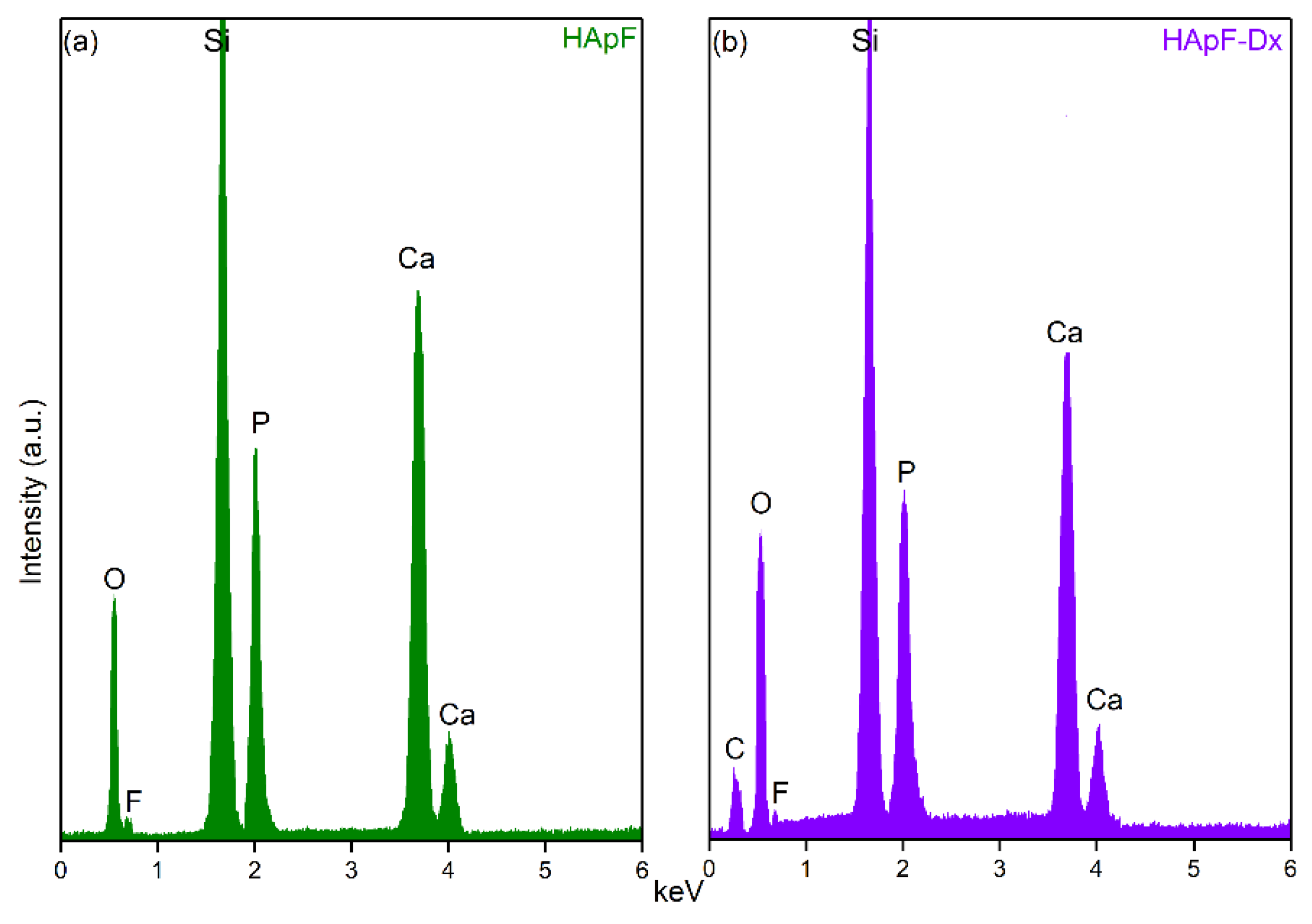
Data on the chemical composition of the HApF and HApF-Dx thin films were obtained through EDS analysis. The EDS spectra obtained on the HApF thin films, illustrated in
Figure 9a, reveal the presence of chemical elements that belong to the HApF structure, specifically Ca, P, O, and F. In
Figure 9b, the EDS spectra of HApF-Dx thin films are presented. Thus, in addition to the chemical elements specific to the HApF structure (Ca, P, O, and F), it can be observed that a C line also appears, which is most likely due to the presence of dextran in the HApF-Dx composition. Additionally, in both spectra appears a Si line, attributed to the substrate on which thin films were deposited. Moreover, no other lines appear in the EDS spectra, indicating the absence of impurities and underlining the purity of the synthesized thin films. These are in good agreement with the one obtained by other XPS studies (reported in this work) and with the one previously reported [
46] fact that highlights that the method used for the development of the thin films allows for the obtaining of pure specimens.
