CRISPR-Cas: Converting A Bacterial Defence Mechanism into A State-of-the-Art Genetic Manipulation Tool



Abstract
1. Introduction
2. What Is Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)?
3. Structure of CRISPR Loci
4. Steps of CRISPR-Cas Adaptive Immunity
4.1. Adaptation
4.2. CRISPR RNA Biogenesis
4.3. Interference
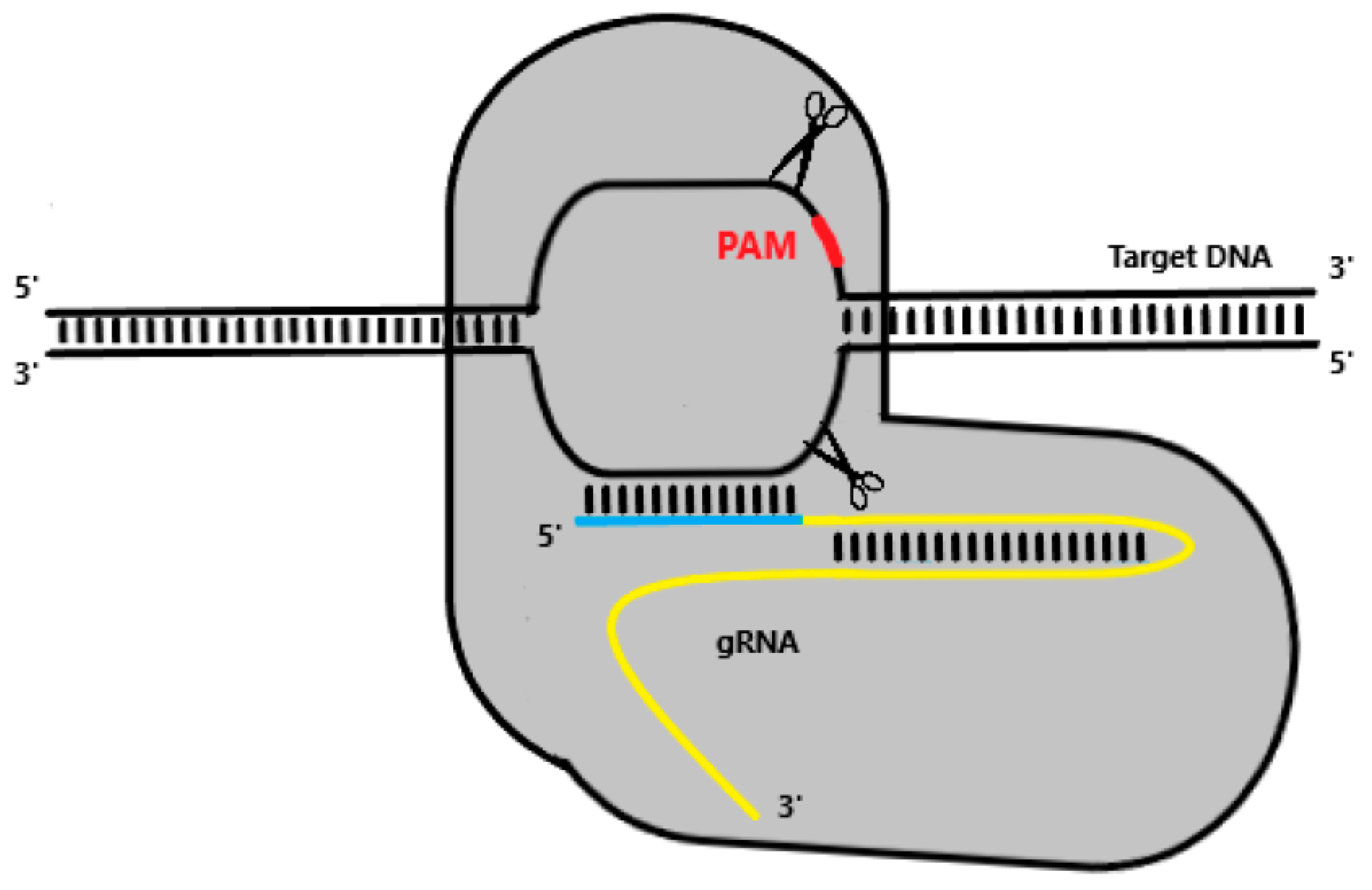
5. CRISPR-Cas Systems as a Gene-Editing Tool
Repurposing CRISPR for Genetic Engineering
6. Advantages of CRISPR Relative to Other Techniques
7. Limitations of CRISPR Systems
8. Novel and Enhanced CRISPR/Cas Systems
8.1. Cas12a (Cpf1)
8.2. Cas13a (C2c2)
8.3. Cas9n
8.4. dCas9
8.5. eSpCas9, SpCas9-HF1, and HypaCas9
9. Delivering CRISPR Systems into the Cell
10. Applications of CRISPR-Cas Systems
10.1. Oncology
10.2. Genetic Diseases
10.3. Viral Diseases
10.4. Bacterial Infections
10.5. Crop Industry
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hu, H.; Xiong, L. Genetic Engineering and Breeding of Drought-Resistant Crops. Annu. Rev. Plant Biol. 2014, 65, 715–741. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.N.; Chang, A.C.Y.; Boyer, H.W.; Helling, R.B. Construction of Biologically Functional Bacterial Plasmids In Vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science (80-.) 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.; Gonnet, M.; Le Romancer, M.; Nicolas, J. CRISPI: A CRISPR interactive database. Bioinformatics 2009, 25, 3317–3318. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Marraffini, L.A. CRISPR-cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell 2014, 54, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Pul, Ü.; Wurm, R.; Arslan, Z.; Geißen, R.; Hofmann, N.; Wagner, R. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 2010, 75, 1495–1512. [Google Scholar] [CrossRef] [PubMed]
- Yosef, I.; Goren, M.G.; Qimron, U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012, 40, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Chylinski, K.; Makarova, K.S.; Charpentier, E.; Koonin, E.V. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014, 42, 6091–6105. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Nuñez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006, 1, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Pougach, K.; Tikhonov, A.; Wanner, B.L.; Severinov, K.; Semenova, E. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012, 3, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, J.K.; Harrington, L.B.; Kranzusch, P.J.; Engelman, A.N.; Doudna, J.A. Foreign DNA capture during CRISPR-Cas adaptive immunity. Nature 2015, 527, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.; Beloglazova, N.; Flick, R.; Graham, C.; Skarina, T.; Nocek, B.; Gagarinova, A.; Pogoutse, O.; Brown, G.; Binkowski, A.; et al. A dual function of the CRISPR-Cas system in bacterial antivirus immunity and DNA repair. Mol. Microbiol. 2011, 79, 484–502. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Zhou, K.; Jinek, M.; Coyle, S.M.; Ma, W.; Doudna, J.A. Structural Basis for DNase Activity of a Conserved Protein Implicated in CRISPR-Mediated Genome Defense. Structure 2009, 17, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, J.K.; Kranzusch, P.J.; Noeske, J.; Wright, A.V.; Davies, C.W.; Doudna, J.A. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014, 21, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Erdmann, S.; Mojica, F.J.M.; Garrett, R.A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013, 10, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Zhao, H.; Sheng, G.; Wang, M.; Yin, M.; Wang, Y. Structural and Mechanistic Basis of PAM-Dependent Spacer Acquisition in CRISPR-Cas Systems. Cell 2015, 163, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, J.K.; Lee, A.S.Y.; Engelman, A.; Doudna, J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature 2015, 519, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Savitskaya, E.; Semenova, E.; Logacheva, M.D.; Datsenko, K.A.; Severinov, K. Pervasive generation of oppositely oriented spacers during CRISPR adaptation. Nucleic Acids Res. 2014, 42, 5907–5916. