
Fused-Ring Oxazolopyrrolopyridopyrimidine Systems with Gram-Negative Activity


Abstract
:1. Introduction
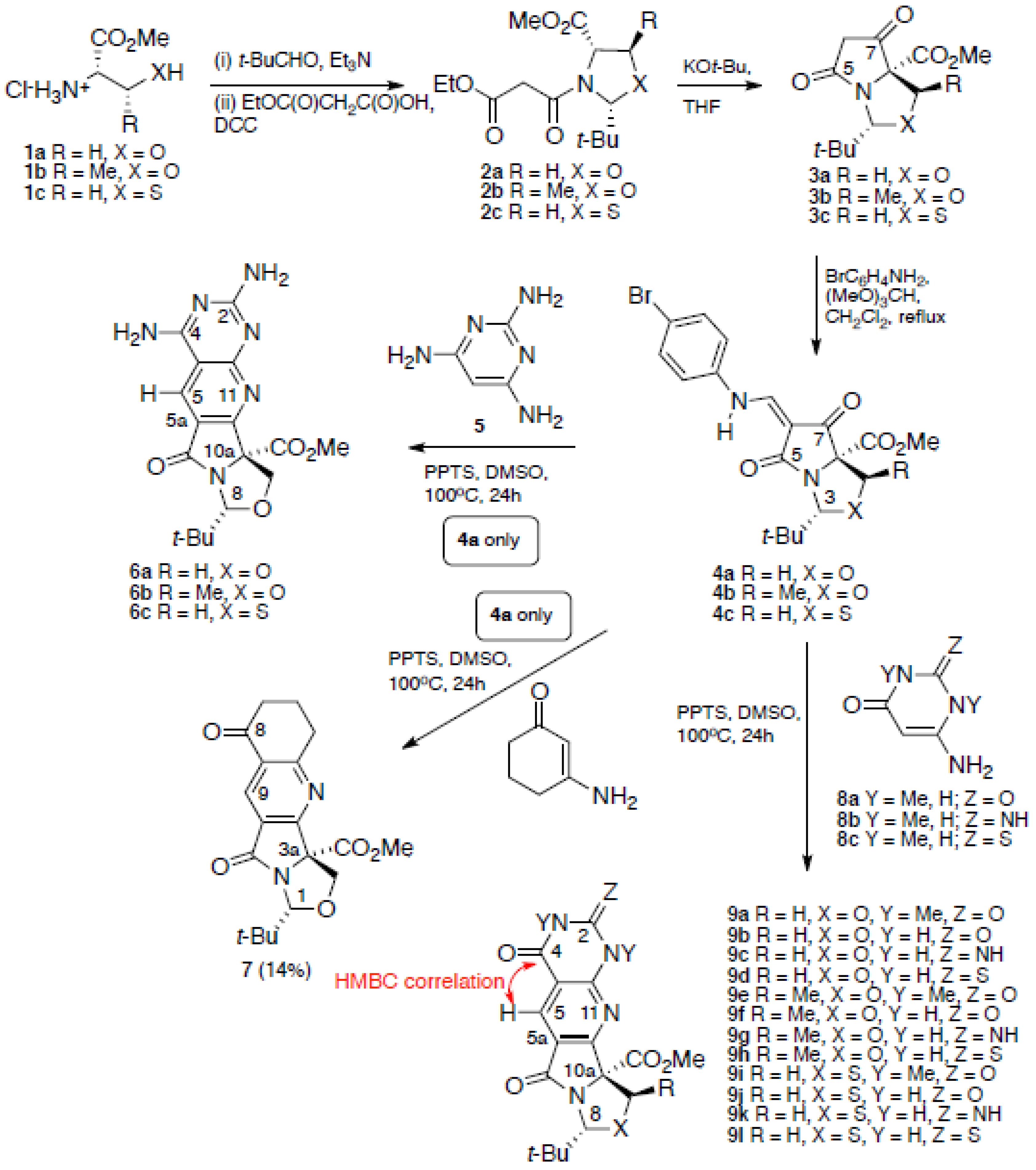
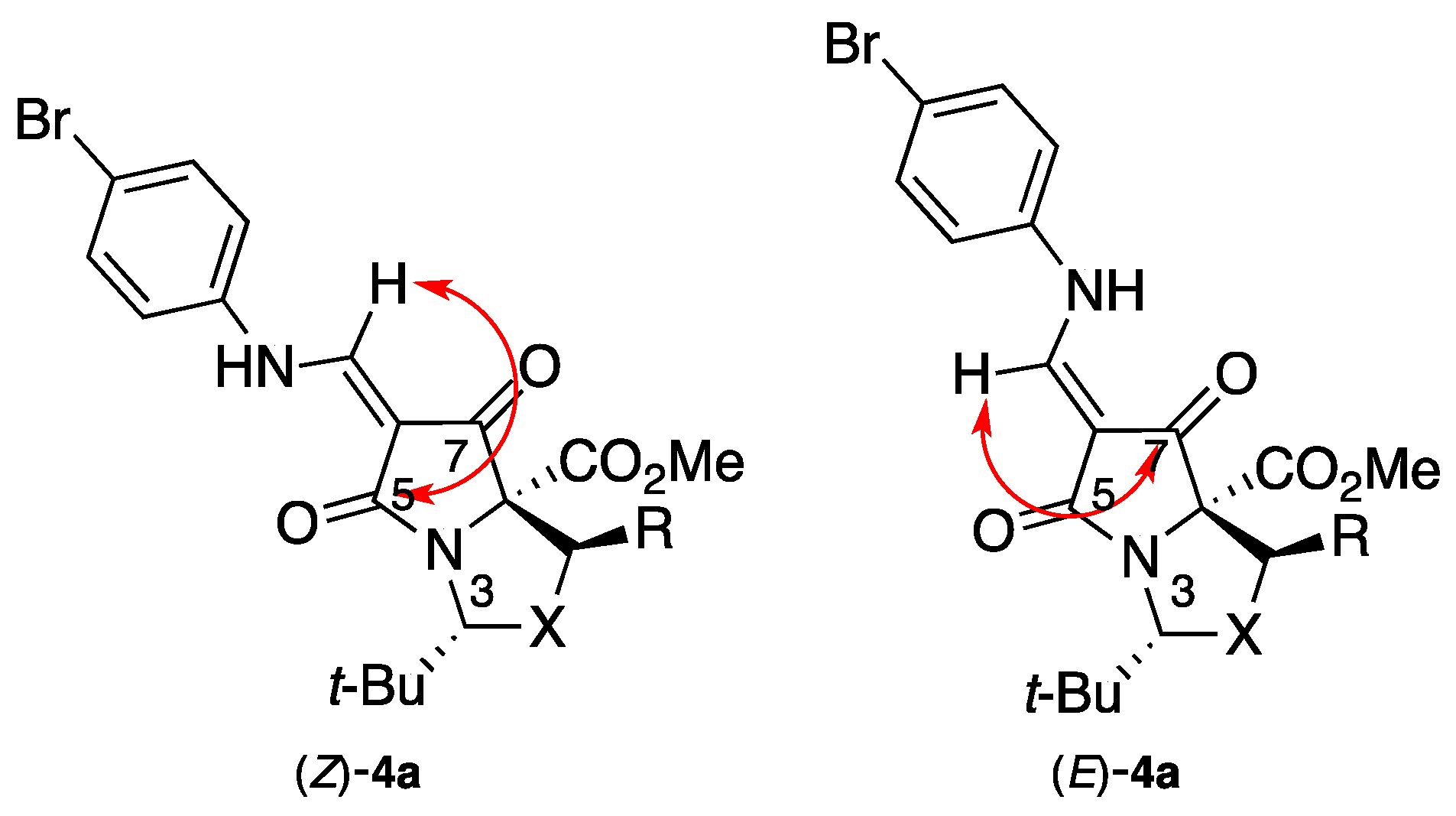
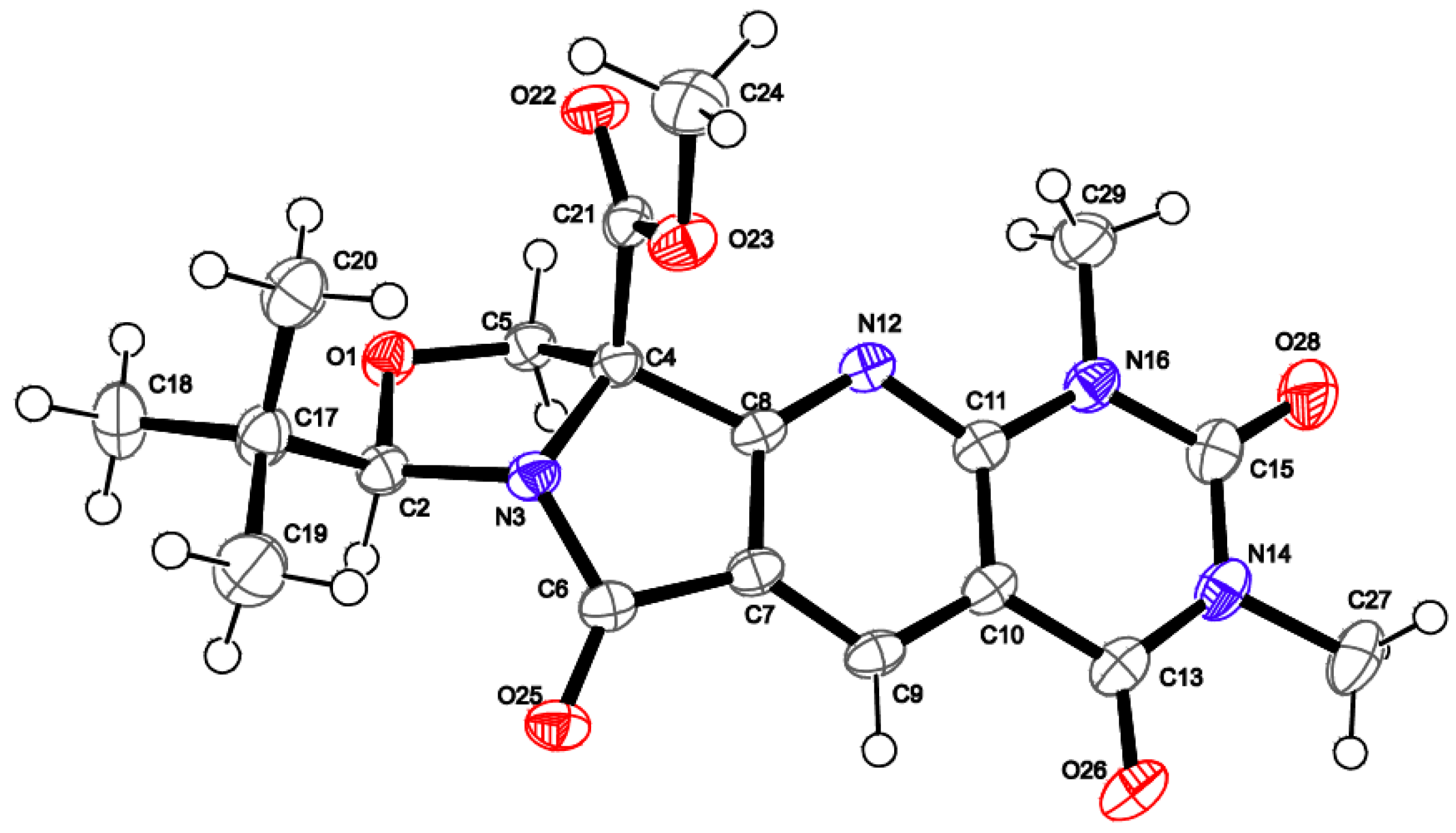
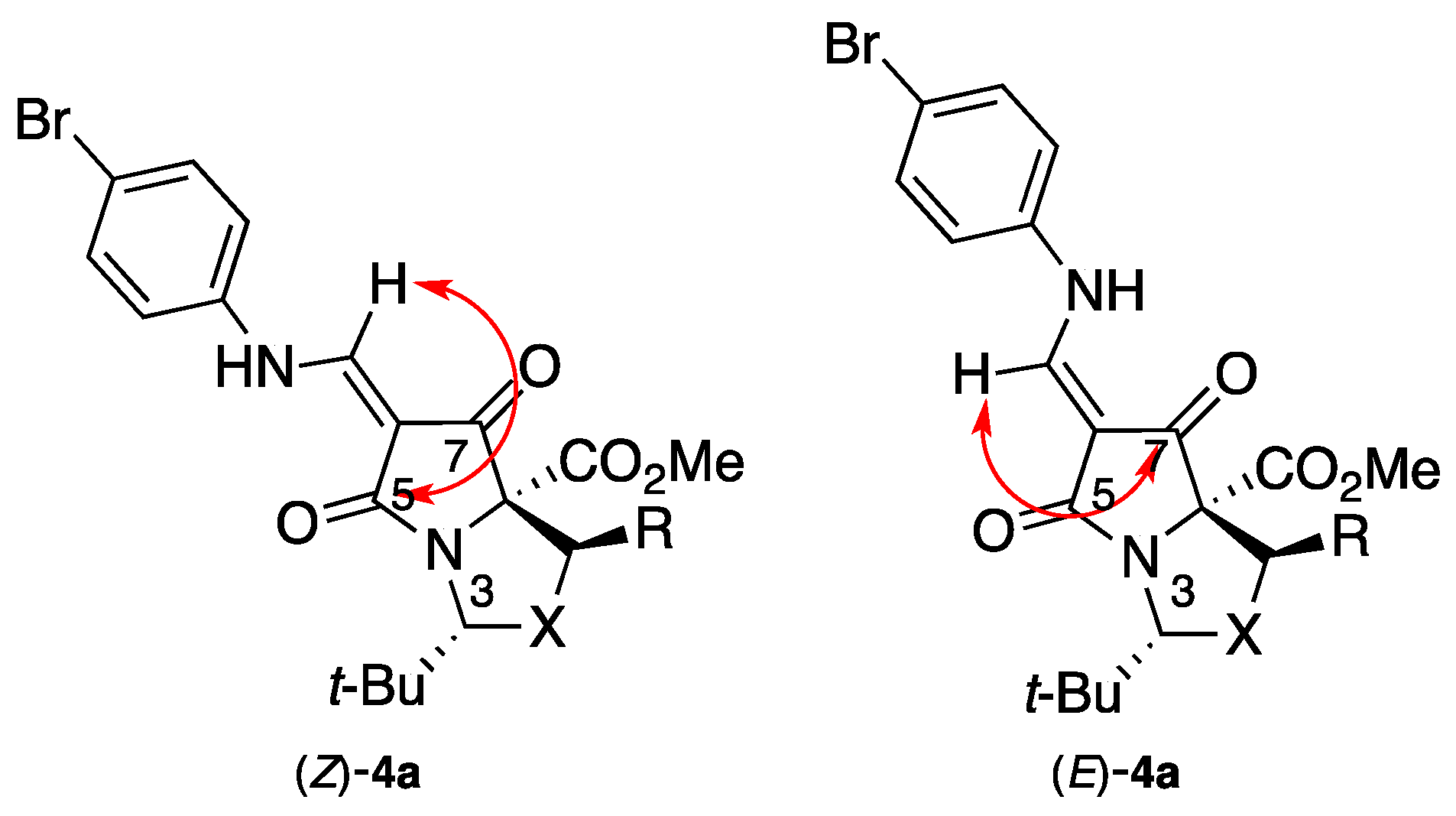
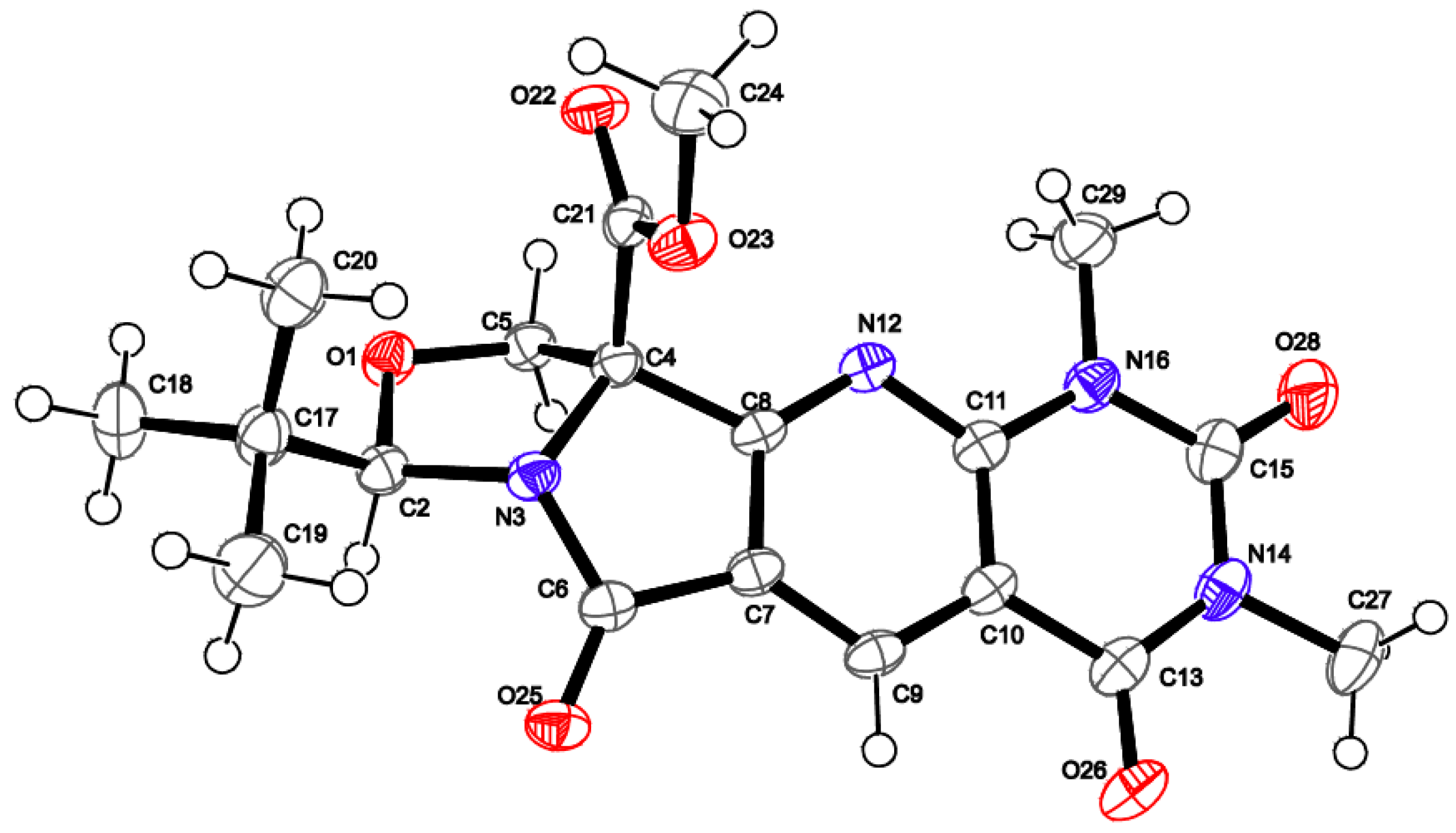
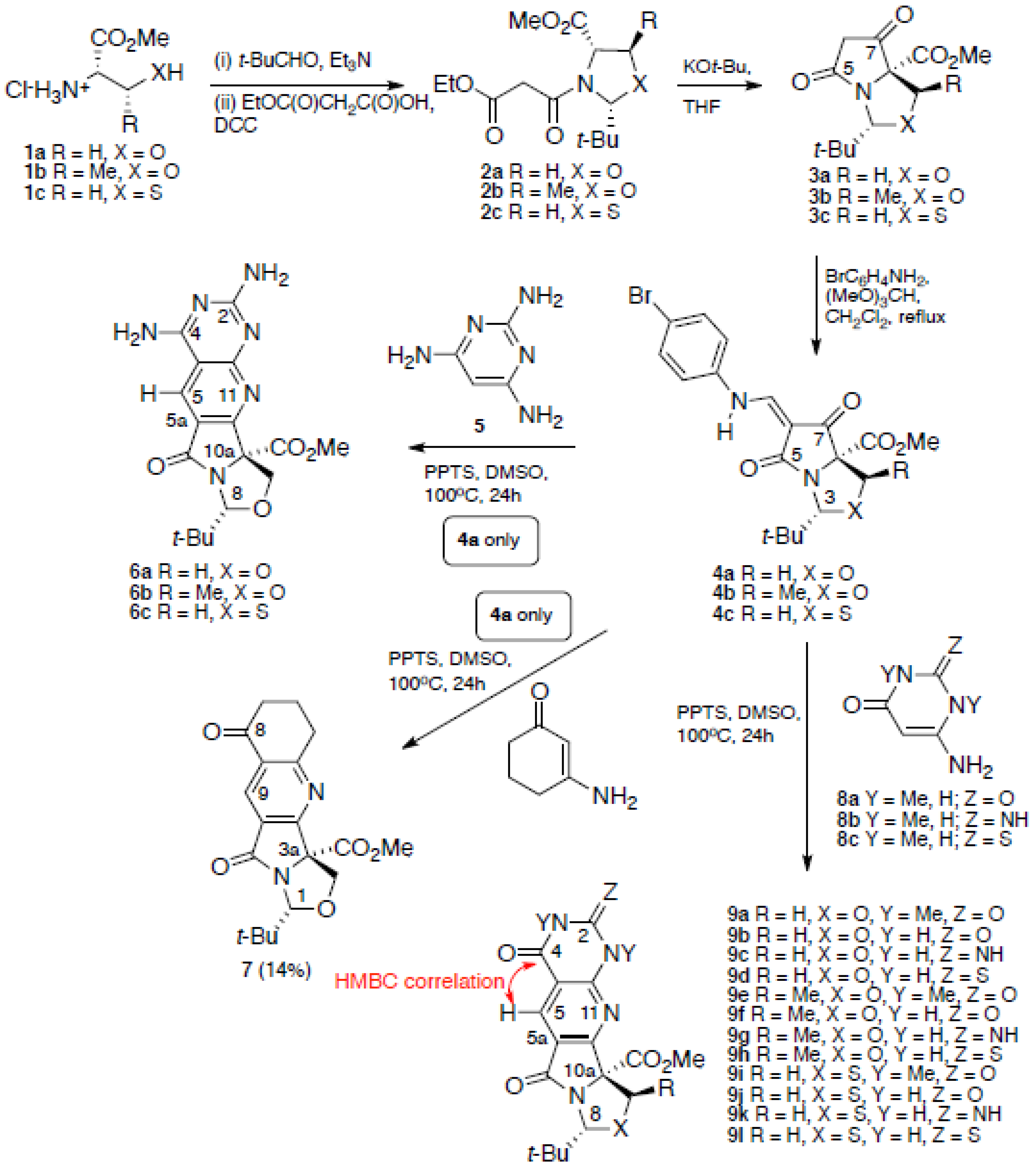
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for Synthesis of Enamines
Methyl (3R,7aR)-6-{[(4-bromophenyl)amino]methylene}-3-(tert-butyl)-5,7-dioxodihydro-1H,3H-pyrrolo[1,2-c]oxazole-7a(5H)-carboxylate 4a
Methyl (1R,3R,7aR)-6-{[(4-bromophenyl)amino]methylene}-3-(tert-butyl)-1-methyl-5,7-dioxodihydro-1H,3H-pyrrolo[1,2-c]oxazole-7a(5H)-carboxylate 4b
Methyl (3R,7aR)-6-{[(4-bromophenyl)amino]methylene}-3-(tert-butyl)-5,7-dioxodihydro-1H,3H-pyrrolo[1,2-c]thiazole-7a(5H)-carboxylate 4c
3.2. General Procedure for Annulation Process
Methyl (8R,10aS)-2,4-diamino-8-(tert-butyl)-6-oxo-6H,8H-oxazolo[3′′,4′′:1′,5′] pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 6a
Methyl (8R,10R,10aS)-2,4-diamino-8-(tert-butyl)-10-methyl-6-oxo-6H,8H-oxazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 6b
Methyl (8R,10aR)-2,4-diamino-8-(tert-butyl)-6-oxo-6H,8H-thiazolo[3′′,4′′:1′,5′] pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 6c
Methyl-(1S,3aS)-1-(tert-butyl)-8,10-dioxo-5,7,8,10-tetrahydro-1H,3H-oxazolo [3′,4′:1,2]pyrrolo[3,4-b]quinoline-3a(6H)-carboxylate 7