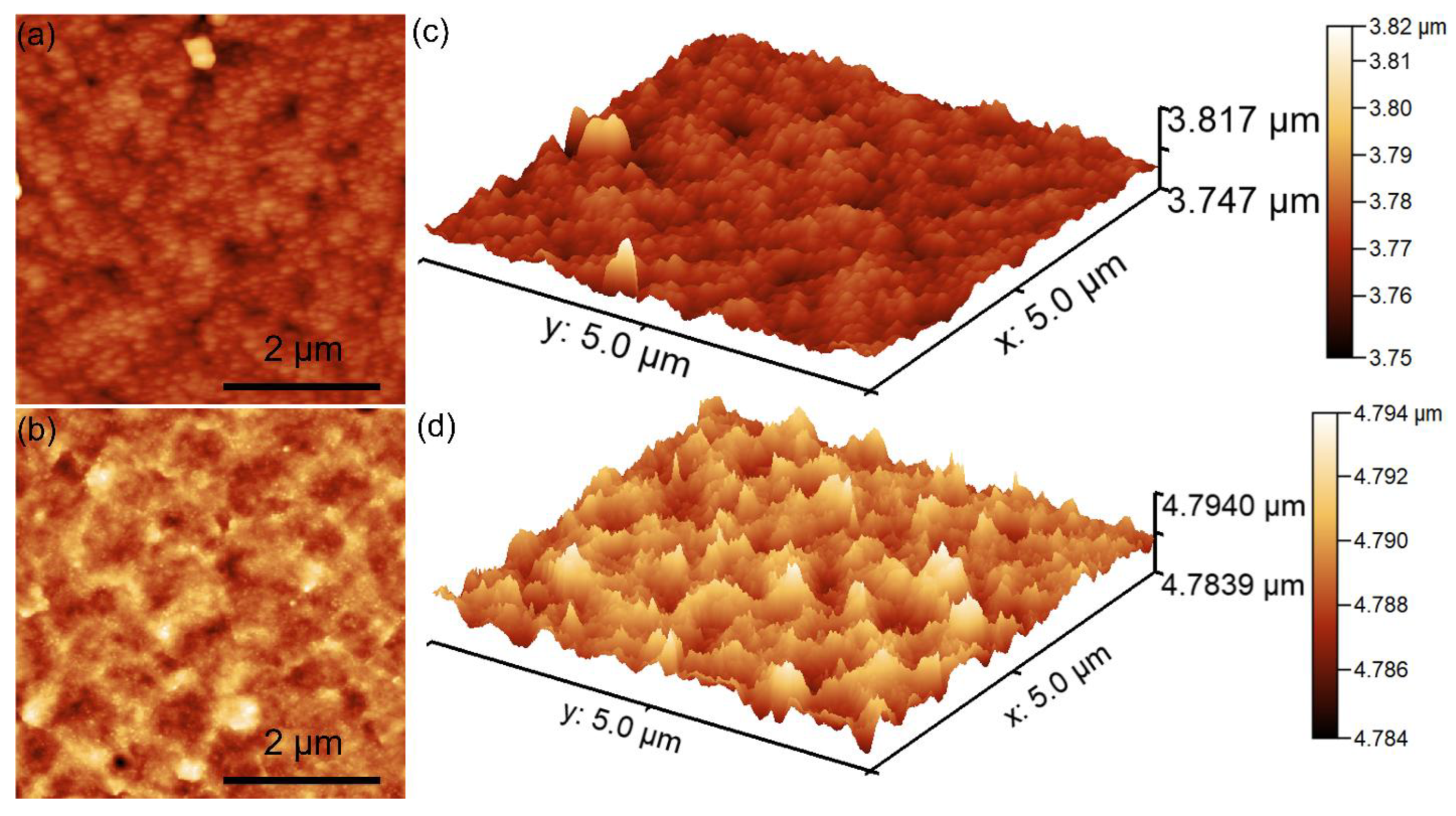
The surface topography recorded on an area of 5 × 5 µm
2 of the HApF and HApF-Dx coatings is depicted in
Figure 10a,d.