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, S.; Garrett, R.A. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Mol. Microbiol. 2012, 85, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Silas, S.; Mohr, G.; Sidote, D.J.; Markham, L.M.; Sanchez-Amat, A.; Bhaya, D.; Lambowitz, A.M.; Fire, A.Z. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science (80-.) 2016, 351, aad4234. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Gesner, E.M.; Schellenberg, M.J.; Garside, E.L.; George, M.M.; MacMillan, A.M. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat. Struct. Mol. Biol. 2011, 18, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, O.; Jinek, M.; Doudna, J.A. Evolution of CRISPR RNA recognition and processing by Cas6 endonucleases. Nucleic Acids Res. 2014, 42, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Chylinski, K.; Le Rhun, A.; Charpentier, E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013, 10, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with gRNA and target DNA. 2015, 156, 935–949. [Google Scholar]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; BratoviÄ, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef] [PubMed]
- East-Seletsky, A.; O’Connell, M.R.; Burstein, D.; Knott, G.J.; Doudna, J.A. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol. Cell 2017, 66, 373–383.e3. [Google Scholar] [CrossRef] [PubMed]
- Hatoum-Aslan, A.; Maniv, I.; Marraffini, L.A. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. USA 2011, 108, 21218–21222. [Google Scholar] [CrossRef] [PubMed]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science (80-.) 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Luo, M.; Hayes, R.P.; Kim, J.; Ng, S.; Ding, F.; Liao, M.; Ke, A. Structure Basis for Directional R-loop Formation and Substrate Handover Mechanisms in Type I CRISPR-Cas System. Cell 2017, 170, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.P.; Xiao, Y.; Ding, F.; Van Erp, P.B.G.; Rajashankar, K.; Bailey, S.; Wiedenheft, B.; Ke, A. Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature 2016, 530, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Redding, S.; Sternberg, S.H.; Marshall, M.; Gibb, B.; Bhat, P.; Guegler, C.K.; Wiedenheft, B.; Doudna, J.A.; Greene, E.C. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell 2015, 163, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Mulepati, S.; Bailey, S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA Target. J. Biol. Chem. 2013, 288, 22184–22192. [Google Scholar] [CrossRef] [PubMed]
- Sinkunas, T.; Gasiunas, G.; Waghmare, S.P.; Dickman, M.J.; Barrangou, R.; Horvath, P.; Siksnys, V. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013, 32, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Inanaga, H.; Sato, C.; Numata, T. Crystal structure of the crispr-cas RNA silencing cmr complex bound to a target analog. Mol. Cell 2015, 58, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Rouillon, C.; Zhou, M.; Zhang, J.; Politis, A.; Beilsten-Edmands, V.; Cannone, G.; Graham, S.; Robinson, C.V.; Spagnolo, L.; White, M.F. Structure of the CRISPR interference complex CSM reveals key similarities with cascade. Mol. Cell 2013, 52, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskiene, M.; Tamulaitis, G.; Kostiuk, G.; Venclovas, Č.; Siksnys, V. Spatiotemporal Control of Type III-A CRISPR-Cas Immunity: Coupling DNA Degradation with the Target RNA Recognition. Mol. Cell 2016, 62, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Samai, P.; Pyenson, N.; Jiang, W.; Goldberg, G.W.; Hatoum-Aslan, A.; Marraffini, L.A. Co-transcriptional DNA and RNA cleavage during type III CRISPR-cas immunity. Cell 2015, 161, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskiene, M.; Kostiuk, G.; Venclovas, Č.; Tamulaitis, G.; Siksnys, V. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science (80-.) 2017, 357, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Niewoehner, O.; Garcia-Doval, C.; Rostøl, J.T.; Berk, C.; Schwede, F.; Bigler, L.; Hall, J.; Marraffini, L.A.; Jinek, M. Type III CRISPR–Cas systems produce cyclic oligoadenylate second messengers. Nature 2017, 548, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (80-.) 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ren, K.; Qiu, X.; Zheng, J.; Guo, M.; Guan, X.; Liu, H.; Li, N.; Zhang, B.; Yang, D.; et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 2016, 532, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Alcón, P.; Montoya, G. Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 2017, 546, 559–563. [Google Scholar] [PubMed]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.D.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science (80-.) 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Ma, J.; Li, Z.; You, L.; Wang, J.; Wang, M.; Zhang, X.; Wang, Y. The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 2017, 170, 714–726.e10. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Regan, S.; Xia, Y.; Oostrom, L.; Cowan, C.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem 2013, 12, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [PubMed]
- Generoso, W.C.; Gottardi, M.; Oreb, M.; Boles, E. Simplified CRISPR-Cas genome editing for Saccharomyces cerevisiae. J. Microbiol. Methods 2016, 127, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Homma, A.; Sayadi, J.; Yang, S.; Ohashi, J.; Takumi, T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.K.; Haber, J.E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Gunn, A.; Cheng, A.; Hasty, P.; Stark, J.M. Limiting the Persistence of a Chromosome Break Diminishes Its Mutagenic Potential. PLoS Genet. 2009, 5, e1000683. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.A.; Strande, N.T.; Pryor, J.M.; Strom, C.N.; Mieczkowski, P.; Burkhalter, M.D.; Oh, S.; Qaqish, B.F.; Moore, D.T.; Hendrickson, E.A.; et al. The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat. Commun. 2014, 5, 4286. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gómez-González, B.; Aguilera, A. DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, G.; Andersen, T.; Zhou, P.; Pu, W.T. Optimization of Genome Engineering Approaches with the CRISPR/Cas9 System. PLoS ONE 2014, 9, e105779. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.S.; Wong, S.; Peters, J.M.; Almeida, R.; Qi, L.S. Targeted transcriptional repression in bacteria using CRISPR interference (CRISPRi). Methods Mol. Biol. 2015, 1311, 349–362. [Google Scholar] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Qi, L. Genetic and epigenetic control of gene expression by CRISPR–Cas systems. F1000Research 2017, 6, 747. [Google Scholar] [CrossRef] [PubMed]
- Amabile, A.; Migliara, A.; Capasso, P.; Biffi, M.; Cittaro, D.; Naldini, L.; Lombardo, A. Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 2016, 167, 219–232.e14. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science (80-.) 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Segal, D.J.; Ghiara, J.B.; Barbas, C.F. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5525–5530. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science (80-.) 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell. Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.; Arlotta, P. Programmable Sequence-Specific Transcriptional Regulation of Mammalian Genome Using Designer TAL Effectors. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Dreier, B.; Barbas, C.F., 3rd. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 2000, 97, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.J.; Dreier, B.; Beerli, R.R.; Barbas, C.F. Toward controlling gene expression at will: Selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc. Natl. Acad. Sci. USA 1999, 96, 2758–2763. [Google Scholar] [CrossRef] [PubMed]