Methyl (8R,10aS)-8-(tert-butyl)-1,3-dimethyl-2,4,6-trioxo-1,3,4,6-tetrahydro- 2H,8H-oxazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9a
Methyl (8R,10aS)-8-(tert-butyl)-1,3-dimethyl-2,4,6-trioxo-1,3,4,6-tetrahydro-2H,8H-oxazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9b
Methyl-(8R,10aS)-2-amino-8-(tert-butyl)-4-hydroxy-6-oxo-6H,8H-oxazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9c
Methyl (8R,10aS)-8-(tert-butyl)-4,6-dioxo-2-thioxo-1,3,4,6-tetrahydro-2H,8H-oxazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9d
Methyl (8R,10R,10aS)-8-(tert-butyl)-1,3,10-trimethyl-2,4,6-trioxo-1,3,4,6-tetrahydro-2H,8H-oxazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9e
Methyl (8R,10aR)-8-(tert-butyl)-1,3-dimethyl-2,4,6-trioxo-1,3,4,6-tetrahydro-2H,8H-thiazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9i
Methyl (8R,10aR)-8-(tert-butyl)-2,4,6-trioxo-1,3,4,6-tetrahydro-2H,8H-thiazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6] pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9j
Methyl (8R,10aR)-8-(tert-butyl)-4,6-dioxo-2-thioxo-1,3,4,6-tetrahydro-2H,8H-thiazolo[3′′,4′′:1′,5′]pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a(10H)-carboxylate 9l
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Bush, K.; Courvalin, P.; Dantas, G.; Davies, J.; Eisenstein, B.; Huovinen, P.; Jacoby, G.A.; Kishony, R.; Kreiswirth, B.N.; Kutter, E.; et al. Tackling antibiotic resistance. Nat. Rev. Microbiol. 