The AFM topography highlighted that the surface topography of the HApF coatings exhibits the appearance of a uniformly and continuously deposited layer. More than that, both the 2D AFM surface topography as well as the 3D representation highlights the presence of a homogenous surface with particle conglomerates uniformly and evenly distributed on its surface. The 2D AFM images suggest the absence of noticeable irregularities on the surface of the HApF coatings. Additionally, the 3D representation of the surface topography of the HApF coatings depicted in
Figure 10c further confirms the presence of a uniformly deposited layer with evenly distributed particle conglomerates of nanometric sizes. Furthermore, the AFM 2D topography of the surface HApF-Dx coatings highlight that the presence of the dextran matrix exhibited a noticeable influence on the surface morphology of the HApF coatings. The 2D AFM images of the HApF-Dx coating’s surface showed the presence of a uniformly deposited layer with a more pronounced arrangement of particle conglomerates. The 3D representation of the HApF-Dx coating’s surface topographies also confirms the presence of a homogenous and uniformly deposited layer with nanometric-size particle aggregates. In addition, information regarding the roughness of the HApF and HApF-Dx coatings’ surface was determined by quantifying the R
RMS roughness parameters for the entire recorded surface. The value of the R
RMS parameters determined for HApF was 4.37 nm and for HApF-Dx was 26.08 nm. The data revealed that the presence of the dextran matrix increased the roughness of the HApF-Dx compared to the HApF coating’s surface. These results are in good agreement with the reported literature regarding the influence of particle size on the roughness of the coatings’ surface [
57,
58,
59,
60]. The correlation between particle size and surface roughness in the case of coatings was reported to be intricate and to be influenced by various factors such as particle distribution, agglomeration behavior, and, nonetheless, by the method used to obtain the coating. Although smaller particles can often promote smoother surfaces through improved packing and flow, the smaller particle size can also contribute to an increase in surface roughness if the particles have the tendency to agglomerate or if the process used for obtaining the coating has a limited leveling capability. Therefore, a surface coating that is composed of smaller particles often exhibits a higher surface roughness compared to the surface of a coating composed of larger particles. This could be attributed to how the particles have the ability to pack and interact. The smaller particles possess a higher surface area-to-volume ratio and have the tendency to agglomerate, which can lead to the appearance of irregularities on the coated surface. In contrast, the larger particles, even though they are individually more prominent, have the tendency to settle more evenly and are less likely to form densely clustered rough areas. As a result, coatings that exhibit a smaller particle size could lead to a rougher, more uneven surface texture at the microscopic level.
The surface of the HApF and HApF-Dx thin films was evaluated by metallographic microscopy (MM) studies. The 2D MM images of the HApF and HApF-Dx thin films are shown in
Figure 11a,b. The 2D images were captured using the 20× objective of the metallographic microscope. Consistent with the SEM analysis, the MM images confirmed that both thin films were uniformly deposited, and that no visible cracks or fissures could be observed. Additionally, the MM images revealed that both thin films have a nanostructured surface. Furthermore, the presence of dextran in the HApF-Dx thin films slightly influences the surface morphology features. Additionally, the MM images indicated that the surfaces of the examined thin films (HApF and HApF-Dx) are nanostructured. Taking into consideration the results of the US studies, we have proved that the HApF-Dx suspensions are more stable than the HApF suspension, and the SEM, AFM, and MM images showed that this behavior allowed for the development of more homogeneous and uniform HApF-Dx thin films.
