A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters

 , ,
, ,  ,
,  and
and 
Abstract
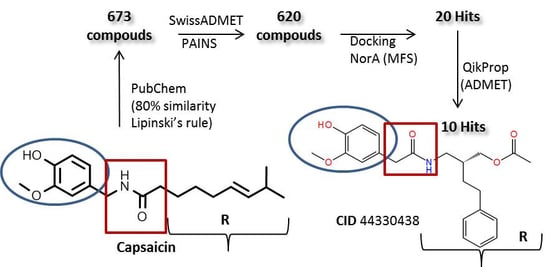
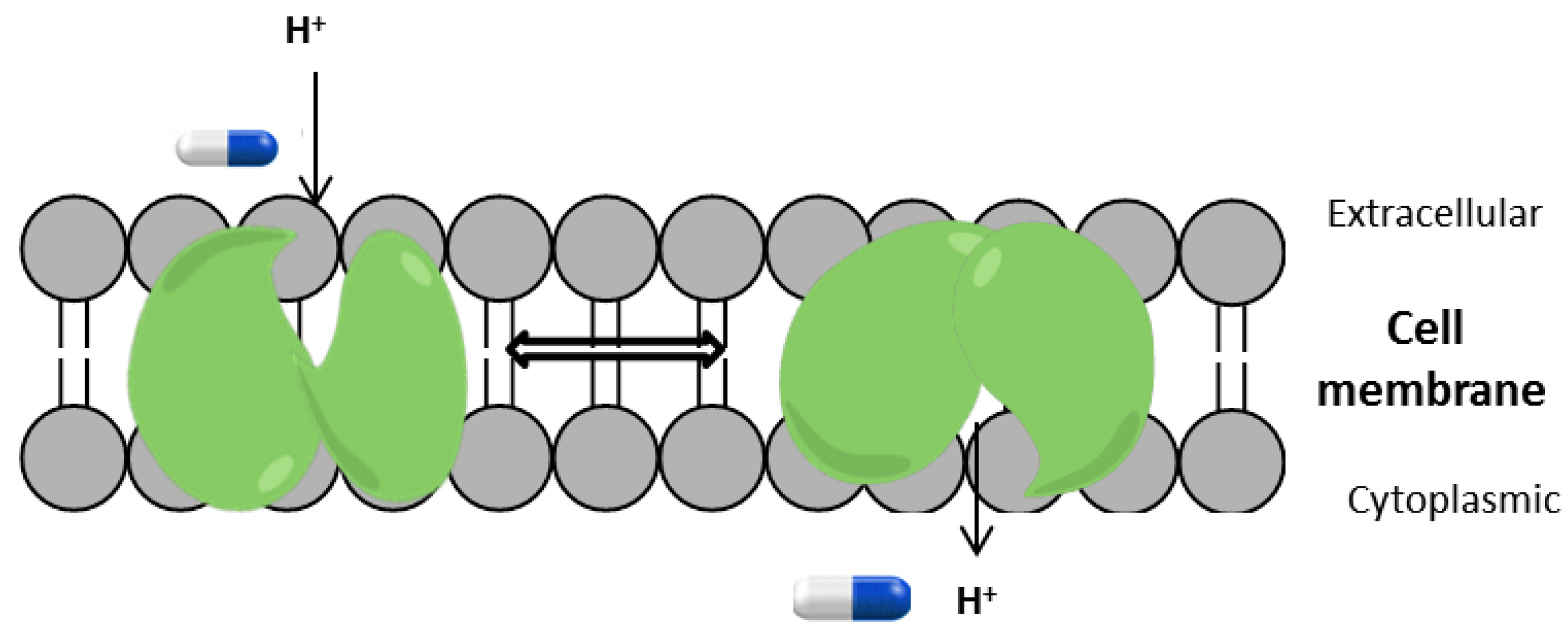
1. Introduction
2. Results and Discussion
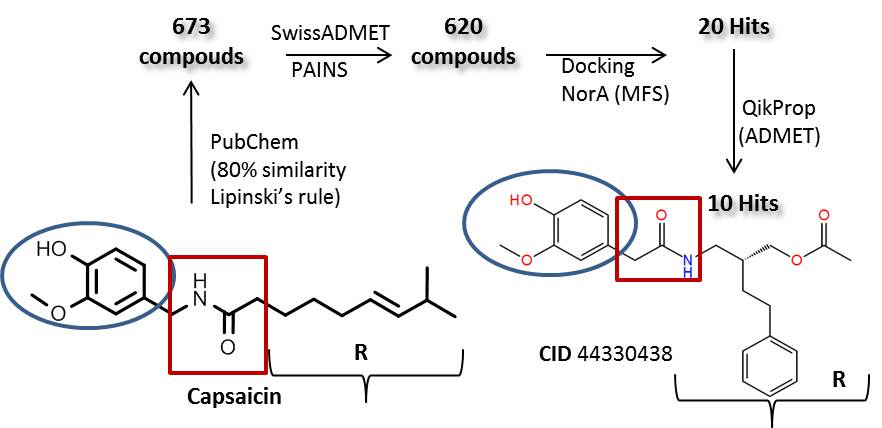
2.1. Preparation of the NorA Target
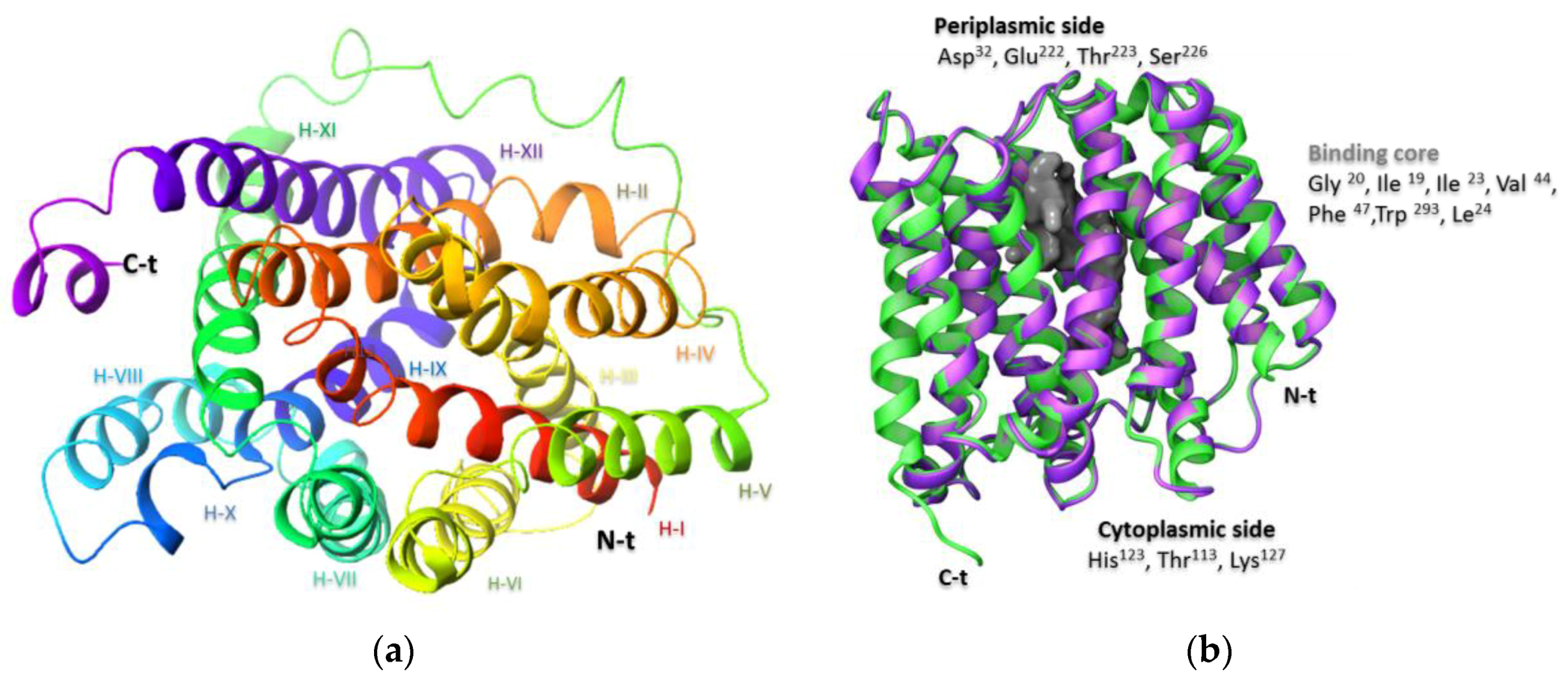
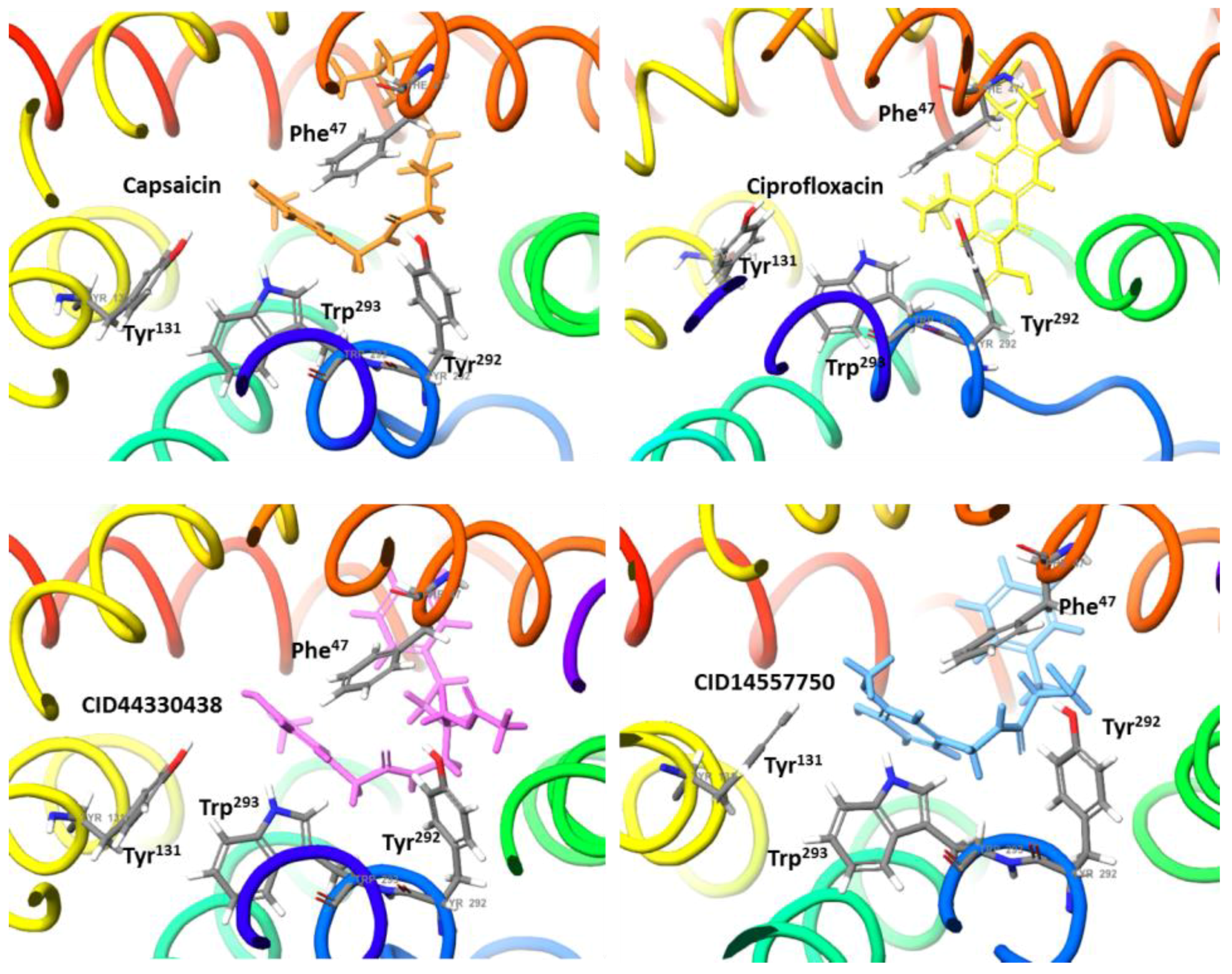
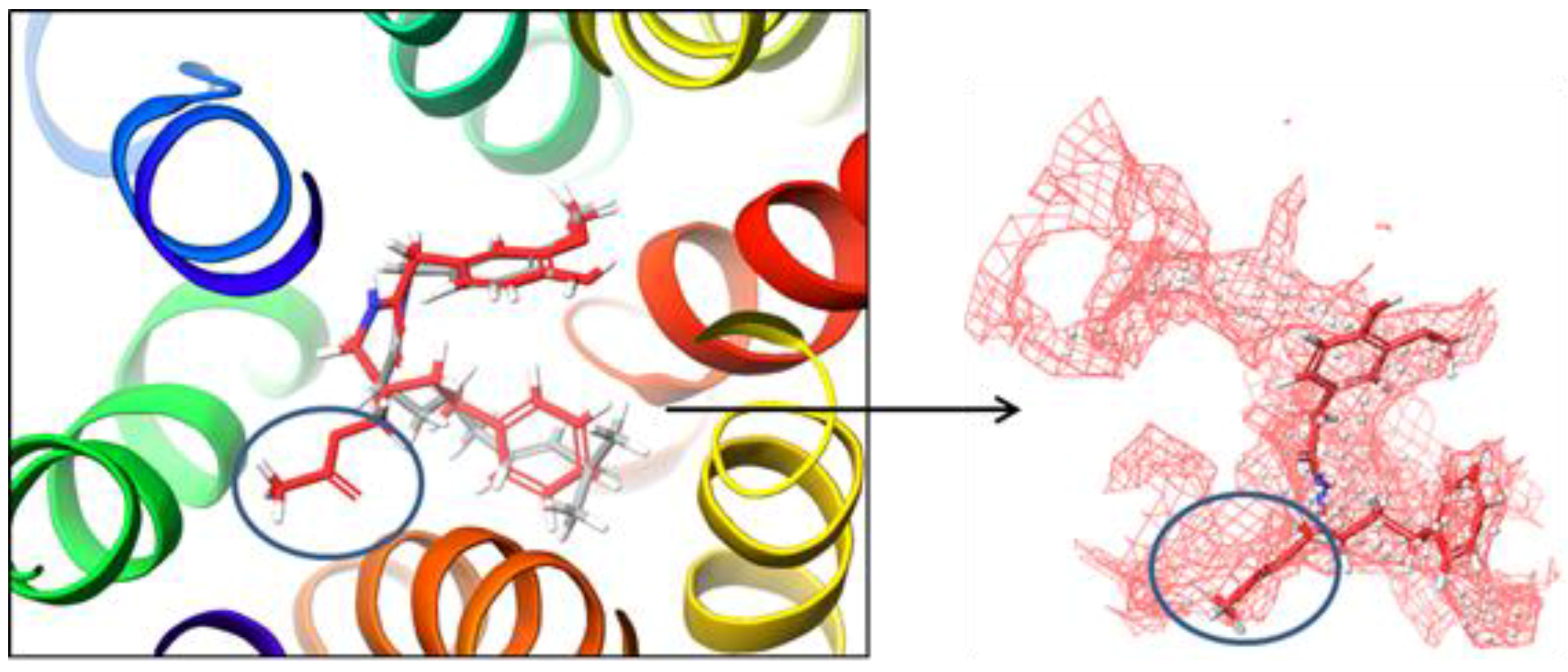
2.2. Molecular Docking Simulation of NorA Capsaicin and Ciprofloxacin Binding
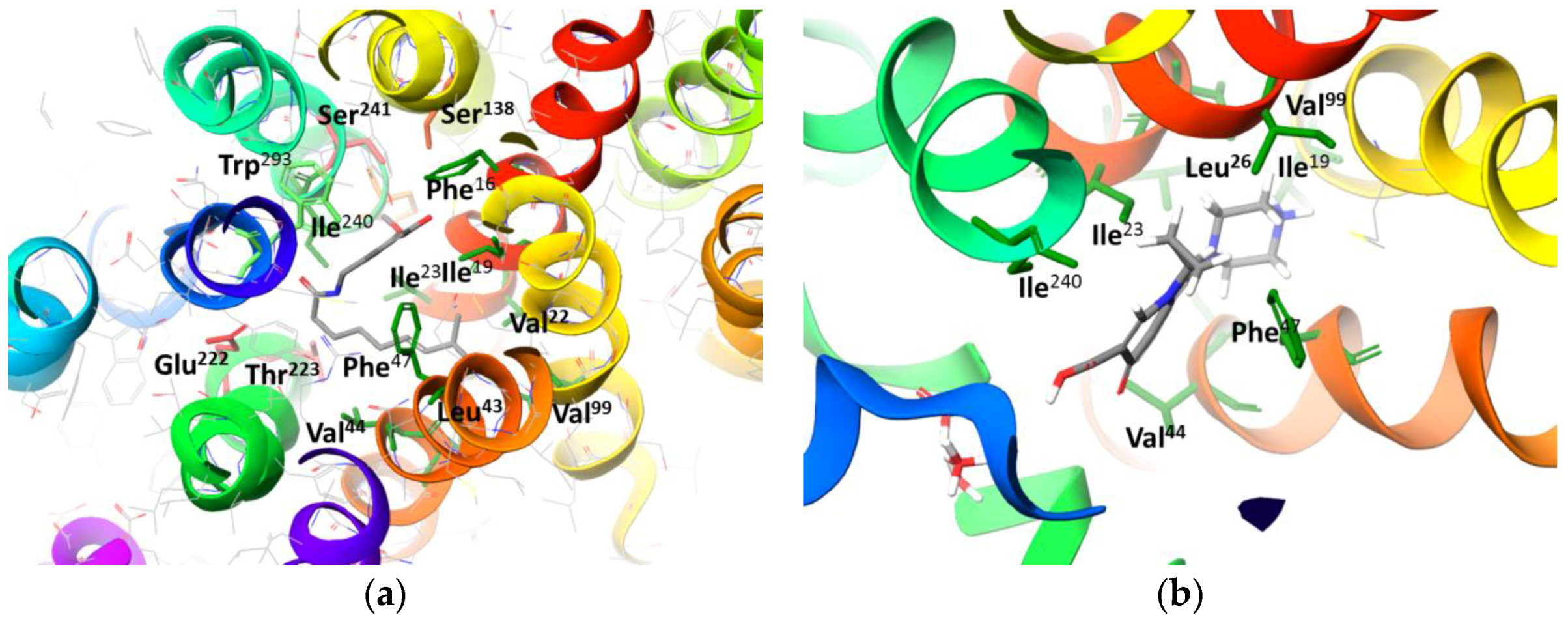
2.3. Molecular Docking Simulation of Potential Novel NorA Inhibitors
3. Materials and Methods
3.1. Protein Modeling: Preparation of NorA Efflux Pump
3.2. Procedure for Molecular Docking Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Spengler, G.; Kincses, A.; Gajdács, M.; Amaral, L. New Roads leading to old Destinations: Efflux Pumps as targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; Bhatt, K.; Misra, P.; Chakraborti, P.K. Involvement of a natural transport system in the process of efflux- mediated drug resistance in Mycobacterium smegmatis. Mol. Gen. Genet. 2000, 262, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.A.; Piddock, L.J.V. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Ambudkar, S.V. The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm. Sin. B 2014, 4, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Quistgaard, E.M.; Low, C.; Guettou, F.; Nordlund, P. Understanding transport by the major facilitator superfamily (MFS): Structures pave the way. Mol. Cell Biol. 2016, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug efflux pumps in Staphylococcus aureus: An update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Hirakata, Y.; Kondo, A.; Hoshino, K.; Yano, H.; Arai, K.; Hirotani, A.; Kunishima, H.; Yamamoto, N.; Hatta, M.; Kitagawa, M.; et al. Efflux pump inhibitors reduce the invasiveness of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2009, 34, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Kumari, N.; Pahwa, S.; Agrahari, U.C.; Bhutani, K.K.; Jachak, H.; Nandanwar, S.M. NorA efflux pump inhibitory activity of coumarins from Mesua ferrea. Fitoterapia 2013, 90, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Holler, J.G.; Slotved, H.-C.; Mølgaard, P.; Olsen, C.E.; Christensen, S.B. Chalcone inhibitors of the NorA efflux pump in Staphylococcus aureus whole cells and enriched everted membrane vesicles. Bioorg. Med. Chem. 2012, 20, 4514–4521. [Google Scholar] [CrossRef] [PubMed]
- Hequet, A.; Burchak, O.N.; Jeanty, M.; Guinchard, X.; Le Pihive, E.; Maigre, L.; Bouhours, P.; Schneider, D.; Maurin, M.; Paris, J.M.; et al. 1-(1Hindol-3-yl) ethanamine derivatives as potent Staphylococcus aureus NorA efflux pump inhibitors. ChemMedChem 