2011, 9, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Pollastri, M.P.; Schiffer, C.A.; Peet, N.P. The challenge of developing robust drugs to overcome resistance. Drug Discovery Today 2011, 16, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Hurdle, J.G.; O′Neill, A.J.; Chopra, I.; Lee, R.E. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 2011, 9, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Molecular mechanisms of antibiotic resistance. Chem. Commun. 2011, 47, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D. 10 × ′20 Progress—Development of new drugs active against gram-negative bacilli: An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Royles, B.J.L. Naturally-occurring tetramic acids—Structure, isolation, and synthesis. Chem. Rev. 1995, 95, 1981–2001. [Google Scholar] [CrossRef]
- Schobert, R.; Schlenk, A. Tetramic and tetronic acids: An update on new derivatives and biological aspects. Bioorg. Med. Chem. 2008, 16, 4203–4221. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-C.; Moloney, M.G. Tetramic acids as scaffolds: Synthesis, tautomeric and antibacterial behaviour. Synlett 2009, 15, 2487–2491. [Google Scholar]
- Jeong, Y.-C.; Moloney, M.G.; Bikadi, Z.; Hazai, E. A detailed study of antibacterial 3-acyltetramic acids and 3-acylpiperidine-2,4-diones. ChemMedChem 2014, 9, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-C.; Anwar, M.; Moloney, M.G.; Bikadi, Z.; Hazai, E. Natural product inspired antibacterial tetramic acid libraries with dual enzyme target activity. Chem. Sci. 2013, 4, 1008–1015. [Google Scholar] [CrossRef]
