
The Desotamide Family of Antibiotics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Members of the Desotamide Family of Antibiotics and Their Bioactivities
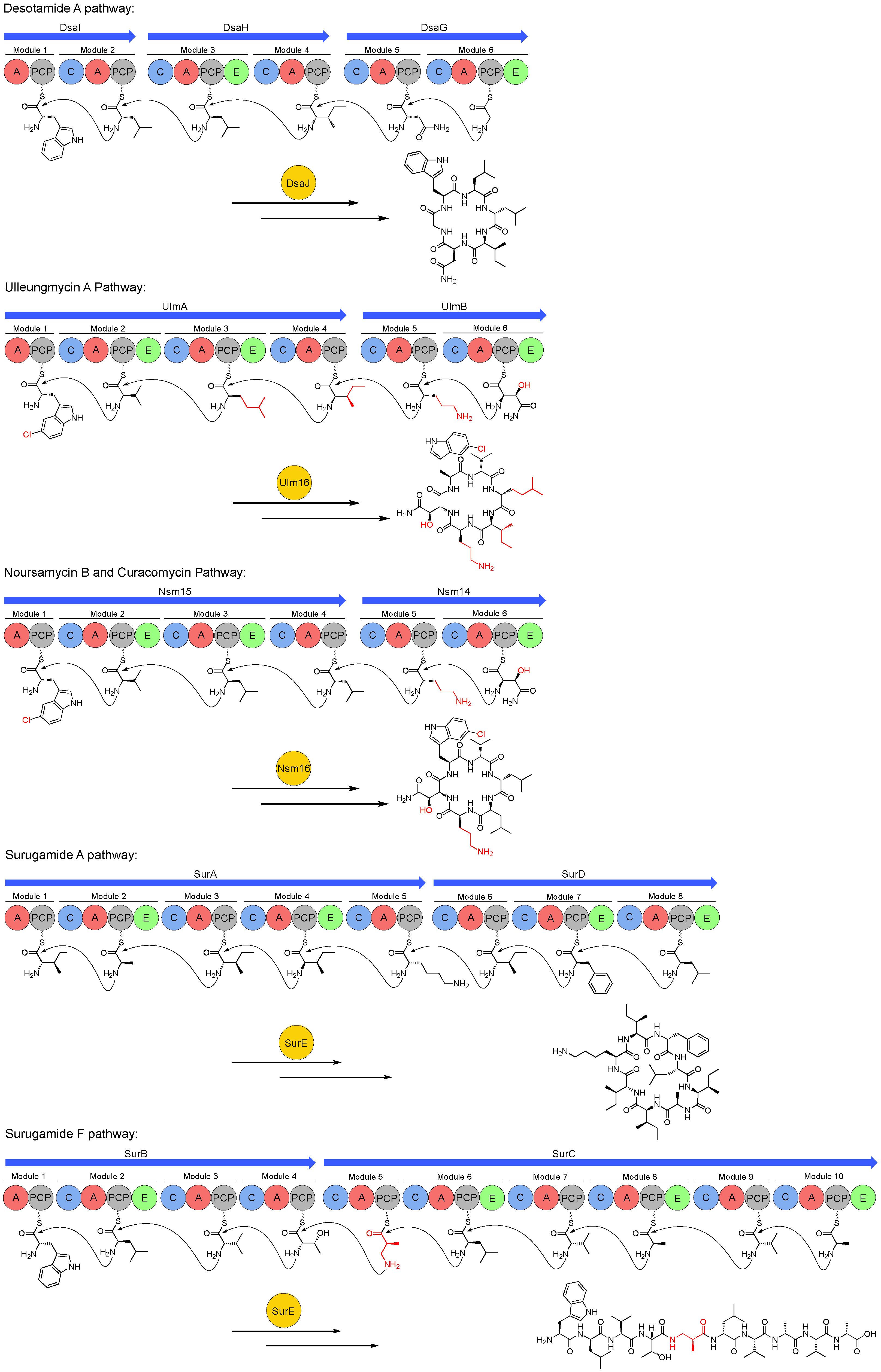
3. Biosynthetic Gene Clusters of the Desotamide Family
4. BGC-Encoded Precursors and Modified Amino Acids
4.1. Ornithine
4.2. Kynurenine and N-Formyl-l-Kynurenine
4.3. Allo-Isoleucine and Homoleucine
4.4. 3-amino-2-methylpropionic Acid (AMPA)
4.5. Chlorotryptophan
4.6. β-Hydroxyasparagine
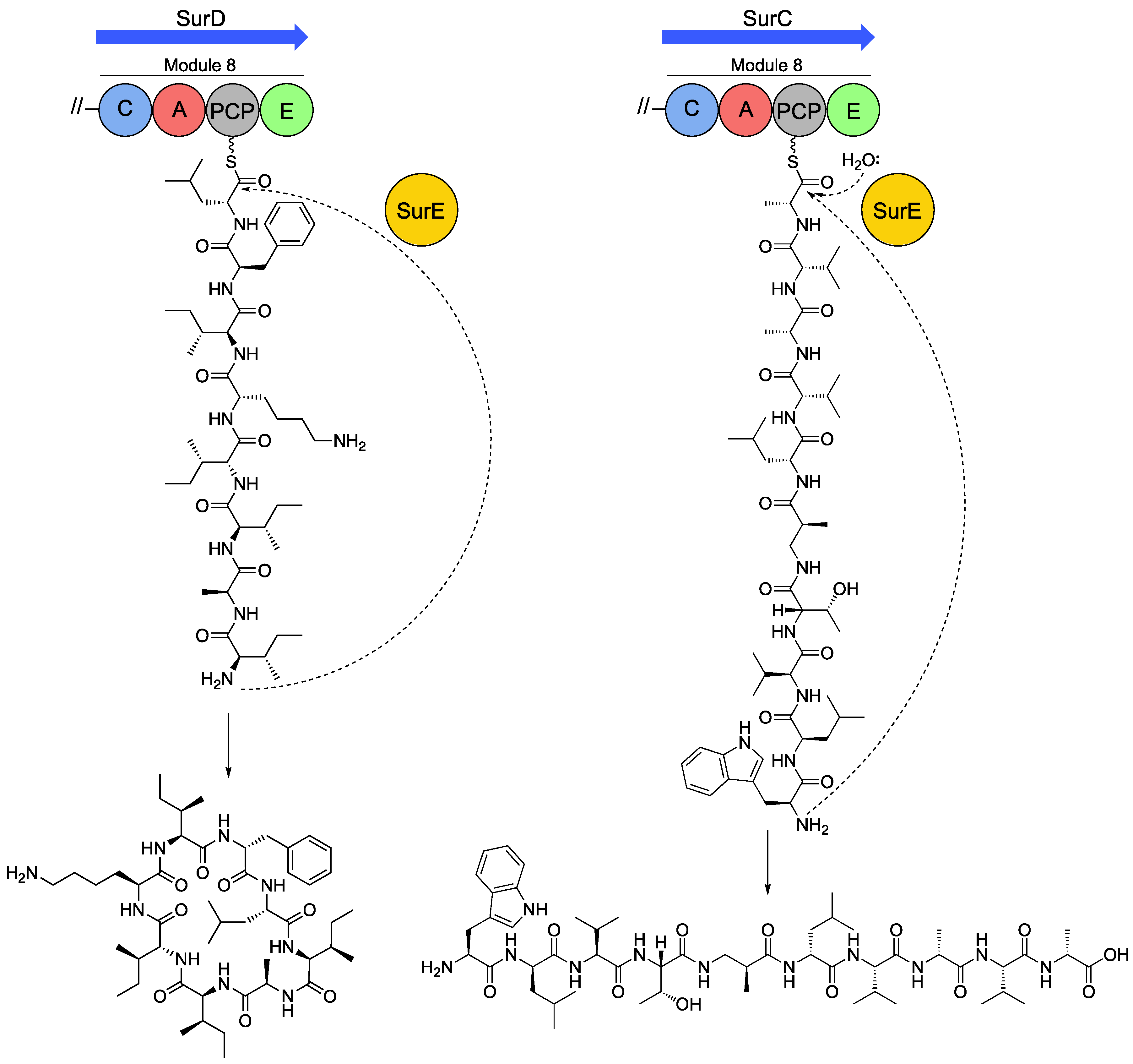
5. Peptide Offloading within the Desotamide Family by a Novel Class of Standalone Cyclases
5.1. Discovery and Characterisation of a Standalone Peptide Cyclase from the Sur BGC
5.2. SurE Cyclases are Widespread
6. Conclusions and Further Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Flärdh, K.; Buttner, M.J. Streptomyces morphogenetics: Dissecting differentiation in a filamentous bacterium. Nat. Rev. Microbiol. 2009, 7, 36–49. [Google Scholar] [CrossRef]
- Van der Heul, H.U.; Bilyk, B.L.; McDowall, K.J.; Seipke, R.F.; Van Wezel, G.P. Regulation of antibiotic production in Actinobacteria: New perspectives from the post-genomic era. Nat. Prod. Rep. 2018, 35, 575–604. [Google Scholar] [CrossRef]
- Romero, D.; Traxler, M.F.; López, D.; Kolter, R. Antibiotics as signal molecules. Chem. Rev. 2011, 111, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 2016, 33, 127–135. [Google Scholar] [CrossRef]
- Mullowney, M.W.; McClure, R.A.; Robey, M.T.; Kelleher, N.L.; Thomson, R.J. Natural products from thioester reductase containing biosynthetic pathways. Nat. Prod. Rep. 2018, 35, 847–878. [Google Scholar] [CrossRef]
- Müller, S.; Rachid, S.; Hoffmann, T.; Surup, F.; Volz, C.; Zaburannyi, N.; Müller, R. Biosynthesis of crocacin involves an unusual hydrolytic release domain showing similarity to condensation domains. Chem. Biol. 2014, 21, 855–865. [Google Scholar] [CrossRef]
- Balibar, C.J.; Walsh, C.T. GliP, a multimodular nonribosomal peptide synthetase in Aspergillus fumigatus, makes the diketopiperazine scaffold of gliotoxin. Biochemistry 2006, 45, 15029–15038. [Google Scholar] [CrossRef]
- Liu, X.; Jin, Y.; Cui, Z.; Nonaka, K.; Baba, S.; Funabashi, M.; Yang, Z.; Van Lanen, S.G. The role of a nonribosomal peptide synthetase in L-lysine lactamization during capuramycin biosynthesis. Chembiochem 2016, 17, 804–810. [Google Scholar] [CrossRef]
