
Biotin Protein Ligase Is a Target for New Antibacterials

 ,
, 
Abstract
:1. Introduction
2. Biotin Protein Ligase as a Novel Antibacterial Target
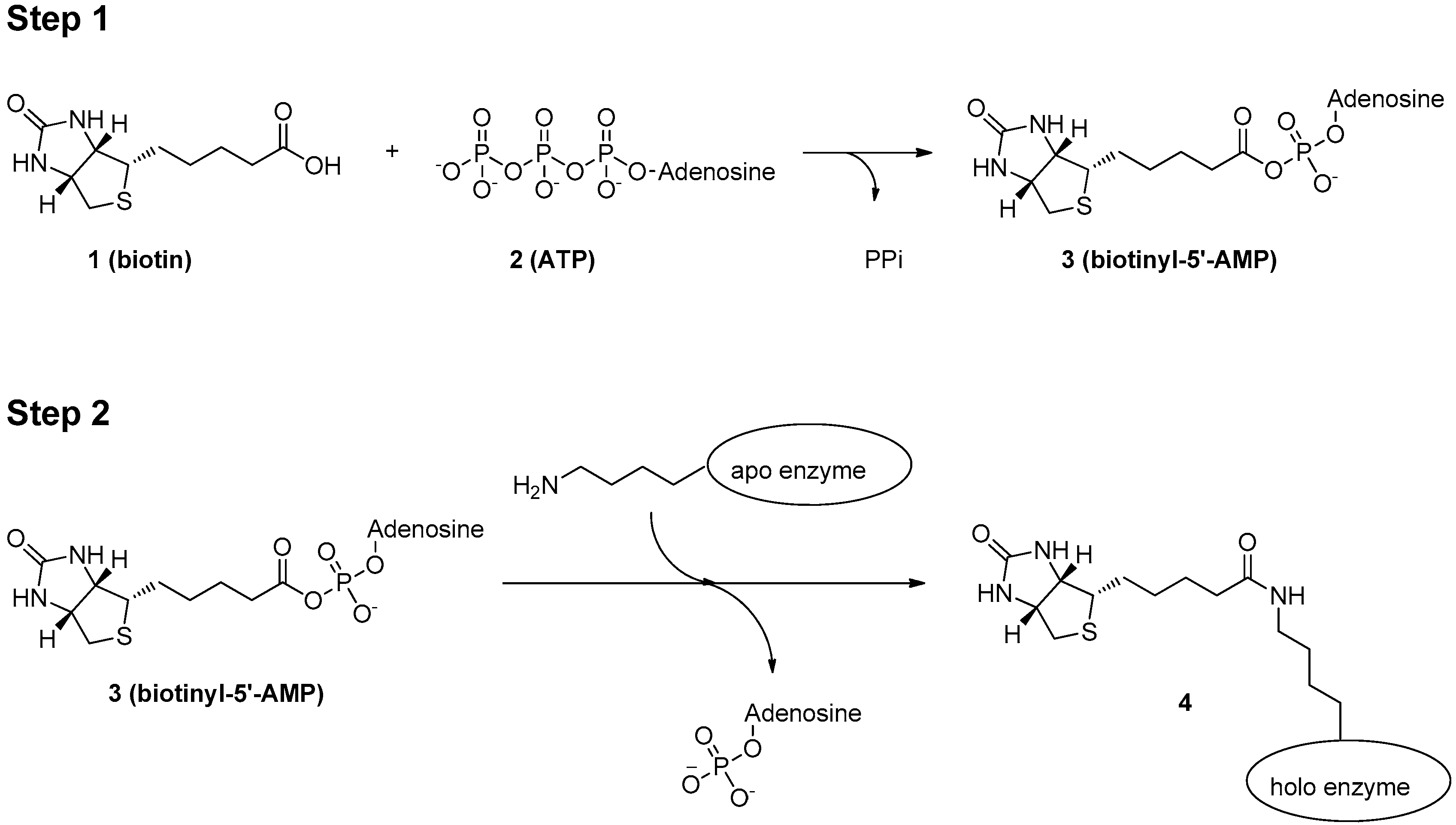
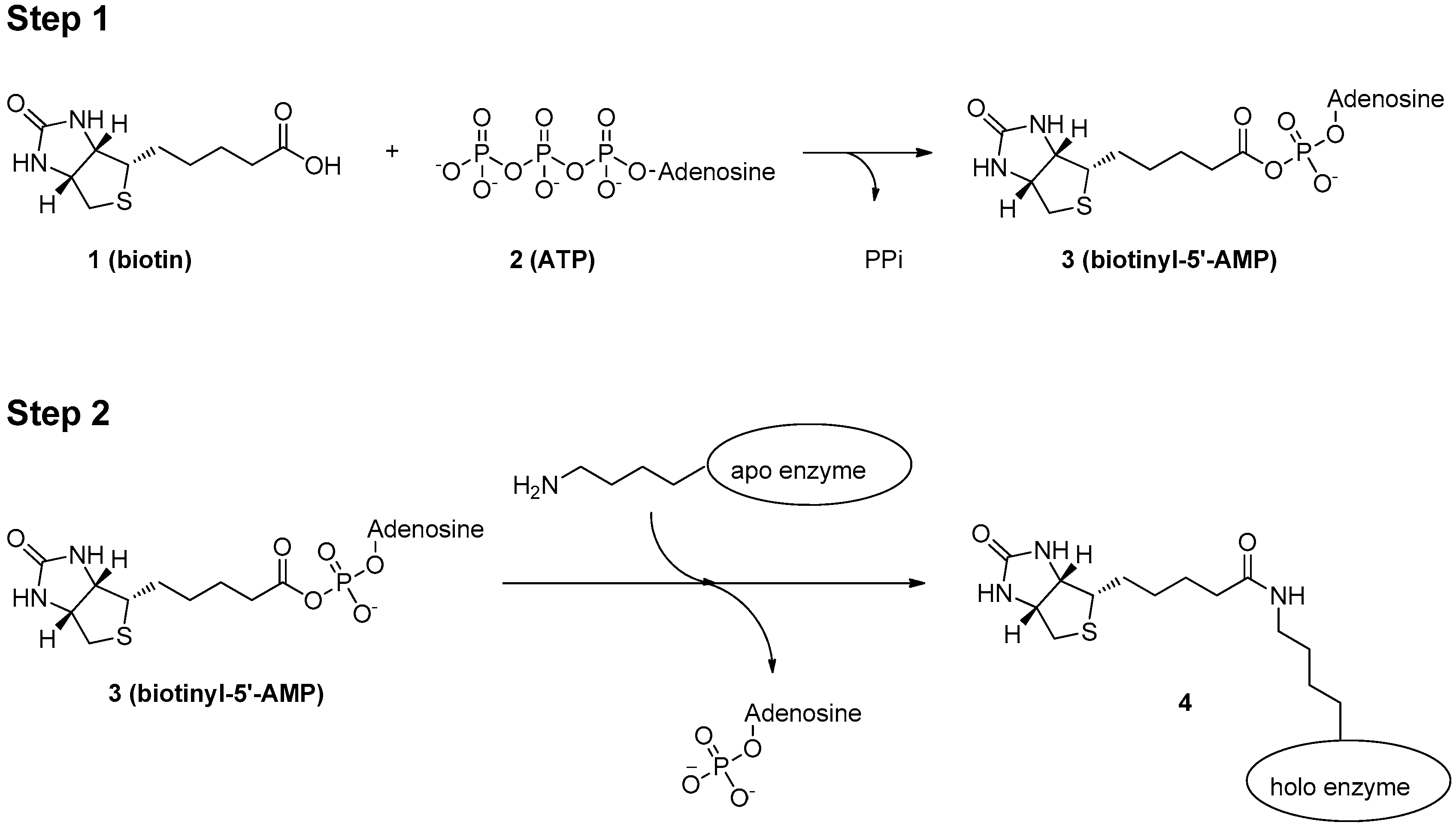
3. Mechanism of Protein Biotinylation
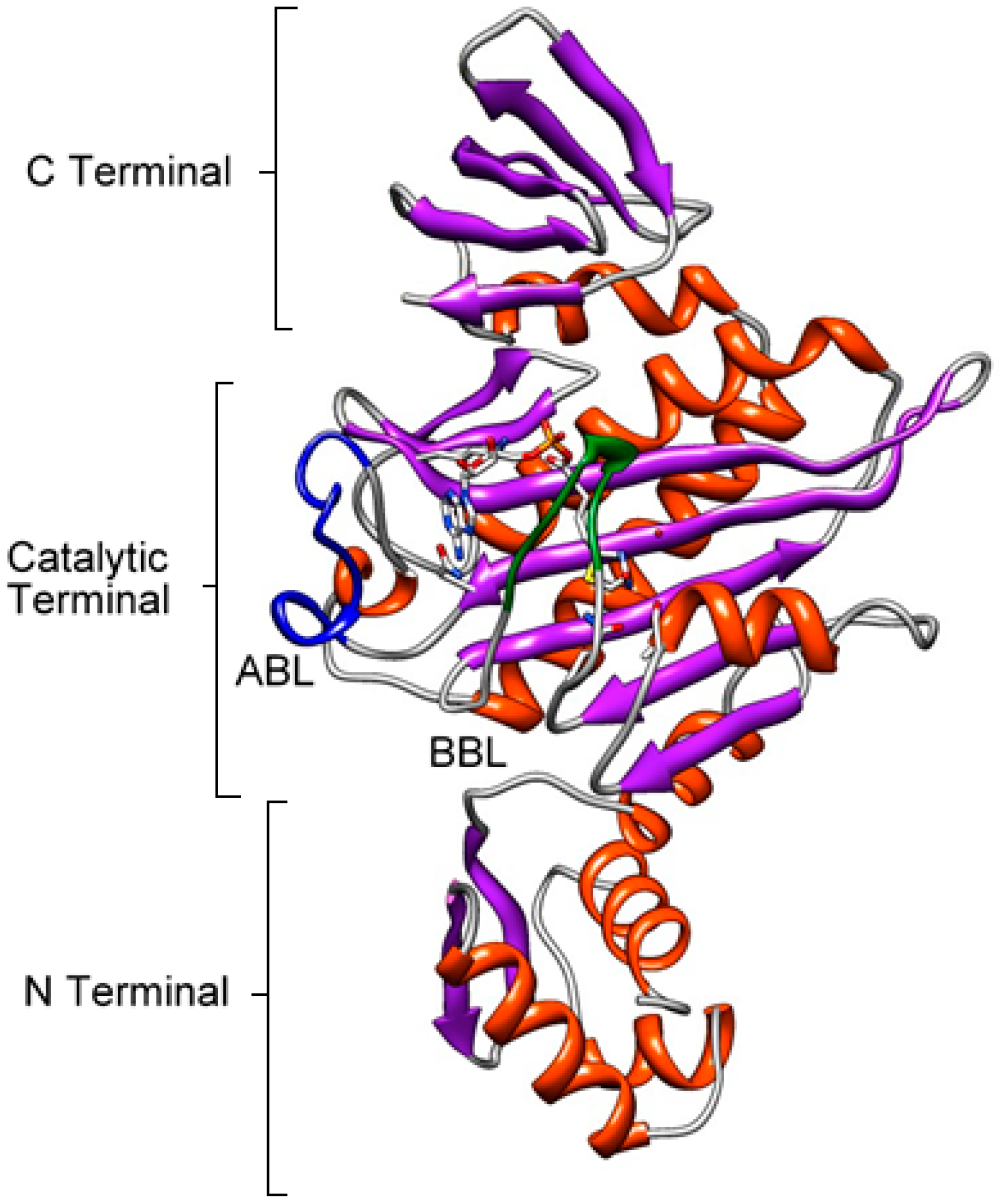
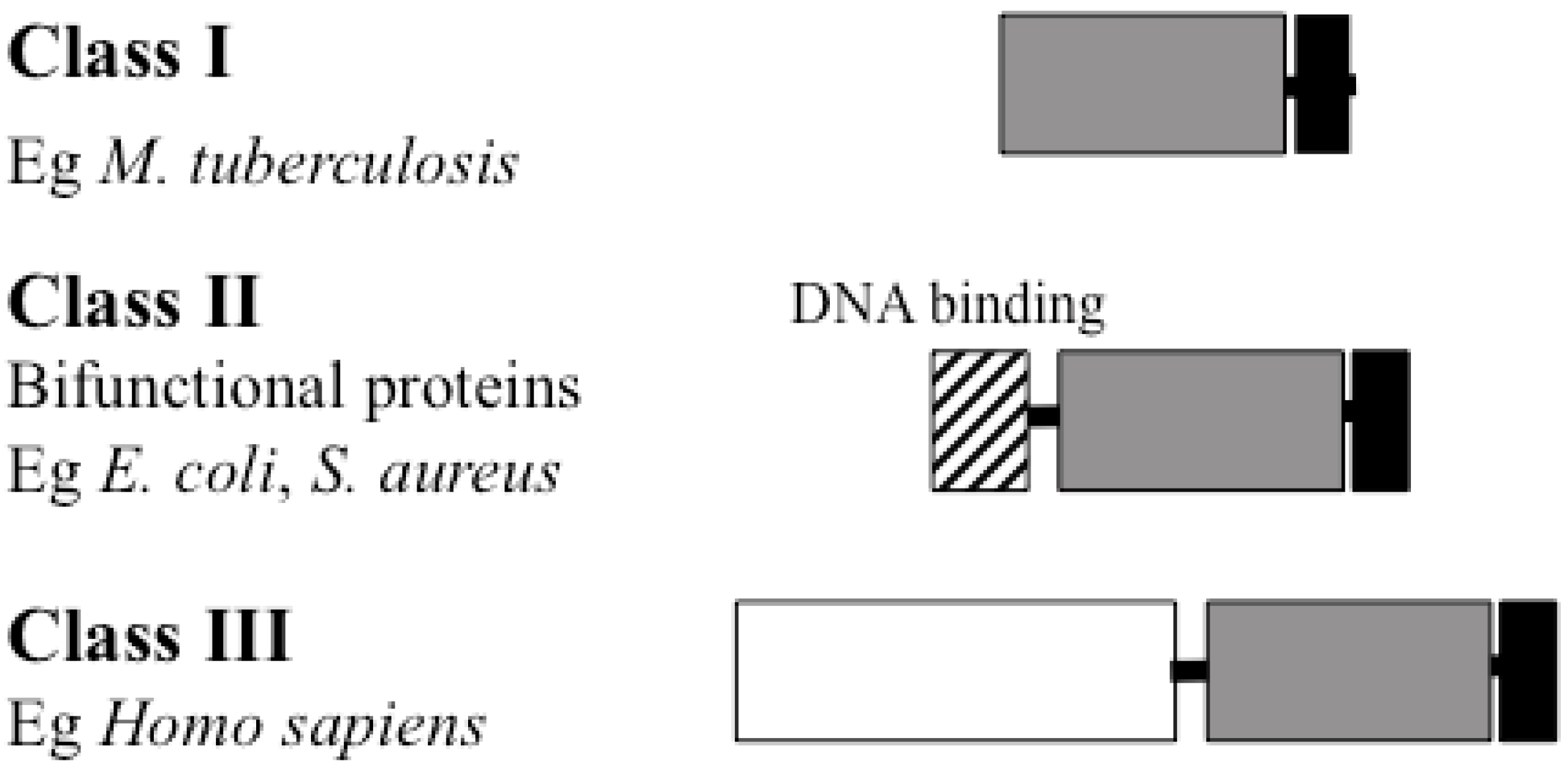
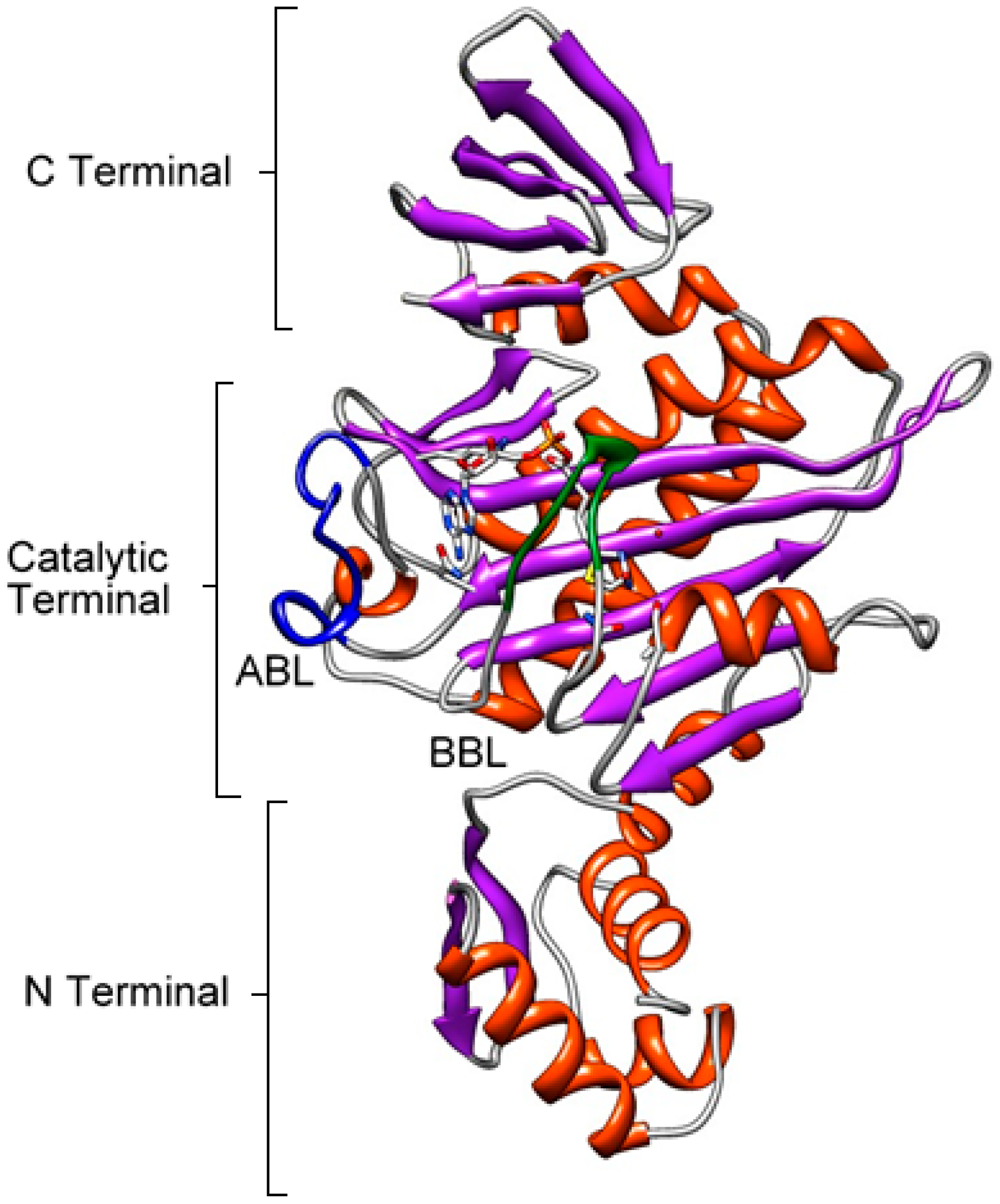
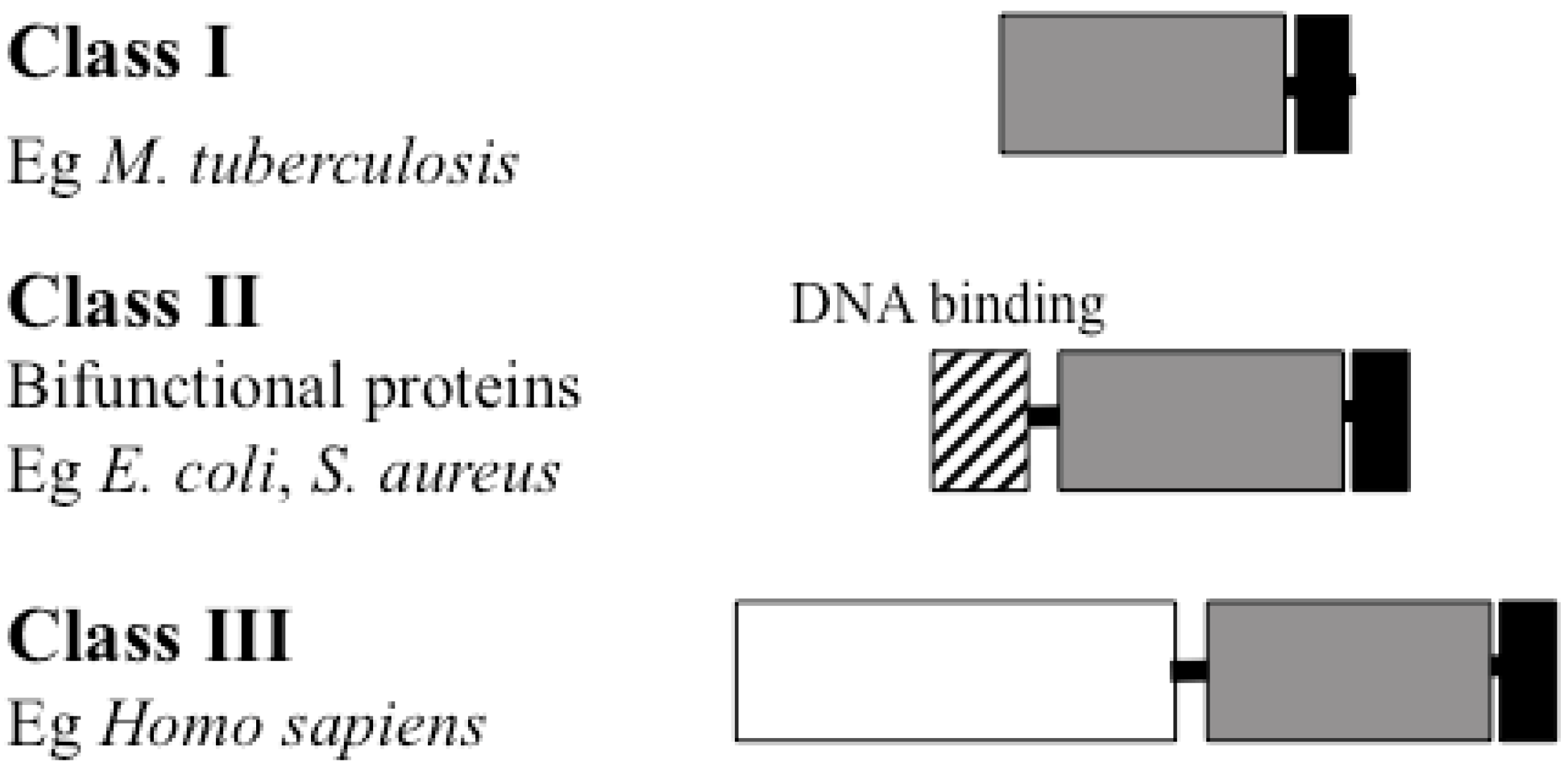
4. BPL Structure