- Weidenmaier, C.; Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 2008, 6, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.W.; Wade, M.M.; Holman, S.C.; Champlin, F.R. Status of methods for assessing bacterial cell surface charge properties based on zeta potential measurements. J. Microbiol. Methods 2001, 43, 153–164. [Google Scholar] [CrossRef]
- O’Shea, R.; Moser, H.E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Xia, L.; Idhayadhulla, A.; Lee, Y.R.; Kim, S.H.; Wee, Y.-J. Microwave-assisted synthesis of diverse pyrrolo[3,4-c]quinoline-1,3- diones and their antibacterial activities. ACS Comb. Sci. 2014, 16, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Moloney, M.G. (8r,10as)-methyl 2,4-diamino-8-(tert-butyl)-6-oxo-6,8,10,10a-tetrahydro-oxazolo-[3′′,4′′:1′,5′]-pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-10a-carboxylate. Molbank 2015. [Google Scholar] [CrossRef]
- Josa-Culleré, L.; Moloney, M.G.; Thompson, A.L. Stereoselectivity in the reduction of bicyclic tetramates. Synlett 2016, 27, 1677–1681. [Google Scholar]
- Anwar, M.; Moloney, M.G. Chiral bicyclic tetramates as non-planar templates for chemical library synthesis. Chem. Biol. Drug Des. 2013, 81, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Oshega, J.S.; Paponov, B.V.; Omelchenko, I.V.; Shishkin, O.V. One-pot three-component synthesis of 3-cyano-4-methyl-2,6-dioxopyridine amino enones. Mendeleev Commun. 2015, 25, 133–134. [Google Scholar] [CrossRef]