2014, 9, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Lowrence, R.C.; Raman, T.; Makala, H.V.; Ulaganathan, V.; Subramaniapillai, S.G.; Kuppuswamy, A.A.; Mani, A.; Chittoor, N.S.; Nagarajan, S. Dithiazole thione derivative as competitive NorA efflux pump inhibitor to curtail multi drug resistant clinical isolate of MRSA in a zebrafish infection model. Appl. Microbiol. Biotechnol. 2016, 100, 9265–9281. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kalia, N.P.; Joshi, P.; Kumar, A.; Sharma, P.R.; Kumar, A.; Bharate, S.B.; Khan, I.A.; Boeravinone, B. A novel dual inhibitor of NorA bacterial efflux pump of Staphylococcus aureus and human P-glycoprotein, reduces the biofilm formation and intracellular invasion of bacteria. Front. Microbiol. 2017, 8, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Gaur, R.; Sharma, A.; Akther, J.; Saini, M.; Bhakuni, R.S.; Pathania, R. A novel bi-functional chalcone inhibits multi-drug resistant Staphylococcus aureus and potentiates the activity of fluoroquinolones. Bioorg. Chem. 2019, 83, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lei, M.; Du, X.; Cui, P.; Zhang, S. Identification of a novel antimicrobial peptide from amphioxus Branchiostoma japonicum by in silico and functional analyses. Sci. Rep. 2015, 5, 18355–18367. [Google Scholar] [CrossRef]
- Bhaskar, B.V.; Muni, T.; Babu, C.; Reddy, N.V.; Rajendra, W. Homology modeling, molecular dynamics, and virtual screening of NorA efflux pump inhibitors of Staphylococcus aureus Drug Design. Dev. Therapy 2016, 10, 3237–3252. [Google Scholar] [CrossRef] [PubMed]
- Kalia, N.P.; Mahajan, P.; Mehra, R.; Nargotra, A.; Sharma, J.P.; Koul, S.; Khan, I.A. Capsaicin, a novel inhibitor of the NorA efflux pump, reduces the intracellular invasion of Staphylococcus aureus. J. Antimicrob. Chemother. 2012, 10, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; He, X.; Szewczyk, P.; Nguyen, T.; Chang, G. Structure of the Multidrug Transporter EmrD from Escherichia coli. Science 2006, 312, 741–744. [Google Scholar] [CrossRef] [PubMed]
- SAVES v5.0. 2019. Available online: http://servicesn.mbi.ucla.edu/SAVES/ (accessed on 10 March 2019).
- Lewinson, O.; Adler, J.; Sigal, N.; Bibi, E. promiscuity in multidrug recognition and transport: The bacterial MFS Mdr transporters. Mol. Microbiol. 2006, 61, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, Z.; Sivaraj, S.; Vaidyanathan, R. Computational docking in silico analysis of potential efflux pump inhibitor punigratane. Int. J. Pharm. Pharm. Sci. 2018, 10, 27–34. [Google Scholar] [CrossRef]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-4: Schrödinger Suite 2018-3 Protein Preparation Wizard; Epik, Schrödinger, LLC/Impact, Schrödinger, LLC: New York, NY, USA, 2018.