Catalytic Domain
5. BPL Inhibitors as New Antibacterials
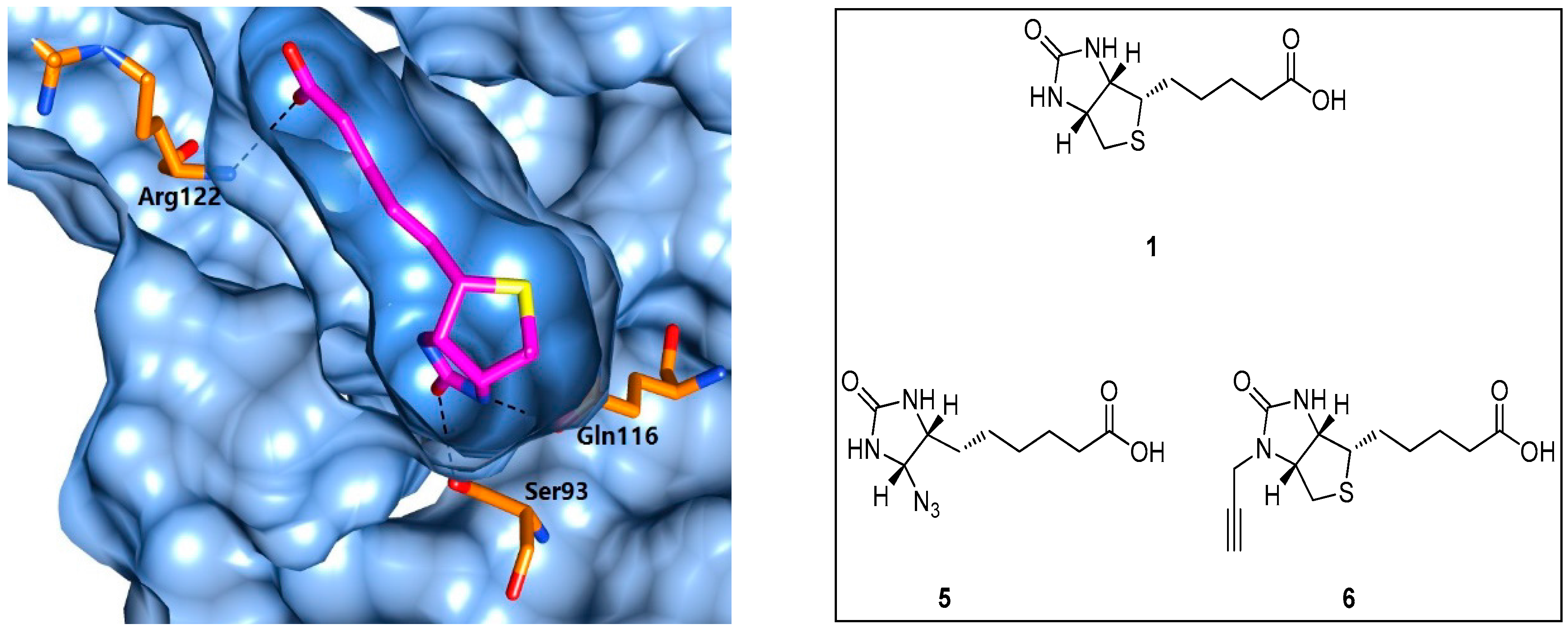
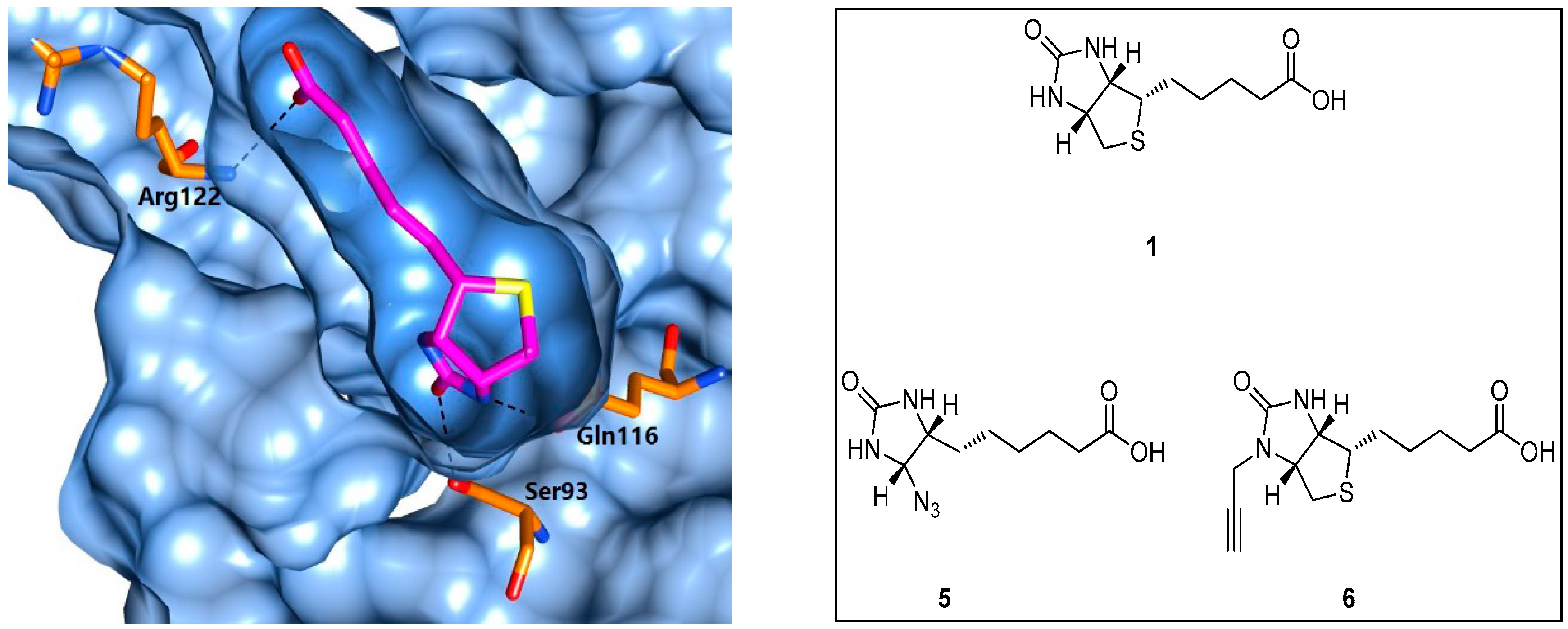
5.1. Biotin Analogues as Antibacterial Agents
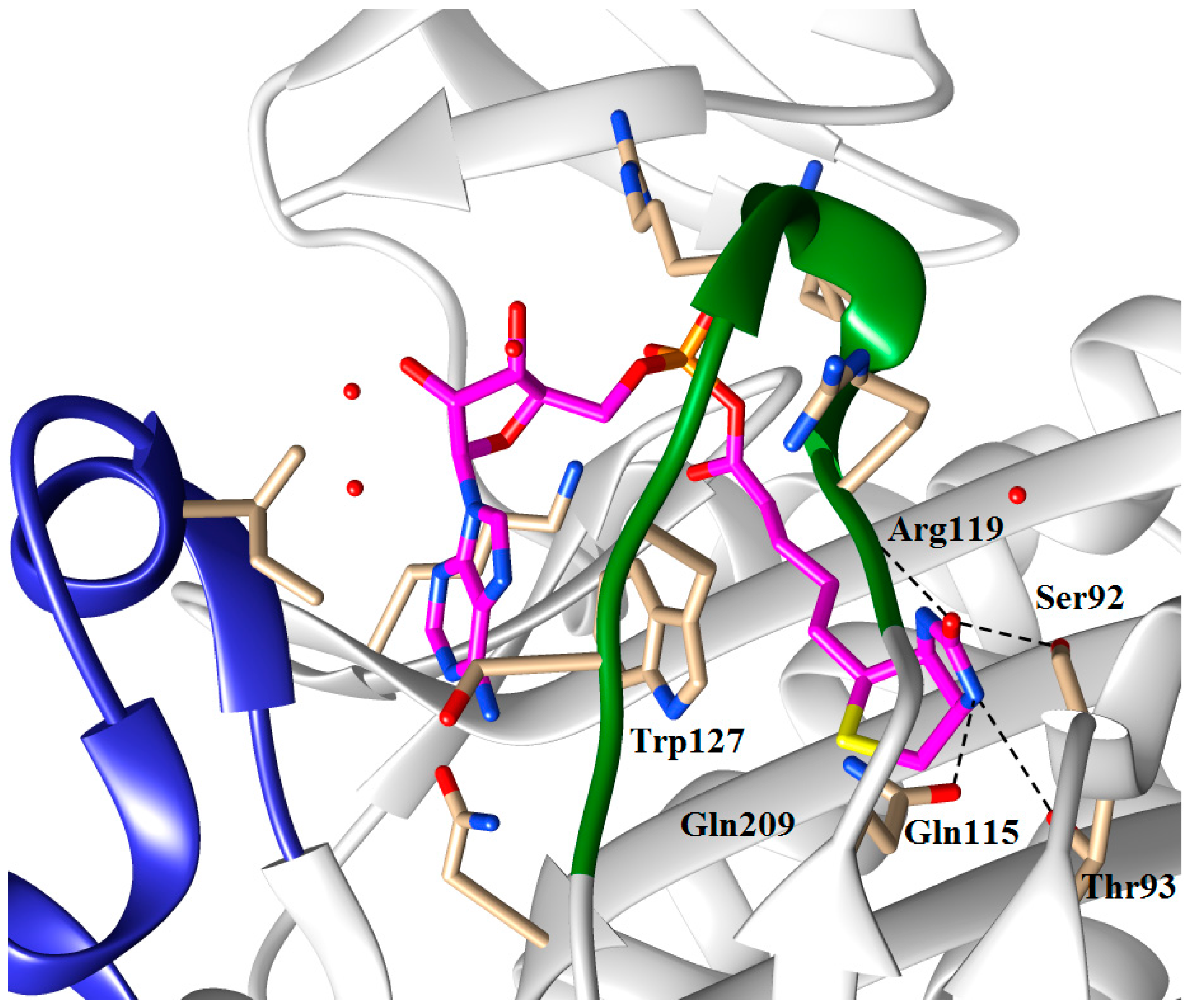
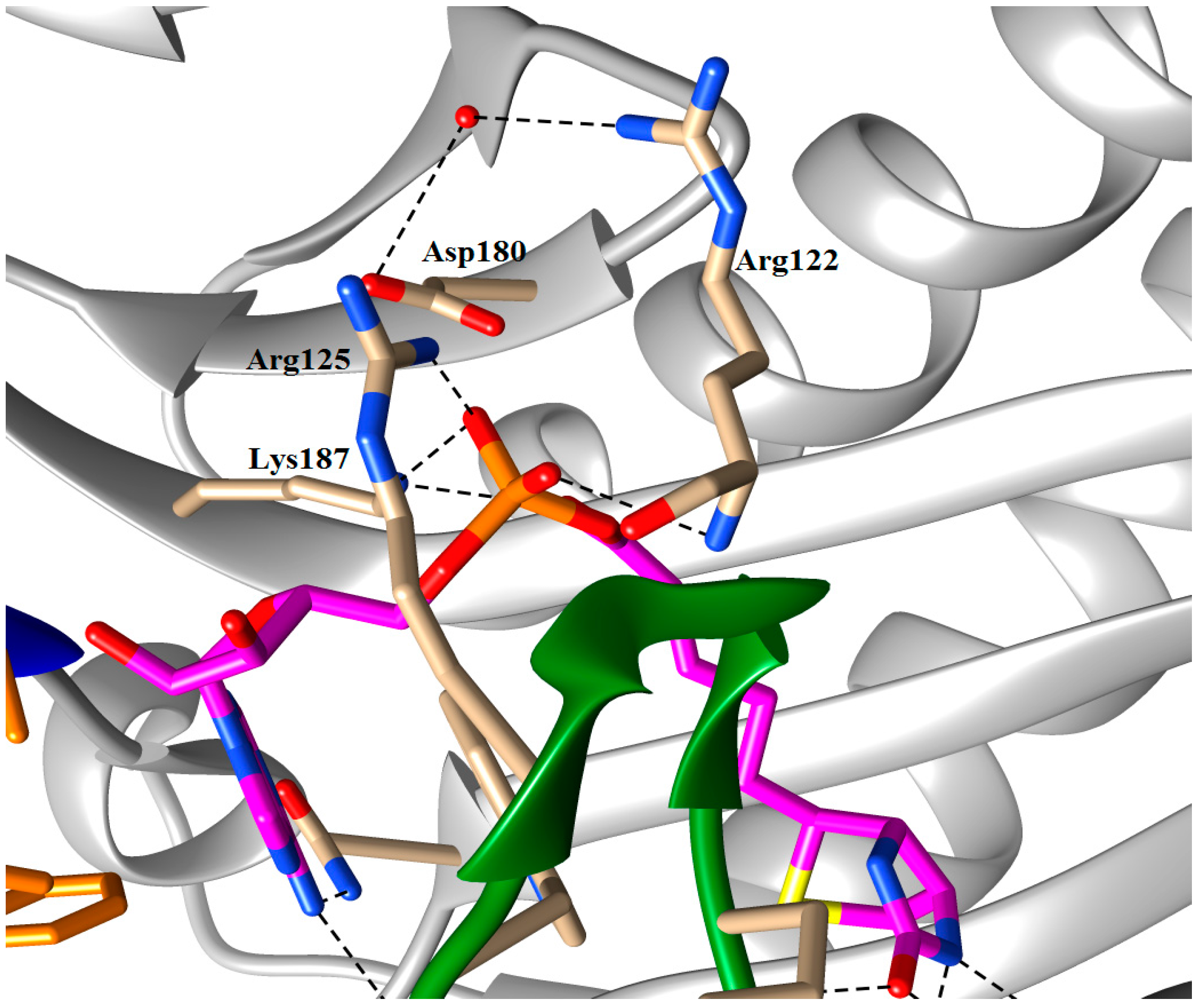
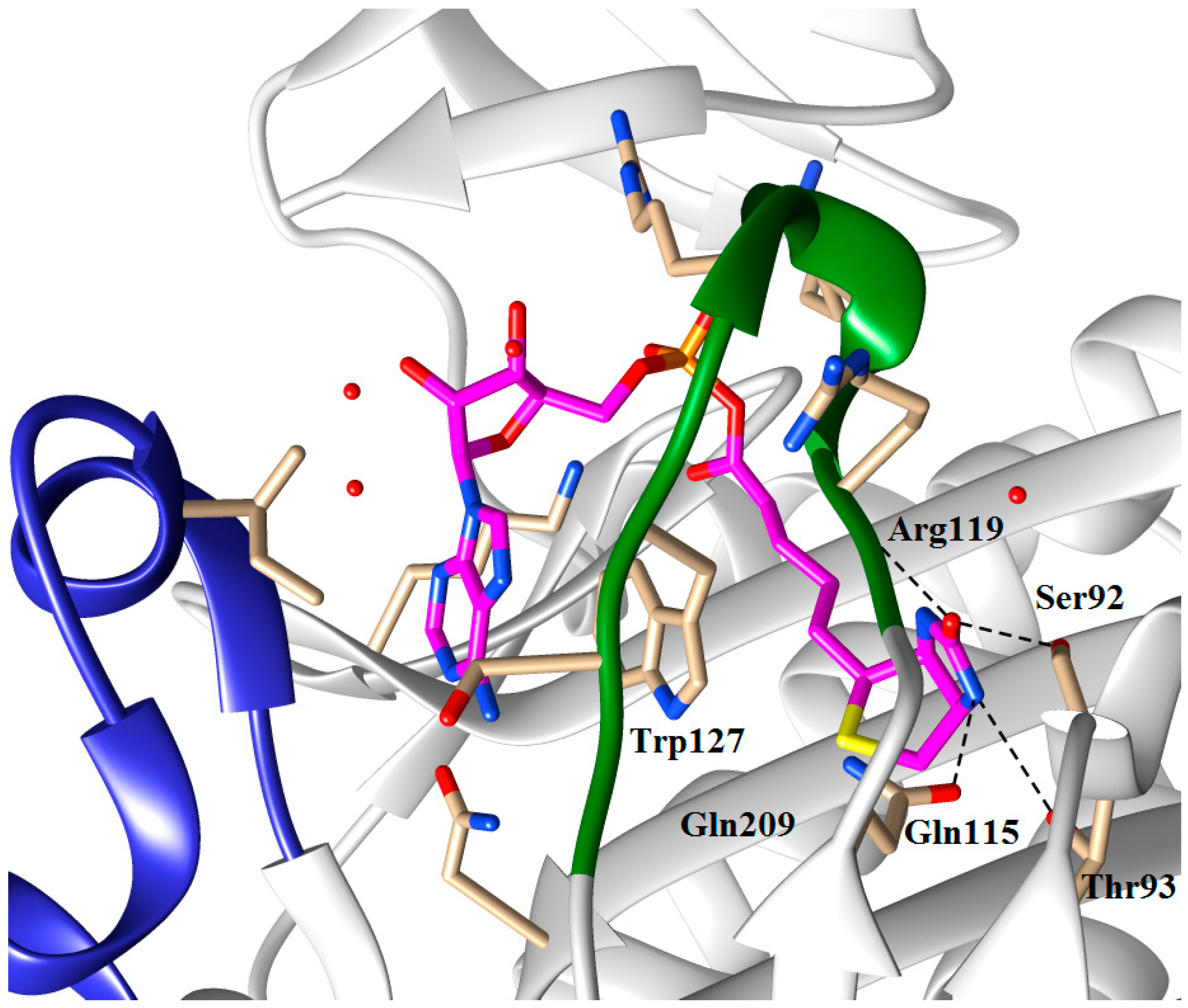
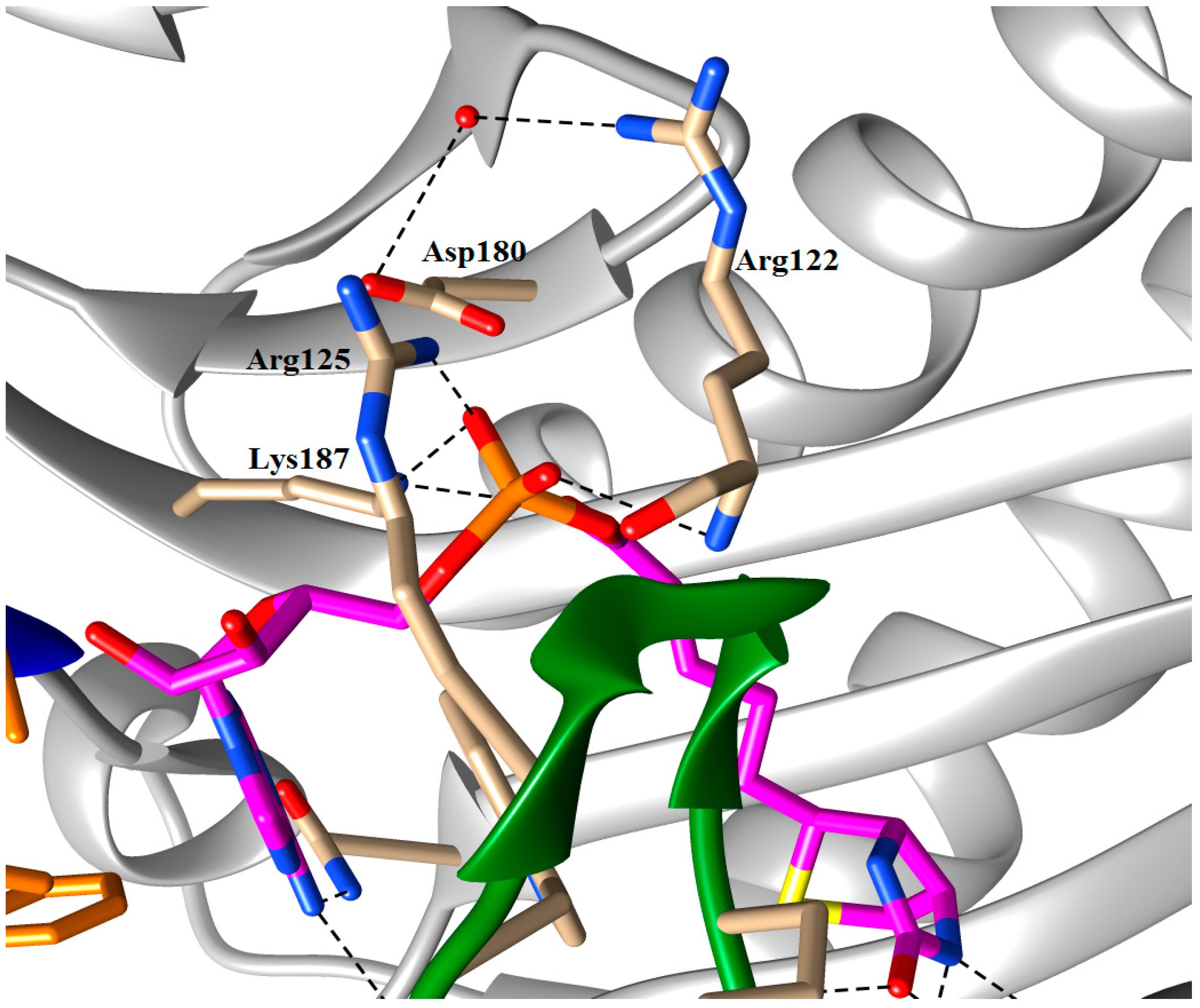
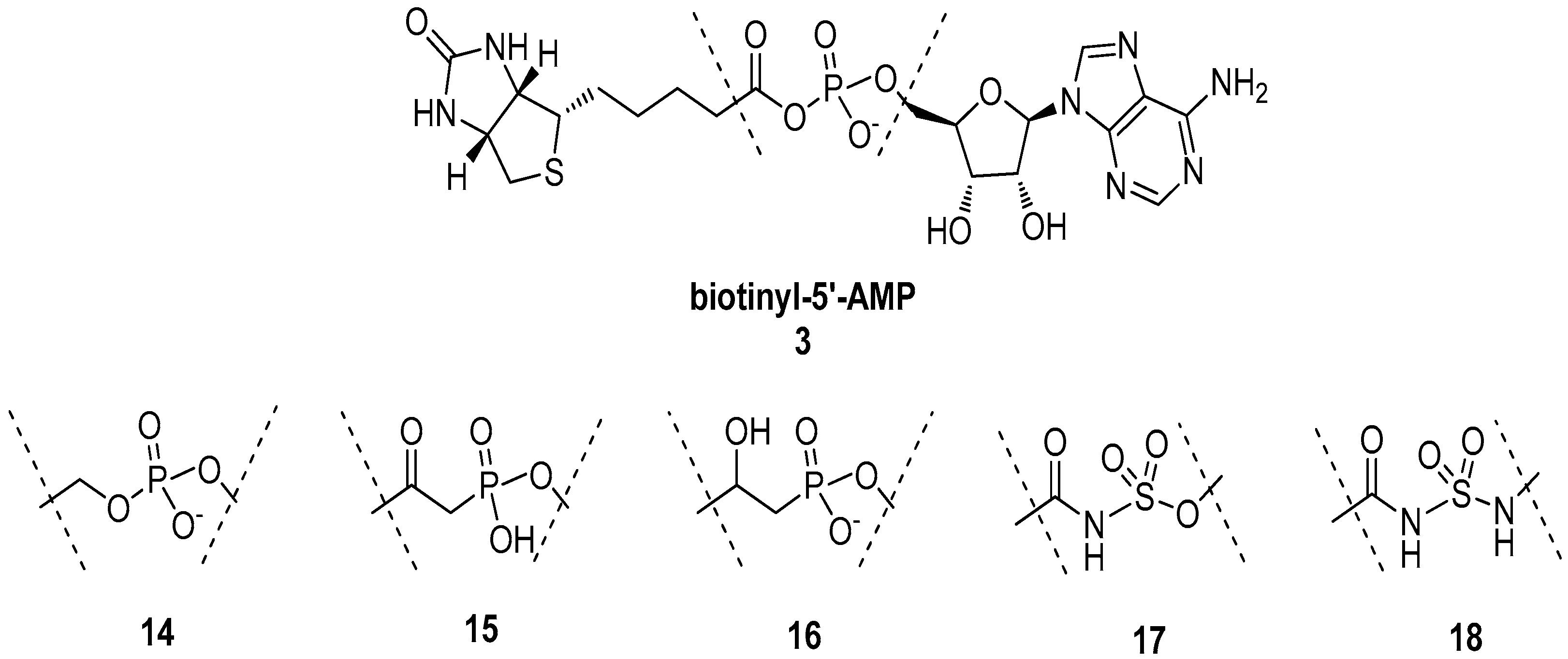
5.2. BPL Reaction Intermediate Analogues as Antibacterial Agents