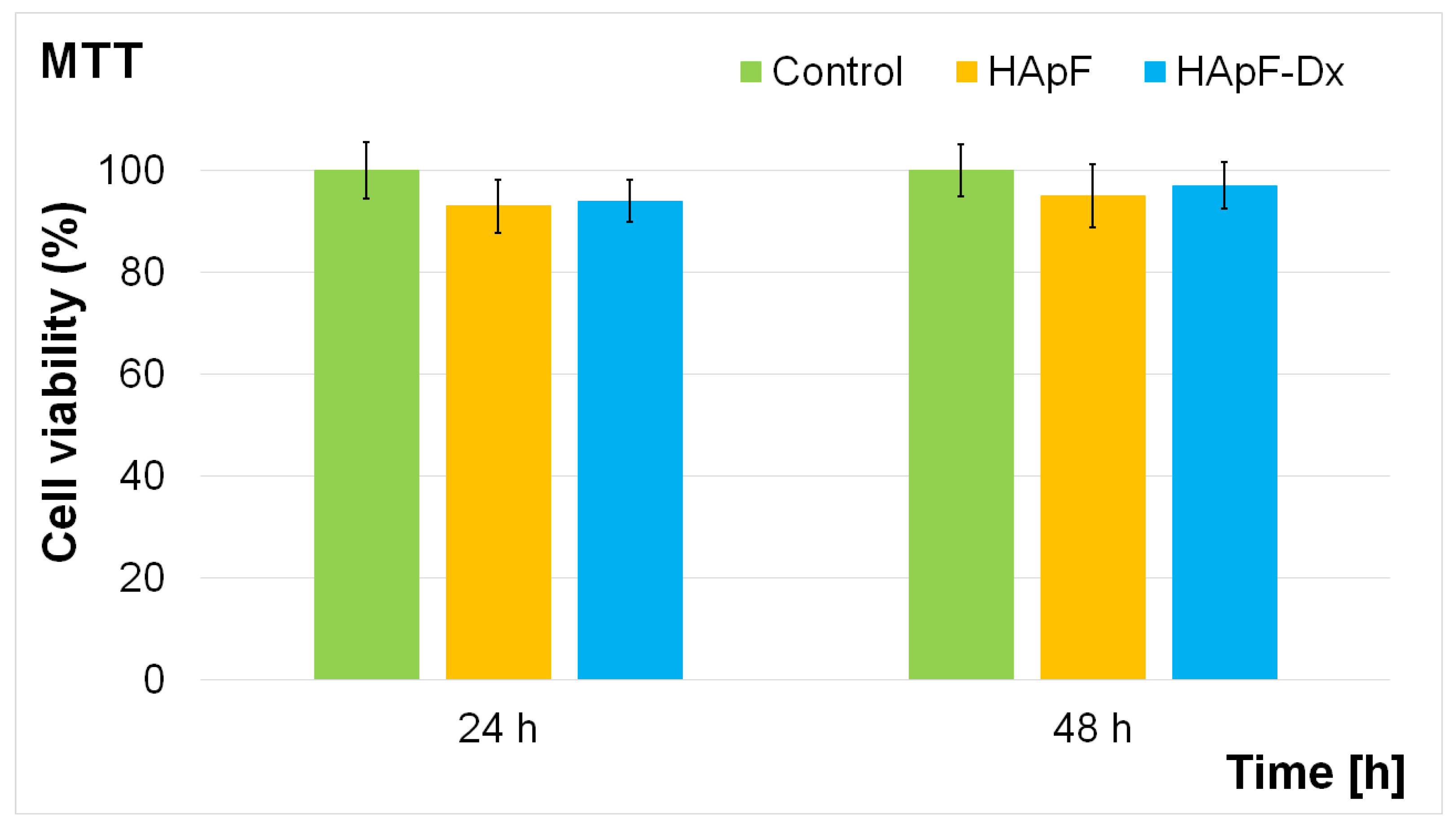
The biocompatibility of the HApF and HApF-Dx coatings was investigated with the aid of the MTT assay. The cells were incubated for 24 and 48 h with the coatings and the cell viability was assessed and graphically represented in
Figure 12. The experiments were performed in triplicate, and the results are presented as mean ± standard deviation. Statistical analysis using a
t-test confirmed that all calculated
p-values were
p < 0.05 when comparing the experimental and control groups. The results of the MTT assays demonstrated that, after 24 h of culturing, the coatings exhibited an increased cell viability for both coatings, indicating a positive effect of fluoride-doped hydroxyapatite on HGF-1 cell viability. After 48 h, both the HApF and HApF-Dx coatings continued to promote an improved cell viability, likely due to the slower release rate of fluoride. On the other hand, the results showed that the cell viability of the cells exposed to HApF-Dx showed a noticeable increase compared to the cell viability obtained for the cells exposed to HApF. Specifically, the cell viability of HGF-1 cells incubated with HApF coatings reached 93% after 24 h, and 95% after 48 h, while the HGF-1 cells incubated with HApF-Dx coatings reached 94% after 24 h, and 97% after 48 h. These results underscore a consistent improvement in cell viability with prolonged incubation time, as evidenced by the in vitro assays. These findings align with previous studies investigating the impact of fluoride-doped hydroxyapatite coatings on cell viability [
46,
47,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72].