- Summers, M.F.; Bax, A. H and 13c assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2d multiple quantum nmr. J. Am. Chem. Soc. 1986, 108, 2093–2094. [Google Scholar]
- Liu, H.; Li, X. Synthesis of protected sugar-amino acid hybrid molecules as platform for further derivatization. Tetrahedron Lett. 2012, 53, 6957–6960. [Google Scholar] [CrossRef]
- Taylor, E.C.; Palmer, D.C.; George, T.J.; Fletcher, S.R.; Tseng, C.P.; Harrington, P.J.; Beardsley, G.P.; Dumas, D.J.; Rosowsky, A.; Wick, M. Synthesis and biological activity of L-5-deazafolic acid and L-deazaaminopterin: synthetic strategies to 5-deazapteridines. J. Org. Chem. 1983, 48, 4852–4860. [Google Scholar] [CrossRef]
- Stark, E.; Breitmaier, E. 5-Desazapteridine, synthese und NMR-spektroskopie. Tetrahedron 1973, 29, 2209–2217. [Google Scholar] [CrossRef]
- Hafiz, A.; Saad, I.; Reheim, M.A.; Ahmed, M.; Mohamed Baker, S.; Mahfouz Ramiz, M. Enaminone in heterocyclic synthesis: synthesis of new pyrazolopyrazole, pyrazolothienooxazine and pyrazolothienopyridine derivatives. J. Chem. Soc. Pak. 2014, 36, 1133–1144. [Google Scholar]
- Abu-Shanab, F.A.; Sherif, S.M.; Mousa, S.A.S. Dimethylformamide dimethyl acetal as a building block in heterocyclic synthesis. J. Heterocycl. Chem. 2009, 46, 801–827. [Google Scholar] [CrossRef]