- Berry, D.; Mace, W.; Grage, K.; Wesche, F.; Gore, S.; Schardl, C.L.; Young, C.A.; Dijkwel, P.P.; Leuchtmann, A.; Bode, H.B.; et al. Efficient nonenzymatic cyclization and domain shuffling drive pyrrolopyrazine diversity from truncated variants of a fungal NRPS. Proc. Natl. Acad. Sci. USA 2019, 116, 25614–25623. [Google Scholar] [CrossRef]
- Zhou, Y.; Lin, X.; Xu, C.; Shen, Y.; Wang, S.-P.; Liao, H.; Li, L.; Deng, H.; Lin, H.-W. Investigation of penicillin binding protein (PBP)-like peptide cyclase and hydrolase in surugamide non-ribosomal peptide biosynthesis. Cell Chem. Biol. 2019, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Kuranaga, T.; Matsuda, K.; Sano, A.; Kobayashi, M.; Ninomiya, A.; Takada, K.; Matsunaga, S.; Wakimoto, T. Total synthesis of a non-ribosomal peptide surugamide B identifies a new offloading cyclase family. Angew. Chem. Int. Engl. 2018, 1–7. [Google Scholar] [CrossRef]
- Thankachan, D.; Fazal, A.; Francis, D.; Song, L.; Webb, M.E.; Seipke, R.F. A trans-acting cyclase offloading strategy for nonribosomal peptide synthetases. ACS Chem. Biol. 2019, 14, 845–849. [Google Scholar] [CrossRef]
- Miao, S.; Anstee, M.R.; LaMarco, K.; Matthew, J.; Huang, L.H.T.; Brasseur, M.M. Inhibition of bacterial RNA polymerases. Peptide metabolites from the cultures of Streptomyces sp. J. Nat. Prod. 1997, 60, 1–4. [Google Scholar] [CrossRef]
- Takada, K.; Ninomiya, A.; Naruse, M.; Sun, Y.; Miyazaki, M.; Nogi, Y.; Okada, S.; Matsunaga, S. Surugamides A–E, cyclic octapeptides with four D-Amino acid residues, from a marine Streptomyces sp.: LC–MS-aided inspection of partial hydrolysates for the distinction of D- and L-amino acid residues in the sequence. J. Org. Chem. 2013, 78, 6746–6750. [Google Scholar] [CrossRef]
- Song, Y.; Li, Q.; Liu, X.; Chen, Y.; Zhang, Y.; Sun, A.; Zhang, W.; Zhang, J.; Ju, J. Cyclic hexapeptides from the deep South China Sea-derived Streptomyces scopuliridis SCSIO ZJ46 active against pathogenic Gram-positive bacteria. J. Nat. Prod. 2014, 77, 1937–1941. [Google Scholar] [CrossRef]
- Ninomiya, A.; Katsuyama, Y.; Kuranaga, T.; Miyazaki, M.; Nogi, Y.; Okada, S.; Wakimoto, T.; Ohnishi, Y.; Matsunaga, S.; Takada, K. Biosynthetic gene cluster for surugamide A encompasses an unrelated decapeptide, surugamide F. Chembiochem 2016, 17, 1709–1712. [Google Scholar] [CrossRef]
- Son, S.; Hong, Y.-S.; Jang, M.; Heo, K.T.; Lee, B.; Jang, J.-P.; Kim, J.-W.; Ryoo, I.-J.; Kim, W.-G.; Ko, S.-K.; et al. Genomics-driven discovery of chlorinated cyclic hexapeptides ulleungmycins A and B from a Streptomyces species. J. Nat. Prod. 2017, 80, 3025–3031. [Google Scholar] [CrossRef]