Additional information about the HGF-1 adherence and development on the surface of the HApF and HApF-Dx coatings was obtained using metallographic microscopy. The results of the metallographic microscopy observations are presented in
Figure 13a,b.
The MM images revealed that the surfaces of both the HApF and HApF-Dx coatings effectively supported HGF-1 cell adhesion and development. Furthermore, no morphological abnormalities were observed in the HGF-1 cells that adhered to the coating surfaces. These findings align well with previously reported literature data [
63,
64,
65,
66,
67,
68,
69,
70,
71,
72] and demonstrate that the viability of HGF-1 cells improved for the HApF-Dx coatings compared to HApF. This increase in cell viability underscores the biocompatibility and bioactivity of the coatings, along with their ability to promote cell adhesion, proliferation, and differentiation. The MM images revealed that both the HApF and HApF-Dx coatings effectively supported HGF-1 cell adhesion and development. Notably, the HApF-Dx coatings exhibited superior biological properties, as evidenced by a visible increase in HGF-1 cells compared to the HApF coatings. This enhanced viability highlights the improved biocompatibility and bioactivity of the HApF-Dx coatings. In addition to increased viability, the MM images revealed that the adhered HGF-1 cells exhibited typical, healthy cellular features for both tested coatings. The MM visualization showed that the cells maintained an elongated, spindle-shaped morphology with extended filopodia, indicative of strong adhesion and active cellular interaction with the substrate. More than that, no morphological abnormalities were observed in the HGF-1 cells on either HApF or HApF-Dx coatings, further confirming the favorable biological response of the coatings. These findings are in good agreement with previously reported data and emphasize the superior performance of the HApF-Dx coatings in promoting cell adhesion, proliferation, and differentiation [
63,
64,
65,
66,
67,
68,
69,
70,
71,
72]. The observed increase in cell viability and healthy cell morphology highlights the potential of the HApF-Dx coatings for biomedical applications requiring enhanced biocompatibility and cellular support.
Additional investigations into HGF-1 cell adhesion, morphology, and development on HApF and HApF-Dx coatings were conducted using scanning electron microscopy (SEM). These analyses aimed to assess the interaction of HGF-1 cells with the surfaces of both coatings, particularly emphasizing the influence of dextran’s presence in enhancing biological properties. After 48 h of incubation, SEM imaging (
Figure 14a,b) revealed excellent cell adhesion on both the HApF and HApF-Dx surfaces.
However, the cells cultured on the HApF-Dx surfaces demonstrated noticeably improved adherence characteristics. The HGF-1 cells on the HApF coatings displayed moderate filopodia extension, with cells spreading uniformly across the surface and establishing stable anchorage. In contrast, the HGF-1 cells adhered to the HApF-Dx surfaces exhibited extensive filopodia and lamellipodia formation, indicating an enhancement in the interactions between the cells and the coatings’ surface. This pronounced cellular development on the HApF-Dx coatings suggests improved biocompatibility and cell adhesion properties attributed to the presence of dextran. The improved biological performance of the HApF-Dx coatings can be linked to the dextran component’s ability to enhance surface hydrophilicity and provide additional binding sites conducive to cell attachment [
73,
74,
75,
76]. These observations are in good agreement with results obtained from the cell viability assays as well as the visual observation by MM.