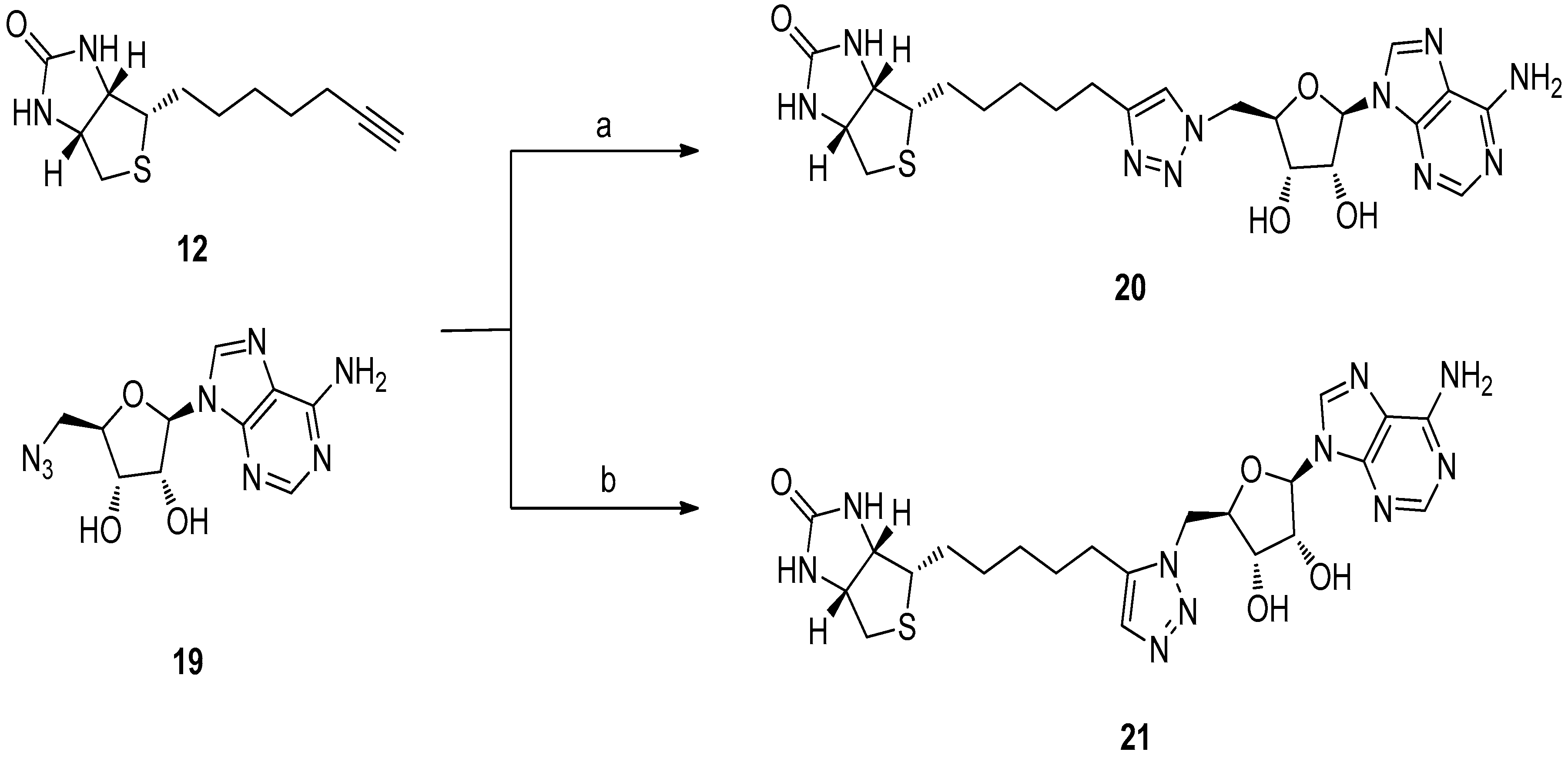
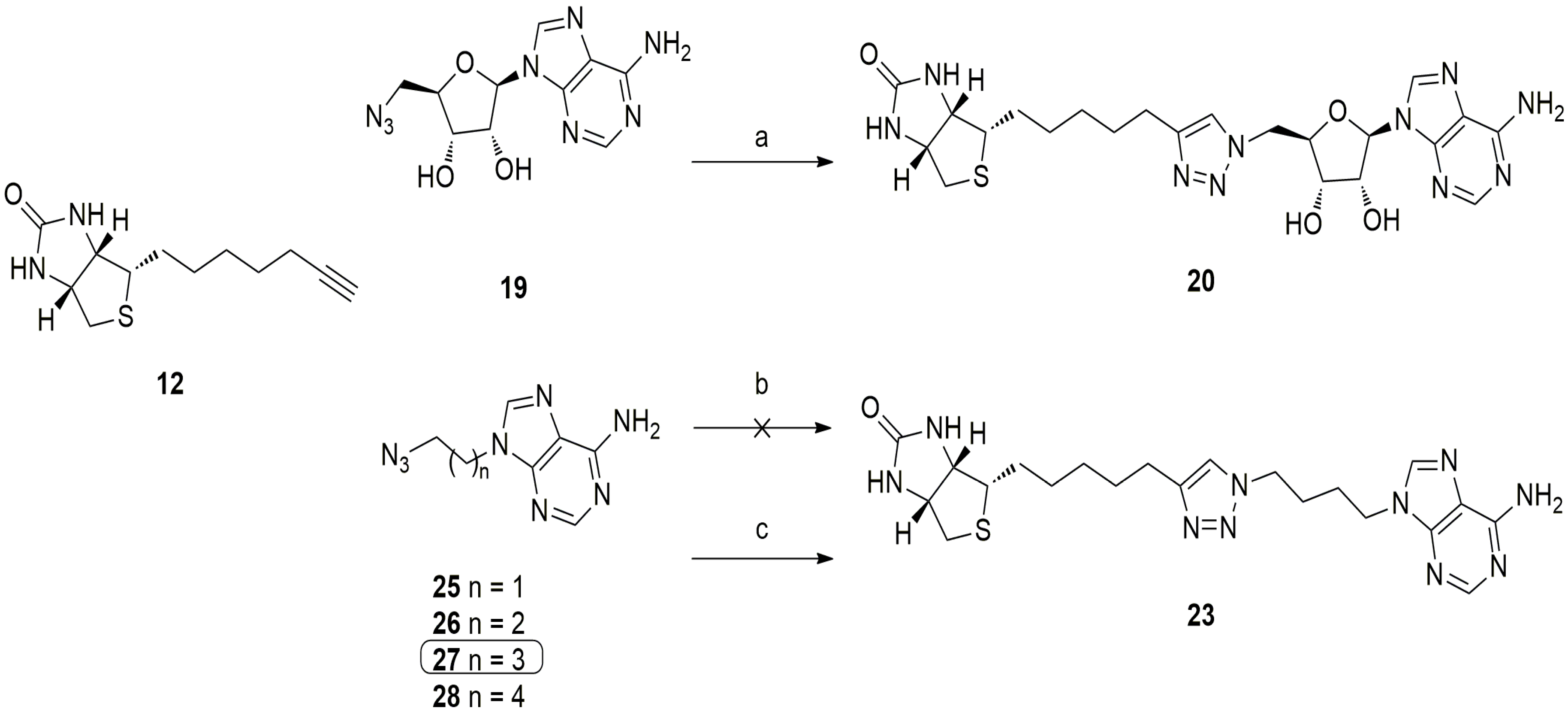
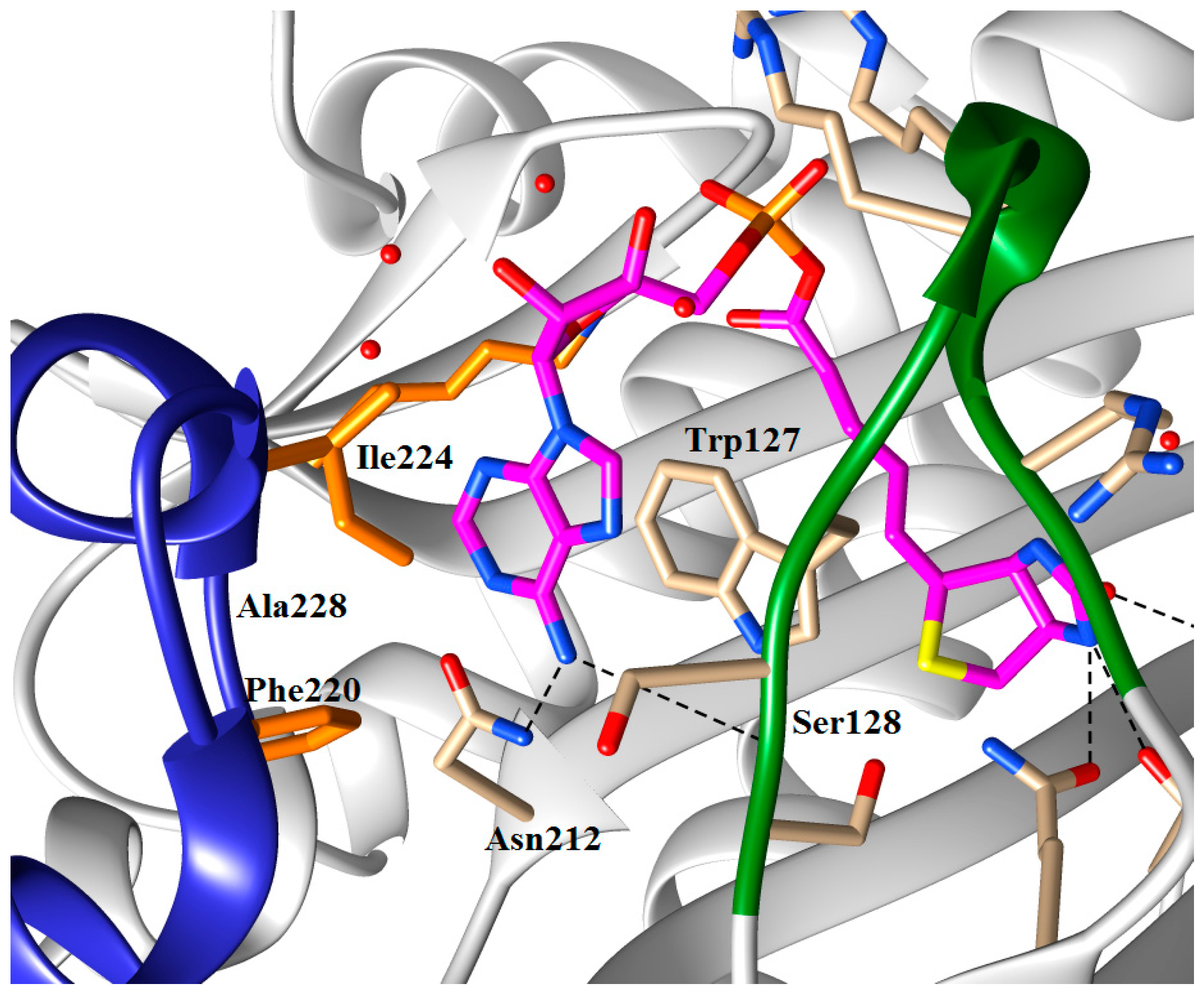
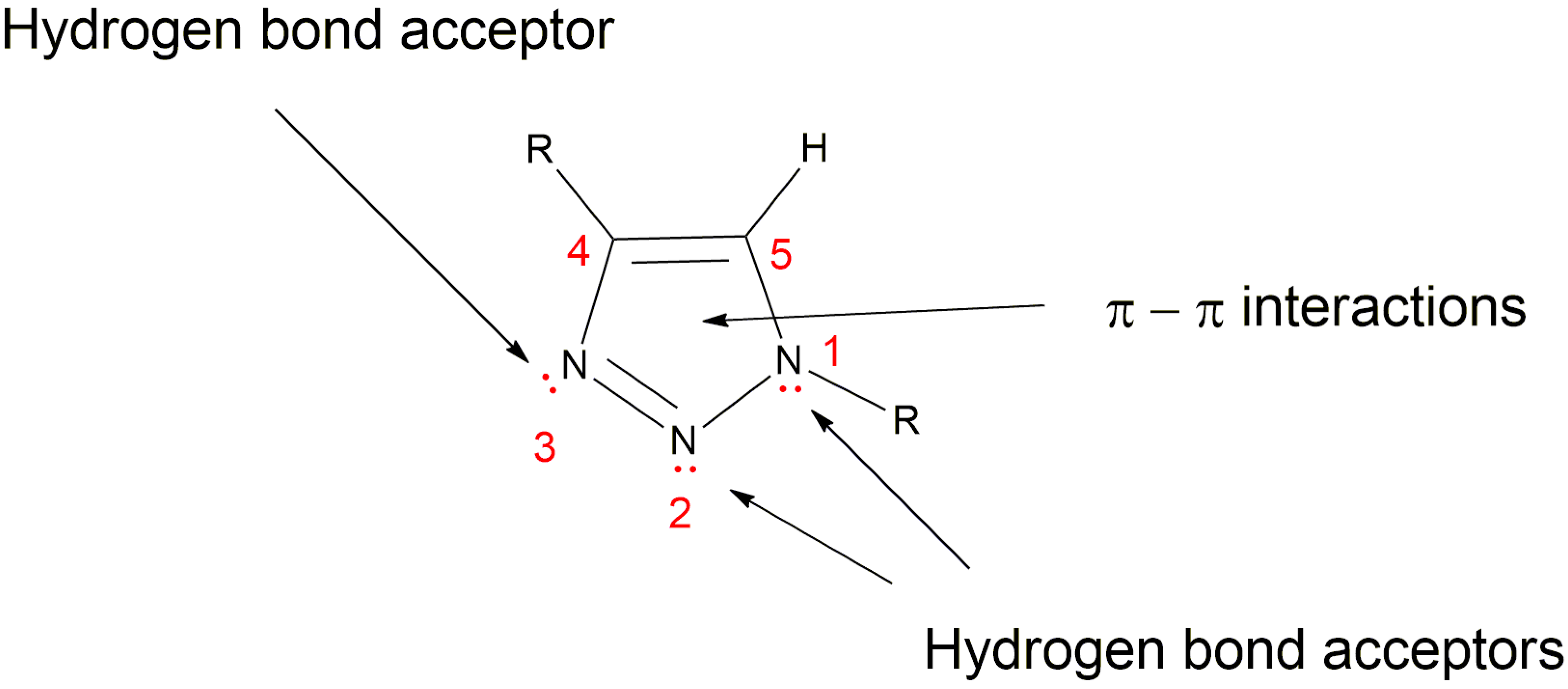
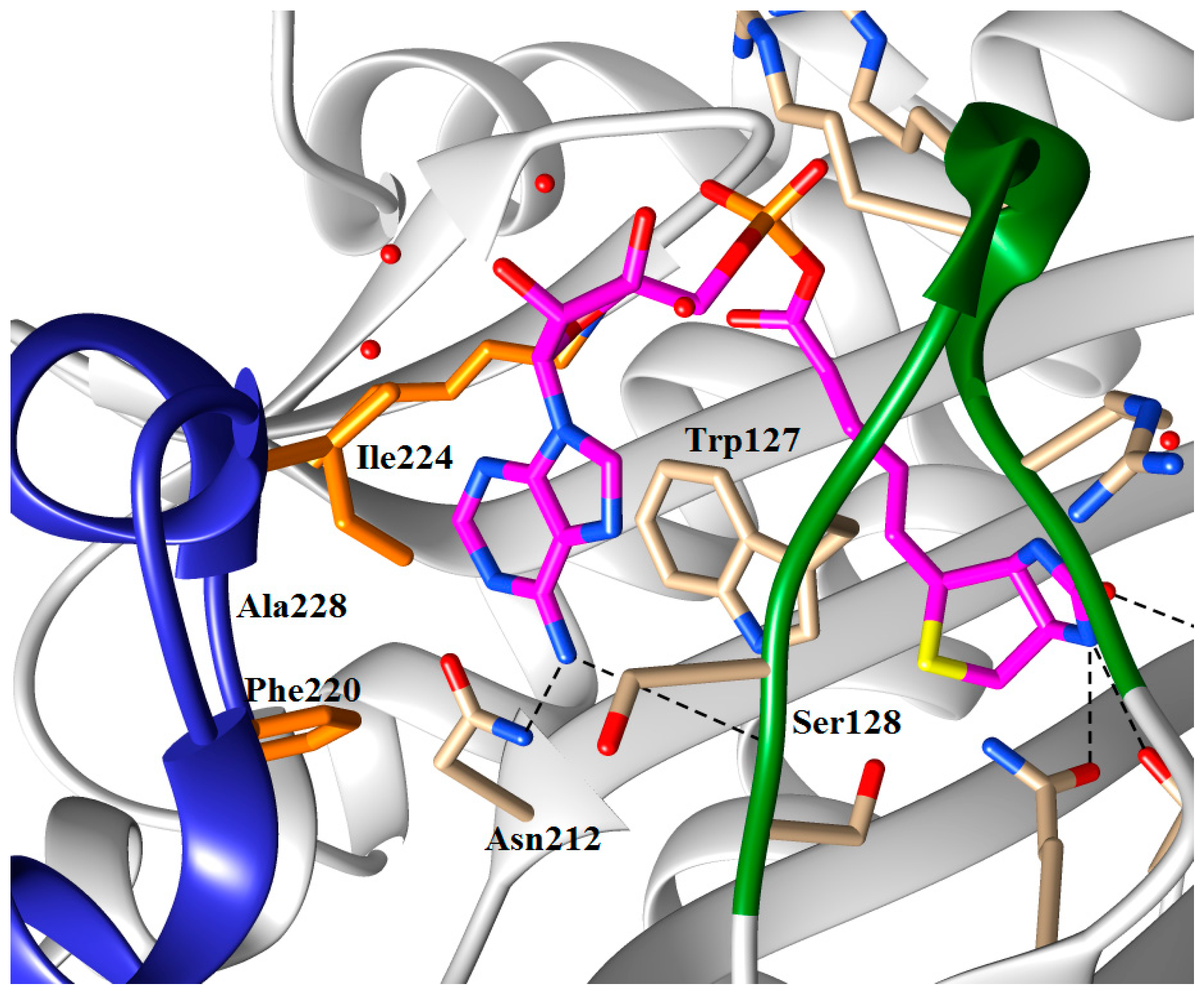
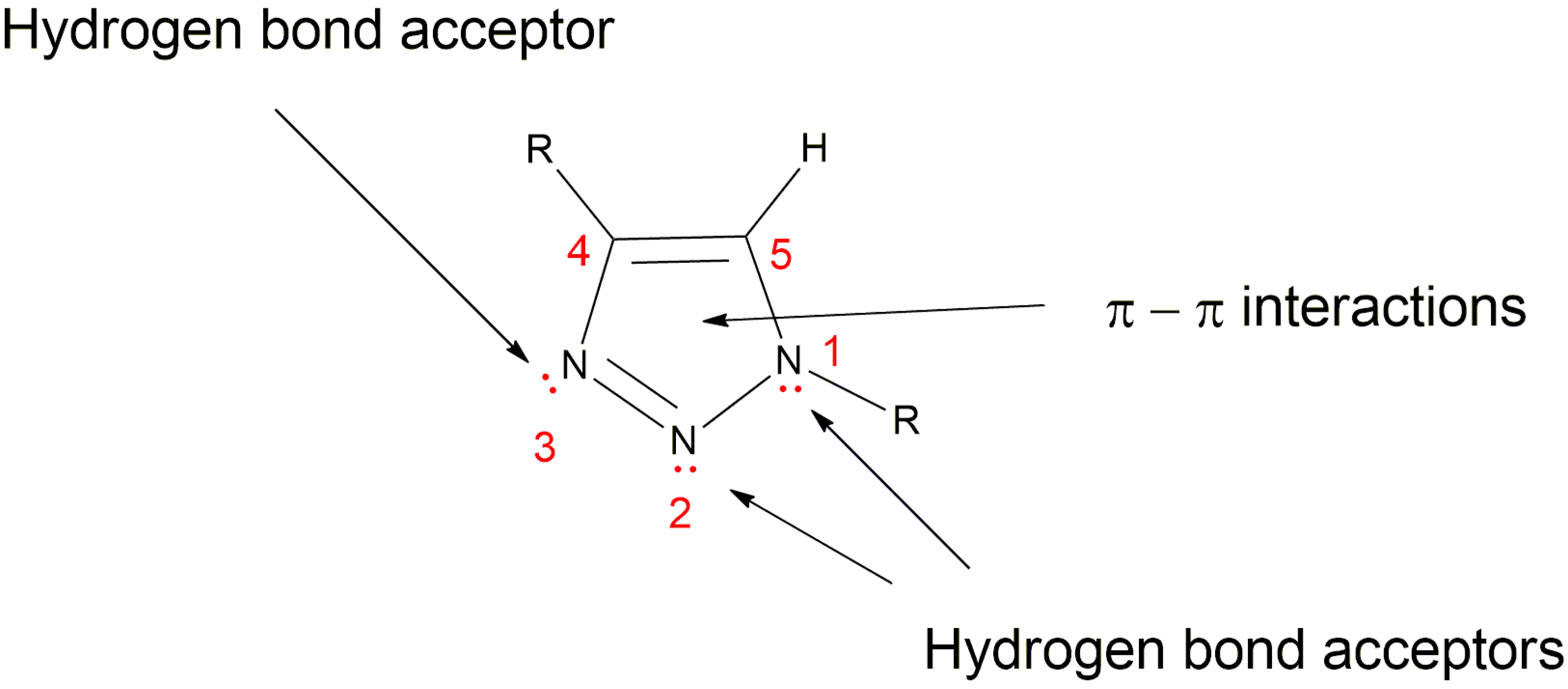
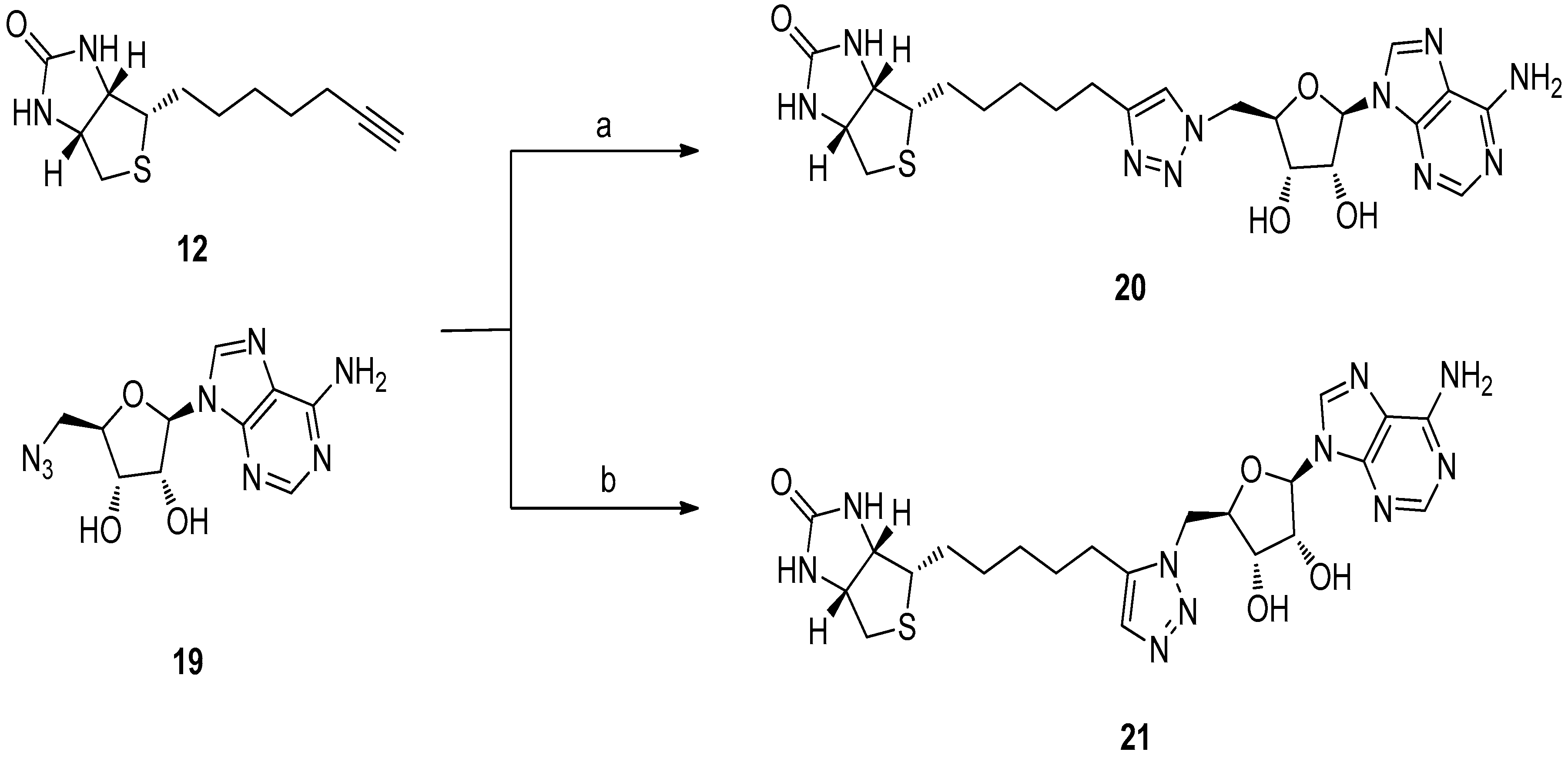
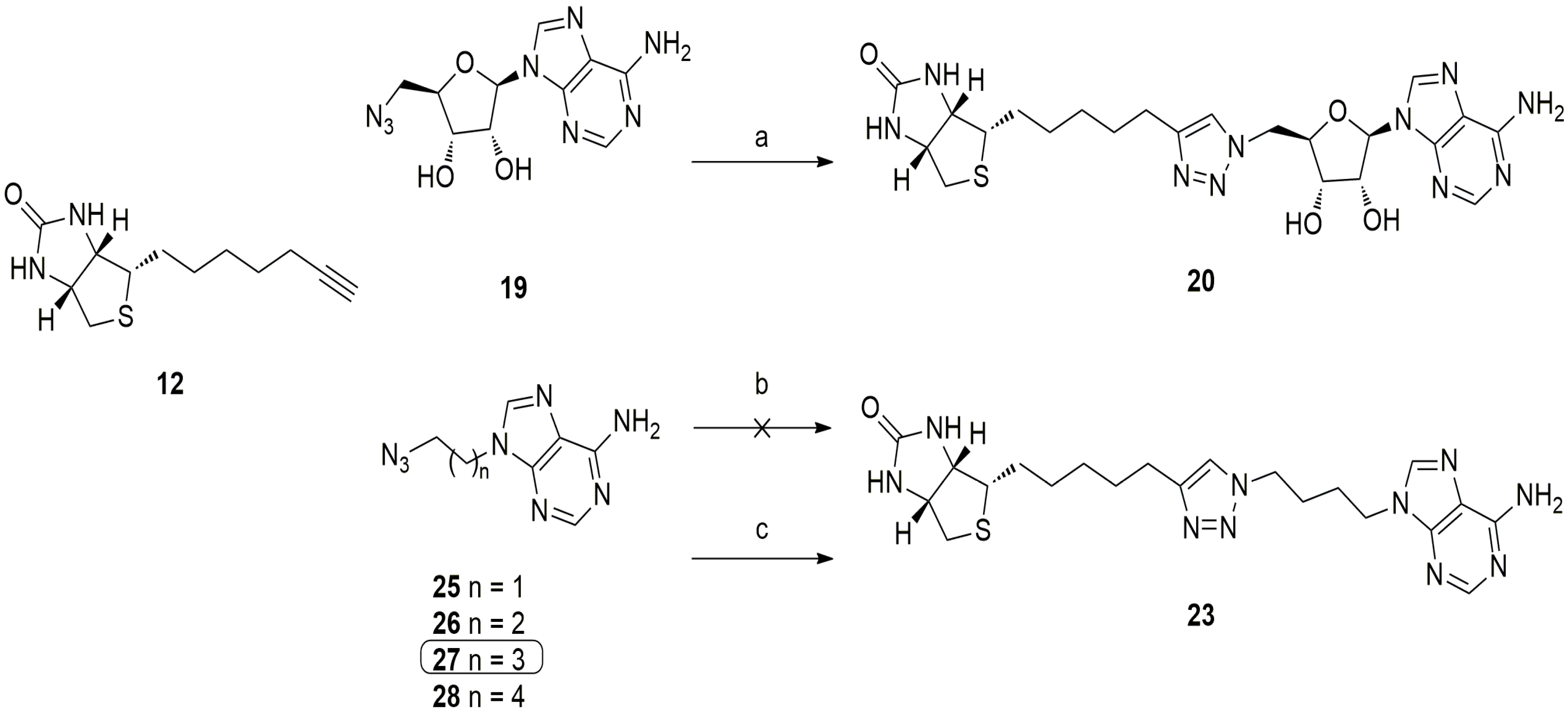
5.3. 1,2,3-Triazole Based Analogues
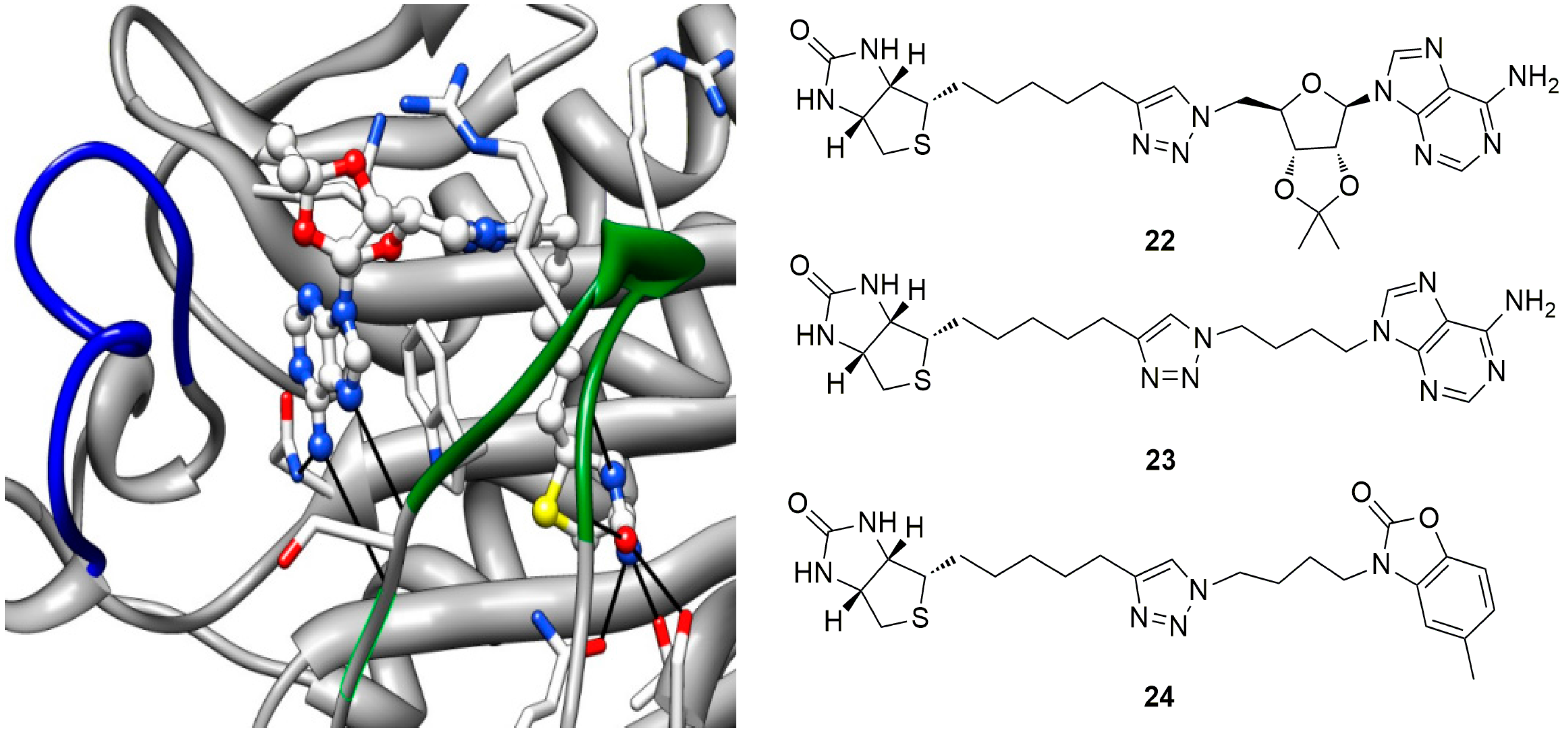
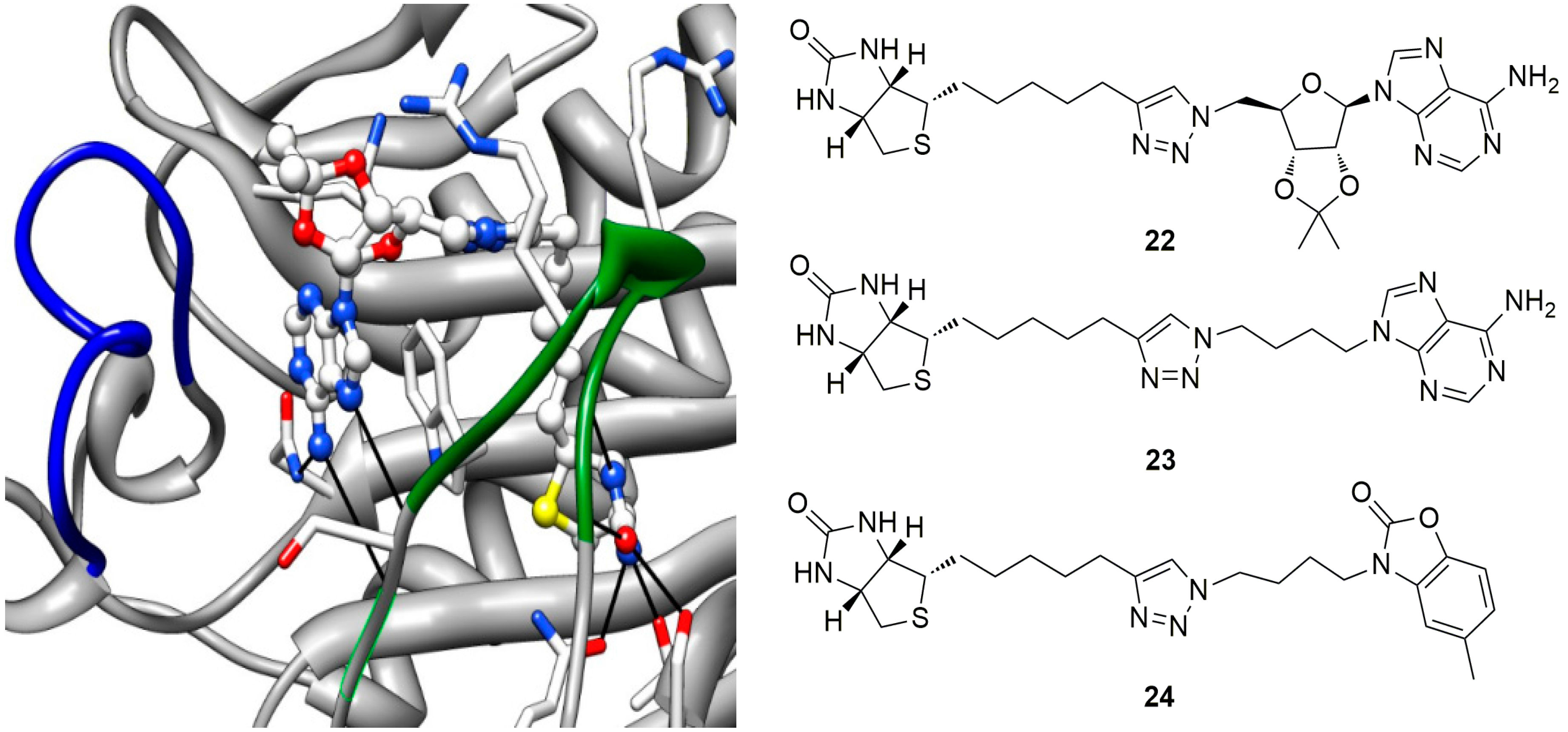