- Mudalungu, C.M.; von Törne, W.J.; Voigt, K.; Rückert, C.; Schmitz, S.; Sekurova, O.N.; Zotchev, S.B.; Süssmuth, R.D. Noursamycins, chlorinated cyclohexapeptides identified from molecular networking of Streptomyces noursei NTR-SR4. J. Nat. Prod. 2019, 82, 1478–1486. [Google Scholar] [CrossRef]
- Kaweewan, I.; Komaki, H.; Hemmi, H.; Kodani, S. Isolation and structure determination of new antibacterial peptide curacomycin based on genome mining. Asian J. Org. Chem. 2017, 6, 1838–1844. [Google Scholar] [CrossRef]
- Khalil, Z.G.; Salim, A.A.; Lacey, E.; Blumenthal, A.; Capon, R.J. Wollamides: Antimycobacterial cyclic hexapeptides from an Australian soil Streptomyces. Org. Lett. 2014, 16, 5120–5123. [Google Scholar] [CrossRef]
- Khalil, Z.G.; Hill, T.A.; De Leon Rodriguez, L.M.; Lohman, R.-J.; Hoang, H.N.; Reiling, N.; Hillemann, D.; Brimble, M.A.; Fairlie, D.P.; Blumenthal, A.; et al. Structure-activity relationships of wollamide cyclic hexapeptides with activity against drug-resistant and intracellular Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e01773-18. [Google Scholar] [CrossRef]
- Li, Q.; Song, Y.; Qin, X.; Zhang, X.; Sun, A.; Ju, J. Identification of the biosynthetic gene cluster for the anti-infective desotamides and production of a new analogue in a heterologous host. J. Nat. Prod. 2015, 78, 944–948. [Google Scholar] [CrossRef]
- Ding, W.; Dong, Y.; Ju, J.; Li, Q. The roles of genes associated with regulation, transportation, and macrocyclization in desotamide biosynthesis in Streptomyces scopuliridis SCSIO ZJ46. Appl. Microbiol. Biotechnol. 2020, 104, 2603–2610. [Google Scholar] [CrossRef]
- Xu, F.; Nazari, B.; Moon, K.; Bushin, L.B.; Seyedsayamdost, M.R. Discovery of a cryptic antifungal compound from Streptomyces albus J1074 using high-throughput elicitor screens. J. Am. Chem. Soc. 2017, 139, 9203–9212. [Google Scholar] [CrossRef]
- Cunin, R.; Glansdorff, N.; Piérard, A.; Stalon, V. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 1986, 50, 314–352. [Google Scholar] [CrossRef]
- Mihali, T.K.; Kellmann, R.; Muenchhoff, J.; Barrow, K.D.; Neilan, B.A. Characterization of the gene cluster responsible for cylindrospermopsin biosynthesis. Appl. Environ. Microbiol. 2008, 74, 716–722. [Google Scholar] [CrossRef]
- Mazmouz, R.; Chapuis-Hugon, F.; Mann, S.; Pichon, V.; Méjean, A.; Ploux, O. Biosynthesis of cylindrospermopsin and 7-epicylindrospermopsin in Oscillatoria sp. strain PCC 6506: Identification of the cyr gene cluster and toxin analysis. Appl. Environ. Microbiol. 2010, 76, 4943–4949. [Google Scholar] [CrossRef]