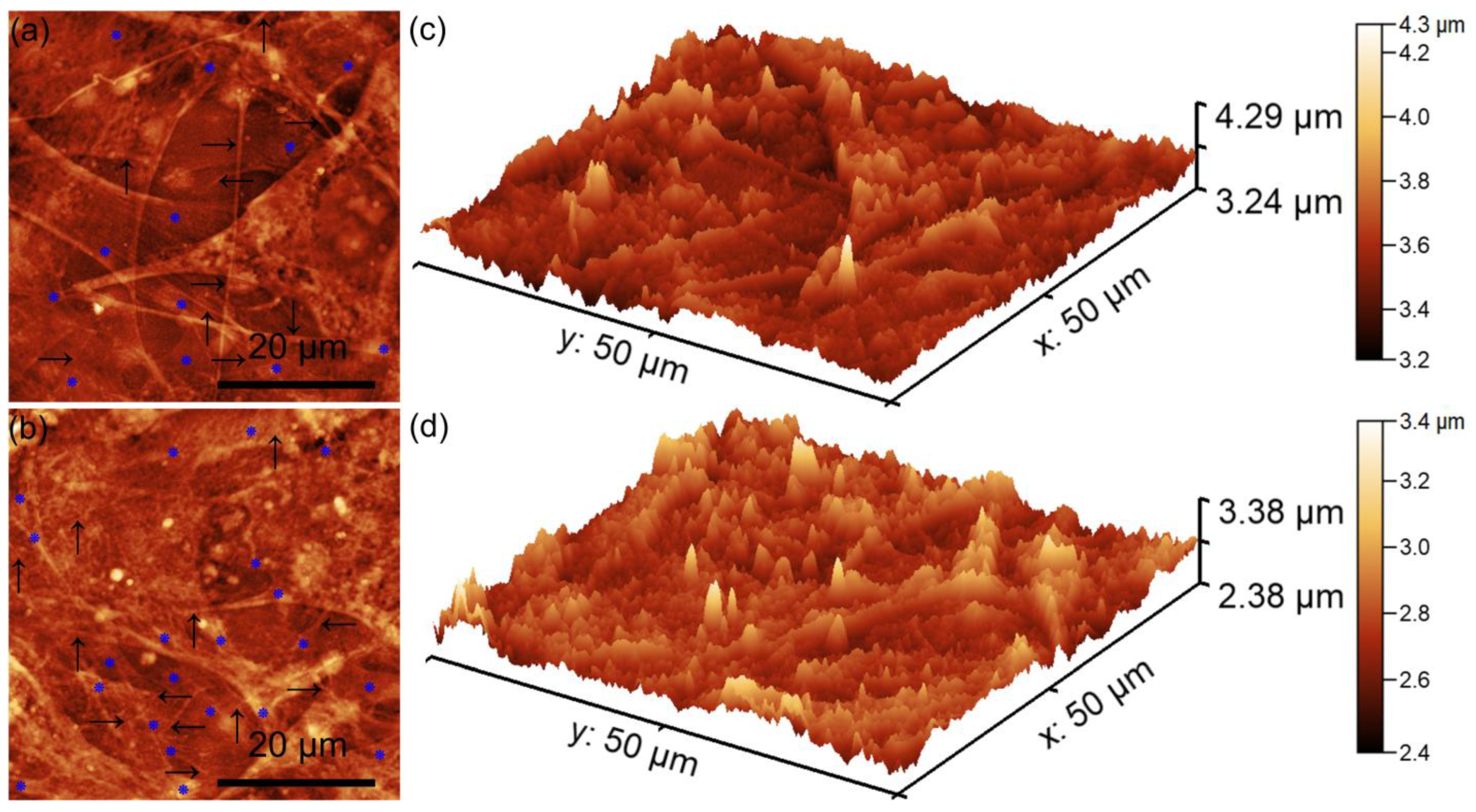
Complementary information about the HGF-1 cells’ adherence and development on the surfaces of the HApF and HApF-Dx coatings was obtained though AFM studies. The 2D atomic force microscopy (AFM) topographies provided detailed insights into the morphological characteristics of the HGF-1 cells adhering to the HApF and HApF-Dx coatings (
Figure 15a,d). The AFM 2D topographies as well as their 3D representations revealed that the HGF-1 cells adhered across the entire surface of the HApF and HApF-Dx coatings. These 2D AFM images highlighted that the adhered cells presented the typical morphological patterns of normal, healthy gingival fibroblasts, which displayed characteristic flattened and elongated cellular shapes. Such morphological features are indicative of healthy cell behavior, confirming the biocompatibility of the HApF and HApF-Dx coatings. Furthermore, the AFM images clearly depicted the development of confluent layers of HGF-1 cells on the HApF and HApF-Dx coatings’ surfaces. The cells appeared to form a continuous cellular network, suggesting their ability to proliferate effectively on the substrate. This observation was particularly evident for the cells adhered to HApF-Dx, where both the 2D AFM topographies and their corresponding 3D representations demonstrated complete surface coverage by the adhered cells.