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P. Going further than Lipinski’s rule in drug design. Expert Opin. Drug Discov. 2012, 7, 99–107. [Google Scholar] [CrossRef] [PubMed]
- SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.L.; Webster, C.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef] [PubMed]
- Xuan-Yu, M.; Hong-Xing, Z.; Mihaly, M.; Meng, C. Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar]
- Lee, Y.H.; Yi, G.S. Prediction of Novel Anoctamin1 (ANO1) Inhibitors Using 3D-QSAR Pharmacophore Modeling and Molecular Docking. Int. J. Mol. Sci. 2018, 17, 19. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CID PubChem | Residues Implicated in the Interaction | Docking Score (kcal/mol) |
|---|---|---|
 Capsaicin (1548943) | Hydrophobic: Phe16, Ile19, Ile23, Ile244 Pi–Pi stacking: Phe47, Trp293 | −7.19 |
 Ciprofloxacin (2764) | Hydrophobic: Val22, Val44, Ile23, Leu26, leu43, Ala46 Pi–Pi stacking: Phe47 | −6.80 |
 44330438 | Hydrophobic: Val22, Ile23, Ala46, Ala49 Pi–Pi stacking: Phe47 | −8.14 |
 14557750 | Hydrophobic: Ile19, Ile 23, Val22, Val44, Leu26, leu43, Ala46 Pi–Pi stacking: Phe47 | −8.02 |
 742523 | Hydrophobic: Met103, Leu43, leu40, leu26, Ile23, Pro27 Pi–Pi stacking: Phe 47 | −7.77 |
 11516039 | H-bond: Thr223 Hydrophobic: Leu43, Leu26, Val44, Val22, Ala46, Phe47, Phe16 | −7.65 |
 790127 | Hydrophobic: Tyr228, Pro27, Leu26, Ile23 | −7.45 |
 5459532 | Hydrophobic: Val44, Val22, Leu43, leu40, Leu26, Ile23 | −7.40 |
 2107051 | Hydrophobic: Tyr292, Met296, Met109, Ile19, Ile23 Pi–Pi stacking: Phe47, Trp293 H-bond: Trp293 | −7.37 |
 754514 | Hydrophobic: Val44, Val22, Leu43, Leu26, Ile23, Ile19 Pi–Pi stacking: Phe47 | −7.37 |
 44316847 | Hydrophobic: Pro27, Leu26, Ile23, Ile19, Trp293, Tyr292 | −7.33 |
 2175449 | Hydrophobic: Tyr131, Tyr292, Ile244, Ile19, Ile23, Ile240, Met103, Val44 Pi–Pi stacking: Phe47 | −7.20 |
| CID PubChem ID | HBA | HBD | QPlogherg | MW | QPlogS | QPlog Kp | QPlog Khsa | QPP Caco | PHOA |
|---|---|---|---|---|---|---|---|---|---|
 Capsaicin (1548943) | 3 | 2 | −3.76 | 305.1 | −4.08 | −1.90 | 0.14 | 1788.6 | 100 |
 Ciprofloxacin (2764) | 7 | 2 | −3.43 | 331.1 | −3.79 | −6.48 | 0.01 | 13.3 | 49 |
 44330438 | 5 | 2 | −4.04 | 385.1 | −4.55 | −2.04 | 0.08 | 504.9 | 95 |
 14557750 | 4 | 3 | −4.12 | 331.1 | −4.09 | −1,31 | 0.11 | 1001.8 | 100 |
 742523 | 4 | 0 | −4.02 | 320.2 | −1.99 | −3,29 | −0.45 | 964.2 | 93 |
 11516039 | 5 | 2 | −1.26 | 361.1 | −3.48 | −2.27 | −0.29 | 193.7 | 86 |
 790127 | 4 | 1 | −4.99 | 285.1 | −3.43 | −1.04 | −0.03 | 3710.8 | 100 |
 5459532 | 3 | 2 | −3.97 | 299.1 | −3.64 | −1.54 | −0.06 | 1907.6 | 100 |
 2107051 | 5 | 1 | −3.18 | 347.1 | −4.14 | −1.04 | 0.18 | 2653.4 | 100 |
 754514 | 4 | 1 | −3.19 | 299.1 | −2.34 | −1.22 | −0.26 | 1725.0 | 100 |
 44316847 | 3 | 2 | −4.01 | 319.0 | −3.51 | −1.41 | −0.06 | 1016.0 | 100 |
 2175449 | 5 | 3 | −5.95 | 313.0 | −4.43 | −3.31 | 0.19 | 156.1 | 82 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zárate, S.G.; Morales, P.; Świderek, K.; Bolanos-Garcia, V.M.; Bastida, A. A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters. Antibiotics 2019, 8, 25. https://doi.org/10.3390/antibiotics8010025
Zárate SG, Morales P, Świderek K, Bolanos-Garcia VM, Bastida A. A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters. Antibiotics. 2019; 8(1):25. https://doi.org/10.3390/antibiotics8010025
Chicago/Turabian StyleZárate, Sandra G., Paula Morales, Katarzyna Świderek, Victor M. Bolanos-Garcia, and Agatha Bastida. 2019. "A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters" Antibiotics 8, no. 1: 25. https://doi.org/10.3390/antibiotics8010025
APA StyleZárate, S. G., Morales, P., Świderek, K., Bolanos-Garcia, V. M., & Bastida, A. (2019). A Molecular Modeling Approach to Identify Novel Inhibitors of the Major Facilitator Superfamily of Efflux Pump Transporters. Antibiotics, 8(1), 25. https://doi.org/10.3390/antibiotics8010025