- Orlando, S.J.; Santiago, Y.; DeKelver, R.C.; Freyvert, Y.; Boydston, E.A.; Moehle, E.A.; Choi, V.M.; Gopalan, S.M.; Lou, J.F.; Li, J.; et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res. 2010, 38, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cristea, S.; Freyvert, Y.; Santiago, Y.; Holmes, M.C.; Urnov, F.D.; Gregory, P.D.; Cost, G.J. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol. Bioeng. 2013, 110, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Jao, L.-E.; Wente, S.R.; Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA 2013, 110, 13904–13909. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, H.; Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. The CRISPR Craze. Science (80-.) 2013, 341, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. Rationally engineered Cas9 nulceases with improved specificity. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Dubois, G.; Valton, J.; Thomas, S.; Stella, S.; Maréchal, A.; Langevin, S.; Benomari, N.; Bertonat, C.; Silva, G.H.; et al. Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res. 2014, 42, 5390–5402. [Google Scholar] [CrossRef] [PubMed]
- Wefers, B.; Panda, S.K.; Ortiz, O.; Brandl, C.; Hensler, S.; Hansen, J.; Wurst, W.; Kühn, R. Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nat. Protoc. 2013, 8, 2355–2379. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Brunet, E.; Simsek, D.; Tomishima, M.; DeKelver, R.; Choi, V.M.; Gregory, P.; Urnov, F.; Weinstock, D.M.; Jasin, M. Chromosomal translocations induced at specified loci in human stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10620–10625. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Tan, C.; Wang, F.; Wang, Y.; Zhou, R.; Cui, D.; You, W.; Zhao, H.; Ren, J.; Feng, B. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016, 44, e85. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cox, D.B.T.; Yan, W.X.; Manteiga, J.C.; Schneider, M.W.; Yamano, T.; Nishimasu, H.; Nureki, O.; Crosetto, N.; Zhang, F. Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 2017, 35, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 2016, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, A.; Phadke, T.; Barrera, L.A.; Margulies, C.M.; Lee, C.S.; Buquicchio, F.; Moss, S.; Abdulkerim, H.S.; Selleck, W.; Jayaram, H.; et al. Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21 ± nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Mahas, A.; Neal Stewart, C.; Mahfouz, M.M. Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnol. Adv. 2018, 36, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Yang, Z.; Mao, D.; Chang, Z.; Qiao, H.-H.; Wang, X.; Sun, J.; Hu, Q.; Cui, Y.; Liu, L.-P.; et al. Performance of the Cas9 Nickase System in Drosophila melanogaster. G3 2014, 4, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Brocken, D.J.W.; Tark-Dame, M.; Dame, R.T. dCas9: A Versatile Tool for Epigenome Editing. Curr. Issues Mol. Biol. 2018, 26, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science (80-.) 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.; Zheng, Z.; Joung, J.K.; Unit, P.; Biology, I.; Hospital, M.G.; et al. High-fidelity CRISPR-Cas9 variants with undetectable genome-wide off-targets. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Crispo, M.; Mulet, A.P.; Tesson, L.; Barrera, N.; Cuadro, F.; Dos Santos-Neto, P.C.; Nguyen, T.H.; Crénéguy, A.; Brusselle, L.; Anegón, I.; et al. Efficient generation of myostatin knock-out sheep using CRISPR/Cas9 technology and microinjection into zygotes. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Arai, Y.; Yamazaki, M.; Morita, S.; Kimura, M.; Itoh, M.; Abe, Y.; Hatada, I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 2014, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Véron, N.; Qu, Z.; Kipen, P.A.S.; Hirst, C.E.; Marcelle, C. CRISPR mediated somatic cell genome engineering in the chicken. Dev. Biol. 2015, 407, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Sakuma, T.; Yamamoto, T.; Mashimo, T. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Sci. Rep. 2014, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, L.C. Single-cell microinjection technology in cell biology. BioEssays 2008, 30, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Valsalakumari, J.; Baby, J.; Bijin, E.; Constantine, I.; Manjila, S.; Pramod, K. Novel gene delivery systems. Int. J. Pharm. Investig. 2013, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, K.; Suda, T.; Zhang, G.; Liu, D. Advances in gene delivery systems. Pharmaceut. Med. 2011, 25, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science (80-.) 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Koike-Yusa, H.; Li, Y.; Tan, E.P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Somia, N.; Verma, I.M. Gene therapy: Trials and tribulations. Nat. Rev. Genet. 2000, 1, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Baricordi, C.; Aiuti, A. Retroviral integrations in gene therapy trials. Mol. Ther. 2012, 20, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Holkers, M.; Maggio, I.; Henriques, S.F.D.; Janssen, J.M.; Cathomen, T.; Gonçalves, M.A.F.V. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods 2014, 11, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Kwaku Dad, A.B.; Beloor, J.; Gopalappa, R.; Lee, S.K.; Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014, 24, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Suresh, B.; Ramakrishna, S.; Kim, H. Eukaryotic Transcriptional and Post-Transcriptional Gene Expression Regulation. Methods Mol. Biol. 2017, 1507, 81–94. [Google Scholar] [PubMed]
- Mout, R.; Ray, M.; Yesilbag Tonga, G.; Lee, Y.W.; Tay, T.; Sasaki, K.; Rotello, V.M. Direct Cytosolic Delivery of CRISPR/Cas9-Ribonucleoprotein for Efficient Gene Editing. ACS Nano 2017, 11, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; Zhang, F.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2016, 160, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Nimura, K.; Nagano, H.; Yamaguchi, S.; Nonomura, N.; Kaneda, Y. CRISPR/Cas9-mediated gene knockout of NANOG and NANOGP8 decreases the malignant potential of prostate cancer cells. Oncotarget 2015, 6, 22361–22374. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shen, J.K.; Li, Z.; Choy, E.; Hornicek, F.J.; Duan, Z. Development and potential applications of CRISPR-Cas9 genome editing technology in sarcoma. Cancer Lett. 2016, 373, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Sebastiani, P. Genetic modifiersof sickle cell disease. Am. J. Hematol. 2012, 87, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Döring, G. Cystic fibrosis. Lancet 2003, 361, 681–689. [Google Scholar] [CrossRef]
- Lanzkron, S.; Carroll, C.P.; Haywood, C., Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Rep. 2013, 128, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Özdoğu, H.; Boğa, C. Erişkin orak hücre hastalığında hematopoietik kök hücre nakli: Problemler ve çözüm önerileri. Turk. J. Hematol. 2015, 32, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; Van Der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ding, L.; Sun, C.W.; Wu, L.C.; Zhou, D.; Pawlik, K.M.; Khodadadi-Jamayran, A.; Westin, E.; Goldman, F.D.; Townes, T.M. Novel HDAd/EBV Reprogramming Vector and Highly Efficient Ad/CRISPR-Cas Sickle Cell Disease Gene Correction. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Lucifora, J.; Mason, W.S.; Sureau, C.; Beck, J.; Levrero, M.; Kann, M.; Knolle, P.A.; Benkirane, M.; Durantel, D.; et al. Towards an HBV cure: State-of-the-art and unresolved questions-report of the ANRS workshop on HBV cure. Gut 2015, 64, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.R.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13 David. Science (80-.) 2017, 0180, 1–15. [Google Scholar]