Moreover, the AFM studies also highlighted the presence of an extracellular matrix (ECM), with a complex network of filopodia extensions. This interconnected network of filopodia was present and more evident for the cells adhered on the HApF-Dx compared to the ones adhered on the HApF coatings, reinforcing the observation of strong cell adhesion and communication. The presence of filopodia is a significant indicator of cellular biocompatibility and successful integration with the material surface. Filopodia play a crucial role in enabling cells to establish contact with both the underlying substrate and neighboring cells, promoting cell-to-cell communication and anchorage. Furthermore, the presence of lamellipodia alongside filopodia offers additional evidence of successful cell adhesion, spreading, and migration across the HApF-Dx coatings’ surfaces. These morphological features collectively indicate that the HApF-Dx coatings provide a conducive environment for cellular attachment and growth without inducing cytotoxic effects.
Enhanced cell spreading was particularly evident on the HApF-Dx surface, emphasizing its superior biocompatibility and improved cellular interaction compared to the HApF coating. AFM analysis further confirmed that HGF-1 cells adhered to both the HApF and HApF-Dx surfaces, displaying a well-spread morphology with prominent cellular extensions. The HApF-Dx surface exhibited increased surface roughness and distinct topographical features that promoted enhanced cellular adhesion and spreading. These characteristics highlight that the HApF-Dx surface showed an improved biocompatibility and enhanced cell-supportive properties compared to the HApF surface.
HGF-1 cells, which are a human gingival fibroblast line, usually exhibit a spindle-shaped, fibroblast-like morphology. This morphology is characterized by the presence of elongated bodies having tapering ends and showing prominent cytoplasmic processes that have the role of enabling the intercellular communication and interaction with the extracellular matrix [
77,
78,
79,
80]. Also, these types of cells present oval, centrally located nuclei with visible nucleoli that depict an active transcription and cellular vitality. When cultured on biocompatible coatings, HGF-1 cells adhere strongly, indicating favorable membrane–surface interactions. In addition, the good adherence and development on a surface is emphasized by the presence of filopodia and lamellipodia [
77,
78,
79,
80] which in our case could be observed in both the MM, SEM, and AFM images. These coatings support cell attachment and spreading, helping the cells retain their shape and proliferative ability. The presence of focal adhesions and a well-organized actin cytoskeleton suggests that the substrate offers a good supportive environment for anchorage and signaling. This behavior that we emphasized in the case of both the HApF and HApF-Dx coatings is an important factor in determining a biomaterial’s suitability for dental or soft tissue engineering, as it demonstrates the material’s potential to integrate with tissues and to be able to support essential regenerative processes.
Over the past few decades, extensive research has been conducted regarding the several aspects of fluoride ion absorption by synthetic hydroxyapatite (HA: Ca
10(PO
4)
6(OH)
2), including the mechanism of fluoride incorporation [
81,
82] and its impact on solubility [
83]. Despite these efforts, no single ion or combination of ions has been identified as possessing all the characteristics needed for the perfect coating. While each study adds valuable insights, the development of a solution with maximum efficiency remains a work in progress. Moreover, the precise control of the morphological, structural, and compositional characteristics of HAp-based materials is crucial for enhancing their bio-functionality as coating materials for implant surfaces. This is because the material’s surface properties play a vital role in influencing the interaction with the cellular coating layer. To improve the mechanical and biological properties of HAp, various surface modification techniques have been implemented.