- Singh, K.; Singh, J.; Singh, H. Carbon transfer reactions of functionalized oxazolidines and their open chain enamine tautomers to enamine nucleophiles. A facile synthesis of substituted pyridines and ring annulated derivatives. Tetrahedron 1998, 54, 935–942. [Google Scholar] [CrossRef]
- Jeong, Y.-C.; Moloney, M.G. Equisetin, reutericyclin and streptolodygin as natural product lead structures for novel antibiotic libraries. Future Med. Chem. 2015, 7, 1861–1877. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.S.W.; Chai, C.L.L.; Moloney, M.G.; Thompson, A.L. Synthesis of mimics of pramanicin from pyroglutamic acid and their antibacterial activity. J. Org. Chem. 2015, 80, 2661–2675. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Warren, S.C.; Newton, G.G.F.; Abraham, E.P. Biosynthesis of penicillin N and cephalosporin C—Antibiotic production and other features of metabolism of a cephalosporium species. Biochem. J. 1967, 103, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Holloway, C.A.; Matthews, C.J.; Moloney, M.G.; Roberts, C.F.; Yaqoob, M. Novel chiral skeletons for drug discovery: Antibacterial tetramic acids. Chem. Biol. Drug Des. 2011, 78, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Cosier, J.; Glazer, A.M. A nitrogen-gas-stream cryostat for general X-ray diffraction studies. J. Appl. Cryst. 1986, 19, 105–107. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. Superflip—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Parois, P.; Cooper, R.I.; Thompson, A.L. Crystal structures of increasingly large molecules: Meeting the