- Chen, Y.; Unger, M.; Ntai, I.; McClure, R.A.; Albright, J.C.; Thomson, R.J.; Kelleher, N.L. Gobichelin A and B: Mixed-ligand siderophores discovered using proteomics. Medchemcomm 2013, 4, 233–238. [Google Scholar] [CrossRef]
- Kurnasov, O.; Jablonski, L.; Polanuyer, B.; Dorrestein, P.; Begley, T.; Osterman, A. Aerobic tryptophan degradation pathway in bacteria: Novel kynurenine formamidase. FEMS Microbiol. Lett. 2003, 227, 219–227. [Google Scholar] [CrossRef]
- Miao, V.; Coëffet-LeGal, M.-F.; Brian, P.; Brost, R.; Penn, J.; Whiting, A.; Martin, S.; Ford, R.; Parr, I.; Bouchard, M.; et al. Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 2005, 151, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Seipke, R.F.; Hutchings, M.I. The regulation and biosynthesis of antimycins. Beilstein J. Org. Chem. 2013, 9, 2556–2563. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.H.; Yeh, E.; Vosburg, D.A.; O’Connor, S.E.; Walsh, C.T. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature 2005, 436, 1191–1194. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Shang, Z.; Lemetre, C.; Ternei, M.A.; Brady, S.F. Cadasides, calcium-Dependent acidic lipopeptides from the soil metagenome that are active against multidrug-resistant bacteria. J. Am. Chem. Soc. 2019, 141, 3910–3919. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qin, X.; Liu, J.; Gui, C.; Wang, B.; Li, J.; Ju, J. Deciphering the biosynthetic origin of L-allo-isoleucine. J. Am. Chem. Soc. 2016, 138, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Bearden, D.W.; Rein, K.S. Biosynthetic origin of the 3-amino-2,5,7,8-tetrahydroxy-10-methylundecanoic acid moiety and absolute configuration of pahayokolides A and B. J. Nat. Prod. 2011, 74, 1535–1538. [Google Scholar] [CrossRef]
- Walsh, C.T.; O’Brien, R.V.; Khosla, C. Nonproteinogenic amino acid building blocks for nonribosomal peptide and hybrid polyketide scaffolds. Angew. Chem. Int. Ed. Engl. 2013, 52, 7098–7124. [Google Scholar] [CrossRef]
- Bills, G.; Li, Y.; Chen, L.; Yue, Q.; Niu, X.-M.; An, Z. New insights into the echinocandins and other fungal non-ribosomal peptides and peptaibiotics. Nat. Prod. Rep. 2014, 31, 1348–1375. [Google Scholar] [CrossRef]
- Zhang, S.; Qiu, Y.; Kakule, T.B.; Lu, Z.; Xu, F.; Lamb, J.G.; Reilly, C.A.; Zheng, Y.; Sham, S.W.S.; Wang, W.; et al. Identification of cyclic depsipeptides and their dedicated synthetase from Hapsidospora irregularis. J. Nat. Prod. 2017, 80, 363–370. [Google Scholar] [CrossRef]