The present study focused on investigating the surface morphology, homogeneity, structural characteristics, and biological properties of the HApF and HApF-Dx thin films obtained by spin-coating method. The findings aim to support the development of advanced coatings for dental implants, with the potential to reduce the risk of post-implant or post-operative complications. The results from the AFM investigation emphasize the strong adhesive properties of both the HApF and HApF-Dx coatings’ surface for HGF-1 cells. Notably, the evidence of widespread cell adherence and proliferation was observed to be more pronounced in the case of HApF-Dx compared to HApF. These results show the favorable surface characteristics of HApF-Dx in supporting cellular adherence and development. The results of the biological assays conducted in our study emphasized that both fluoride as well as dextran played a crucial role in enhancing the bioactivity of hydroxyapatite (HAp) coatings, particularly in improving the adherence and proliferation of human gingival fibroblasts (HGF-1). The fluoride incorporation into HAp enhances its stability by reducing solubility and increasing resistance to degradation in the oral environment, while dextran, which is a hydrophilic polysaccharide, has the ability to improve the surface energy and hydrophilicity, promoting protein adsorption and cell attachment [
73,
74,
75,
76]. The combination of these elements in the HApF-Dx coatings is expected to create a more favorable microenvironment for soft tissue integration around dental implants, which is critical for implant stability and longevity. Existing studies on modified HAp coatings support the potential benefits of fluoride and dextran in influencing cell behavior [
73,
74,
75,
76]. Research on HAp coatings incorporating bioactive agents like zinc oxide and hepatocyte growth factor has demonstrated their ability to enhance cell adhesion and proliferation [
61,
62,
73]. Cao et al. [
61], in their study on “Magnesium and fluoride doped hydroxyapatite coatings grown by pulsed laser deposition for promoting titanium implant cytocompatibility”, highlighted the crucial role of fluoride (F) in enhancing cellular health. After one week of culturing, the coatings significantly improved cell viability, underscoring the beneficial effects of magnesium- and fluoride-doped hydroxyapatite. Their study emphasized that, after two weeks of incubation, the Mg-HA coating demonstrated a noticeable enhancement in osteogenesis, likely due to the controlled release of Mg ions. However, by the third week, a slight decline in cell viability was observed, possibly due to natural cell death and the delayed release of Mg
2+, which may have influenced cell activity [
61]. Furthermore, their results also showed that the Mg-F-HA coatings consistently maintained cell viability comparable to the control sample, emphasizing the synergistic effect of Mg and F doping [
61]. Therefore, their results highlighted that the presence of fluoride played an essential role in sustaining cell proliferation, particularly evident in the enhanced performance of Mg-HA coatings post-immersion. On the other hand, Cheng et al. [
68], in their study regarding “in vitro behavior of osteoblast-like cells on fluoridated hydroxyapatite coatings” reported that fluoridated hydroxyapatite (FHA) coatings with varying fluoride content, prepared using the sol–gel method, exhibited a good cytocompatibility. More than that, they established that a higher fluoride content in FHA coatings could lead to a lower zeta potential, which has the ability to enhance cell attachment. However, they also reported that an increased fluoride level could also reduce Ca
2+ release in the culture medium due to a decreased solubility, thus inhibiting cell proliferation. The results of their study concluded that for clinical applications, a moderate fluoride content is recommended as an optimal balance between cell attachment, proliferation, apatite deposition, and dissolution resistance [
68]. The preliminary results of our study highlighted that dextran has an important role in enhancing the bioactivity of hydroxyapatite–fluoride (HApF) coatings, particularly in promoting human gingival fibroblast cells (HGF-1) cell adherence and development. The results of the biological assays suggested that the incorporation of dextran into HApF coatings (HApF-Dx) leads to a higher surface roughness, which significantly influenced the interactions between the HGF-1 cells and the coating’s surface. This increased roughness provides a more favorable microenvironment for HGF-1 cells, improving initial adhesion and promoting cellular proliferation and differentiation. Compared to the smoother surface exhibited by the HApF coatings, the rougher HApF-Dx surfaces offered enhanced surface properties, facilitating better cell anchorage and cell spreading. Additionally, dextran contributes to a more hydrophilic surface, further supporting cell attachment and growth. Consequently, the combination of dextran and high surface roughness in the HApF-Dx coatings makes them more effective for biomedical applications involving gingival tissue regeneration. While specific studies on HApF-Dx coatings remain limited, these findings suggest that fluoride and dextran modifications could improve the HGF-1 response, leading to better soft tissue integration. Further investigation is needed to optimize their concentrations and understand their combined effects in clinical applications. Future research aimed at comprehending the complex effects will provide valuable insights, enabling the creation of safe and effective coatings for innovative biocompatible devices in tissue engineering and regenerative medicine. The results obtained thus far strongly indicate that HApF-Dx coatings possess considerable potential for future applications in the biomedical field.



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}