challenges with CRYSTALS software. Chem. Cent. J. 2015, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.I.; Thompson, A.L.; Watkin, D.J. CRYSTALS enhancements: Dealing with hydrogen atoms in refinement. J. Appl. Cryst. 2010, 43, 1100–1107. [Google Scholar] [CrossRef]
- Flack, H.D. On enantiomorph-polarity estimation. Acta Crystallogr. 1983, A39, 876–881. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration determinations. J. Appl. Cryst. 2000, 33, 1143–1148. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Cryst. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Compound | Yield (%) | Ratio | Compound | Yield (%) | δH-5 (ppm) | Compound | Yield (%) | δH-5 (ppm) |
|---|---|---|---|---|---|---|---|---|
| 4a | 100 | 1.8:1 | 6a | 68 | 8.53 | 9a | 45 | 8.82 |
| 9b | 82 | 8.61 | ||||||
| 9c | 49 | 8.37 | ||||||
| 9d | 45 | 8.61 | ||||||
| 4b | 88 | 1.8:1 | 6b | 10 | 8.52 | 9e | 53 | 8.81 |
| 9f | - | - | ||||||
| 9g | - | - | ||||||
| 9h | - | - | ||||||
| 4c | 30 | 1.9:1 | 6c | 13 | 8.48 | 9i | 36 | 8.80 |
| 9j | 56 | 8.57 | ||||||
| 9k | - | - | ||||||
| 9l | <10 | 8.58 |
| Compound | Solvent system | Polar Surface Area (PSA) b | Molecular Surface Area (MSA) b | clogP b | PSA/MSA (%) b | Zone size (mm) | |
|---|---|---|---|---|---|---|---|
| E. coli | S. aureus | ||||||
| 6a | DMSO: H2O = 1:1 | 146.55 | 511.28 | 1.22 | 28.7 | 17 | Not active |
| 6b | DMSO: H2O = 1:1 | 146.55 | 540.70 | 1.64 | 27.1 | 17 | Not active |