- Magarvey, N.A.; Beck, Z.Q.; Golakoti, T.; Ding, Y.; Huber, U.; Hemscheidt, T.K.; Abelson, D.; Moore, R.E.; Sherman, D.H. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from Nostoc cyanobionts. ACS Chem. Biol. 2006, 1, 766–779. [Google Scholar] [CrossRef]
- Shiono, Y.; Tsuchinari, M.; Shimanuki, K.; Miyajima, T.; Murayama, T.; Koseki, T.; Laatsch, H.; Funakoshi, T.; Takanami, K.; Suzuki, K. Fusaristatins A and B, two new cyclic lipopeptides from an endophytic Fusarium sp. J. Antibiot 2007, 60, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhang, X.-Y.; Xu, X.-Y.; He, F.; Nong, X.-H.; Qi, S.-H. New cyclic tetrapeptides and asteltoxins from gorgonian-derived fungus Aspergillus sp. SCSGAF 0076. Tetrahedron 2013, 69, 2113–2117. [Google Scholar] [CrossRef]
- Sørensen, J.L.; Sondergaard, T.E.; Covarelli, L.; Fuertes, P.R.; Hansen, F.T.; Frandsen, R.J.N.; Saei, W.; Lukassen, M.B.; Wimmer, R.; Nielsen, K.F.; et al. Identification of the biosynthetic gene clusters for the lipopeptides fusaristatin A and W493 B in Fusarium graminearum and F. pseudograminearum. J. Nat. Prod. 2014, 77, 2619–2625. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.E.; Marquet, A.; Mendel, R.R.; Rébeillé, F.; Smith, A.G. Elucidating biosynthetic pathways for vitamins and cofactors. Nat. Prod. Rep. 2007, 24, 988–1008. [Google Scholar] [CrossRef] [PubMed]
- Beck, Z.Q.; Burr, D.A.; Sherman, D.H. Characterization of the β-Methylaspartate-α-decarboxylase (CrpG) from the cryptophycin biosynthetic pathway. Chembiochem 2007, 8, 1373–1375. [Google Scholar] [CrossRef][Green Version]
- Monteiro, D.C.F.; Patel, V.; Bartlett, C.P.; Nozaki, S.; Grant, T.D.; Gowdy, J.A.; Thompson, G.S.; Kalverda, A.P.; Snell, E.H.; Niki, H.; et al. The structure of the PanD/PanZ protein complex reveals negative feedback regulation of pantothenate biosynthesis by coenzyme A. Chem. Biol. 2015, 22, 492–503. [Google Scholar] [CrossRef]
- Harris, C.M.; Kannan, R.; Kopecka, H.; Harris, T.M. The role of the chlorine substituents in the antibiotic vancomycin: Preparation and characterization of mono- and didechlorovancomycin. J. Am. Chem. Soc. 1985, 107, 6652–6658. [Google Scholar] [CrossRef]
- Zehner, S.; Kotzsch, A.; Bister, B.; Süssmuth, R.D.; Méndez, C.; Salas, J.A.; van Pée, K.-H. A Regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. [Google Scholar] [CrossRef]
- Dong, C.; Flecks, S.; Unversucht, S.; Haupt, C.; van Pée, K.-H.; Naismith, J.H. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. [Google Scholar] [CrossRef]