- Mahas, A.; Mahfouz, M. Engineering virus resistance via CRISPR–Cas systems. Curr. Opin. Virol. 2018, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Thabit, A.K.; Crandon, J.L.; Nicolau, D.P. Antimicrobial resistance: Impact on clinical and economic outcomes and the need for new antimicrobials. Expert Opin. Pharmacother. 2015, 16, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.A.; Tchernov, A.P.; Fodor, I.; Bayev, A.A. Lambda phagemids and their transducing properties. Gene 1984, 28, 29–35. [Google Scholar] [CrossRef]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Su, W.; Fey, P.D.; Liu, F.; Du, L. Yield Improvement of the Anti-MRSA Antibiotics WAP-8294A by CRISPR/dCas9 Combined with Refactoring Self-Protection Genes in Lysobacter enzymogenes OH11. ACS Synth. Biol. 2018, 7, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, J.; Brumin, M.; Wolf, D.; Leibman, D.; Klap, C.; Pearlsman, M.; Sherman, A.; Arazi, T.; Gal-On, A. Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Mol. Plant Pathol. 2016, 17, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, H.; Wang, H.; Lafitte, H.R.; Archibald, R.L.; Yang, M.; Hakimi, S.M.; Mo, H.; Habben, J.E. ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol. J. 2017, 15, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Ueta, R.; Abe, C.; Watanabe, T.; Sugano, S.S.; Ishihara, R.; Ezura, H.; Osakabe, Y.; Osakabe, K. Rapid breeding of parthenocarpic tomato plants using CRISPR/Cas9. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, D.; Ramasamy, K.; Sellamuthu, G.; Jayabalan, S.; Venkataraman, G. CRISPR for Crop Improvement: An Update Review. Front. Plant Sci. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Characteristic | Type I | Type II | Type III | Type IV | Type V | Type VI |
|---|---|---|---|---|---|---|
| Effector complex | Multisubunit (Class 1) | Single unit (Class 2) | Multisubunit (Class 1) | Multisubunit (Class 1) | Single unit (Class 2) | Single unit (Class 2) |
| Signature Protein | Cas3 | Cas9 | Cas10 | Csf1 | Cas12 | Cas13 |
| Target molecule | DNA | DNA | RNA/DNA | ? | DNA | RNA |
| Details | Cleaves ssDNA strands | Originates blunt DSB | Binds to nascent RNA molecules | Most unknown CRISPR system | Originates staggered DSB | RNA-guided RNase |
| Characteristic | ZFN | TALEN | CRISPR/Cas9 |
|---|---|---|---|
| Binding principle | Protein-DNA | Protein-DNA | RNA-DNA |
| Ease of design | Moderate | Easy | Very Easy |
| Assembling | Difficult | Easy | Very Easy |
| Time for construction | 5–7 days | 5–7 days | 1–3 days |
| Cost | High | Moderate | Low |
| Efficiency | Variable | High | High |
| Off-target effects | High but variable | Low | High |
| Single-unit or pair | Pair | Pair | Single-unit |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loureiro, A.; da Silva, G.J. CRISPR-Cas: Converting A Bacterial Defence Mechanism into A State-of-the-Art Genetic Manipulation Tool. Antibiotics 2019, 8, 18. https://doi.org/10.3390/antibiotics8010018
Loureiro A, da Silva GJ. CRISPR-Cas: Converting A Bacterial Defence Mechanism into A State-of-the-Art Genetic Manipulation Tool. Antibiotics. 2019; 8(1):18. https://doi.org/10.3390/antibiotics8010018
Chicago/Turabian StyleLoureiro, Alexandre, and Gabriela Jorge da Silva. 2019. "CRISPR-Cas: Converting A Bacterial Defence Mechanism into A State-of-the-Art Genetic Manipulation Tool" Antibiotics 8, no. 1: 18. https://doi.org/10.3390/antibiotics8010018
APA StyleLoureiro, A., & da Silva, G. J. (2019). CRISPR-Cas: Converting A Bacterial Defence Mechanism into A State-of-the-Art Genetic Manipulation Tool. Antibiotics, 8(1), 18. https://doi.org/10.3390/antibiotics8010018