| 6c | DMSO: H2O = 1:1 | 137.32 | 516.98 | 1.68 | 26.6 | 18.5 | Not active |
| 9a | DMSO: MeOH = 1:1 | 109.35 | 564.05 | 1.29 | 19.4 | 14.5 | 17 |
| 9b | DMSO: H2O = 1:1 | 126.93 | 499.08 | 1.85 | 25.4 | 17.5 | Not active |
| 9c | DMSO | 140.76 | 506.42 | 1.75 | 27.8 | 16 | 15.5 |
| 9d | DMSO: H2O = 7:3 | 109.86 | 504.20 | 2.74 | 21.8 | 15.5 | 15.5 |
| 9e | DMSO: MeOH = 1:1 | 109.35 | 593.44 | 1.70 | 18.4 | 16.5 | Not active |
| 9i | DMSO: H2O = 1:1 | 100.12 | 569.60 | 1.74 | 17.6 | 13 | Not active |
| 9j | DMSO: MeOH = 1:1 | 117.2 | 504.20 | 1.44 | 23.2 | 15 | 12.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Moloney, J.G.; Christensen, K.E.; Moloney, M.G. Fused-Ring Oxazolopyrrolopyridopyrimidine Systems with Gram-Negative Activity. Antibiotics 2017, 6, 2. https://doi.org/10.3390/antibiotics6010002
Chen Y, Moloney JG, Christensen KE, Moloney MG. Fused-Ring Oxazolopyrrolopyridopyrimidine Systems with Gram-Negative Activity. Antibiotics. 2017; 6(1):2. https://doi.org/10.3390/antibiotics6010002
Chicago/Turabian StyleChen, Yiyuan, Jonathan G. Moloney, Kirsten E. Christensen, and Mark G. Moloney. 2017. "Fused-Ring Oxazolopyrrolopyridopyrimidine Systems with Gram-Negative Activity" Antibiotics 6, no. 1: 2. https://doi.org/10.3390/antibiotics6010002
APA StyleChen, Y., Moloney, J. G., Christensen, K. E., & Moloney, M. G. (2017). Fused-Ring Oxazolopyrrolopyridopyrimidine Systems with Gram-Negative Activity. Antibiotics, 6(1), 2. https://doi.org/10.3390/antibiotics6010002