- Yeh, E.; Cole, L.J.; Barr, E.W.; Bollinger, J.M.; Ballou, D.P.; Walsh, C.T. Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry 2006, 45, 7904–7912. [Google Scholar] [CrossRef]
- Yeh, E.; Blasiak, L.C.; Koglin, A.; Drennan, C.L.; Walsh, C.T. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry 2007, 46, 1284–1292. [Google Scholar] [CrossRef]
- Flecks, S.; Patallo, E.P.; Zhu, X.; Ernyei, A.J.; Seifert, G.; Schneider, A.; Dong, C.; Naismith, J.H.; van Pée, K.-H. New insights into the mechanism of enzymatic chlorination of tryptophan. Angew. Chem. Int. Ed. Engl. 2008, 47, 9533–9536. [Google Scholar] [CrossRef]
- Zhu, X.; De Laurentis, W.; Leang, K.; Herrmann, J.; Ihlefeld, K.; van Pée, K.-H.; Naismith, J.H. Structural insights into regioselectivity in the enzymatic chlorination of tryptophan. J. Mol. Biol. 2009, 391, 74–85. [Google Scholar] [CrossRef]
- Kittilä, T.; Kittel, C.; Tailhades, J.; Butz, D.; Schoppet, M.; Büttner, A.; Goode, R.J.A.; Schittenhelm, R.B.; van Pee, K.-H.; Süssmuth, R.D.; et al. Halogenation of glycopeptide antibiotics occurs at the amino acid level during non-ribosomal peptide synthesis. Chem. Sci. 2017, 8, 5992–6004. [Google Scholar] [CrossRef]
- Dorrestein, P.C.; Yeh, E.; Garneau-Tsodikova, S.; Kelleher, N.L.; Walsh, C.T. Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis. Proc. Natl. Acad. Sci. USA 2005, 102, 13843. [Google Scholar] [CrossRef]
- Lin, S.; Van Lanen, S.G.; Shen, B. Regiospecific chlorination of (S)-β-tyrosyl-S-carrier protein catalyzed by SgcC3 in the biosynthesis of the enediyne antitumor antibiotic C-1027. J. Am. Chem. Soc. 2007, 129, 12432–12438. [Google Scholar] [CrossRef]
- Haltli, B.; Tan, Y.; Magarvey, N.A.; Wagenaar, M.; Yin, X.; Greenstein, M.; Hucul, J.A.; Zabriskie, T.M. Investigating β-hydroxyenduracididine formation in the biosynthesis of the mannopeptimycins. Chem. Biol. 2005, 12, 1163–1168. [Google Scholar] [CrossRef][Green Version]
- Vior, N.M.; Lacret, R.; Chandra, G.; Dorai-Raj, S.; Trick, M.; Truman, A.W. Discovery and biosynthesis of the antibiotic bicyclomycin in distantly related bacterial classes. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Patteson, J.B.; Cai, W.; Johnson, R.A.; Santa Maria, K.C.; Li, B. Identification of the biosynthetic pathway for the antibiotic bicyclomycin. Biochemistry 2018, 57, 61–65. [Google Scholar] [CrossRef]
- Ökesli, A.; Cooper, L.E.; Fogle, E.J.; van der Donk, W.A. Nine post-translational modifications during the biosynthesis of cinnamycin. J. Am. Chem. Soc. 2011, 133, 13753–13760. [Google Scholar] [CrossRef]
- Hardy, C.D.; Butler, A. β-Hydroxyaspartic acid in siderophores: Biosynthesis and reactivity. J. Biol. Inorg. Chem. 2018, 23, 957–967. [Google Scholar] [CrossRef]
- Reitz, Z.L.; Hardy, C.D.; Suk, J.; Bouvet, J.; Butler, A. Genomic analysis of siderophore β-hydroxylases reveals divergent stereocontrol and expands the condensation domain family. Proc. Natl. Acad. Sci. USA 2019, 116, 19805–19814. [Google Scholar] [CrossRef]
- Singh, G.M.; Fortin, P.D.; Koglin, A.; Walsh, C.T. β-Hydroxylation of the aspartyl Residue in the phytotoxin syringomycin E: Characterization of two candidate hydroxylases AspH and SyrP in Pseudomonas syringae. Biochemistry 2008, 47, 11310–11320. [Google Scholar] [CrossRef]
- Strieker, M.; Nolan, E.M.; Walsh, C.T.; Marahiel, M.A. Stereospecific synthesis of threo- and erythro-beta-hydroxyglutamic acid during kutzneride biosynthesis. J. Am. Chem. Soc. 2009, 131, 13523–13530. [Google Scholar] [CrossRef]
- Helmetag, V.; Samel, S.A.; Thomas, M.G.; Marahiel, M.A.; Essen, L.-O. Structural basis for the erythro-stereospecificity of the L-arginine oxygenase VioC in viomycin biosynthesis. FEBS J. 2009, 276, 3669–3682. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.-F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem Biol. 2016, 1–27. [Google Scholar] [CrossRef]
- Matsuda, K.; Kobayashi, M.; Kuranaga, T.; Takada, K.; Ikeda, H.; Matsunaga, S.; Wakimoto, T. SurE is a trans-acting thioesterase cyclizing two distinct non-ribosomal peptides. Org. Biomol. Chem. 2019, 17, 1058–1061. [Google Scholar] [CrossRef]
- Matsuda, K.; Zhai, R.; Mori, T.; Kobayashi, M.; Sano, A.; Abe, I.; Wakimoto, T. Heterochiral coupling in non-ribosomal peptide macrolactamization. Nat. Catal. 2020. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fazal, A.; Webb, M.E.; Seipke, R.F. The Desotamide Family of Antibiotics. Antibiotics 2020, 9, 452. https://doi.org/10.3390/antibiotics9080452
Fazal A, Webb ME, Seipke RF. The Desotamide Family of Antibiotics. Antibiotics. 2020; 9(8):452. https://doi.org/10.3390/antibiotics9080452
Chicago/Turabian StyleFazal, Asif, Michael E. Webb, and Ryan F. Seipke. 2020. "The Desotamide Family of Antibiotics" Antibiotics 9, no. 8: 452. https://doi.org/10.3390/antibiotics9080452
APA StyleFazal, A., Webb, M. E., & Seipke, R. F. (2020). The Desotamide Family of Antibiotics. Antibiotics, 9(8), 452. https://doi.org/10.3390/antibiotics9080452