6. In Situ Click Chemistry
7. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACC | Acetyl CoA carboxylase |
| AMP | Adenosine-5′-monophosphate |
| ATP | Adenosine-5′-triphosphate |
| BPL | Biotin protein ligase |
| CuAAC | Copper mediated alkyne azide cycloaddition |
| HPLC | High-performance liquid chromatography |
| HsBPL | Homo sapiens biotin protein ligase |
| Ki | Inhibition constant |
| MRSA | Methicillin resistant S. aureus |
| PC | Pyruvate carboxylase |
| RuAAC | Ruthenium mediated Alkyne Azide cycloaddition |
| SaBPL | Staphylococcus aureus biotin protein ligase |
References
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No eskape! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Antibiotics: Recover the lost art of drug discovery. Nature 2012, 485, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Kallen, A.J.; Mu, Y.; Bulens, S.; Reingold, A.; Petit, S.; Gershman, K.; Ray, S.M.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; et al. Health care-associated invasive MRSA infections, 2005–2008. J. Am. Med. Assoc. 2010, 304, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.; Chronias, A.; Murray, M. Voluntary and mandatory surveillance for methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible S. aureus (MSSA) bacteraemia in England. J. Antimicrob. Chemother. 2009, 64, i11–i17. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J. Healthcare-associated methicillin-resistant Staph aureus (MRSA) control in Australia and New Zealand—2007 Australasian Society for Infectious Diseases (ASID) conference forum convened by healthcare infection control special interest group (HICSIG). Healthc. Infect. 2007, 12, 60–66. [Google Scholar] [CrossRef]
- Klein, E.; Smith, D.; Laxminarayan, R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States. Emerg. Infect. Dis. J. 2007, 13, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N.E.; Turnidge, J.D.; Munckhof, W.J.; Robinson, J.O.; Korman, T.M.; O’Sullivan, M.V.; Anderson, T.L.; Roberts, S.A.; Gao, W.; Christiansen, K.J. Antibiotic choice may not explain poorer outcomes in patients with Staphylococcus aureus bacteremia and high vancomycin minimum inhibitory concentrations. J. Infect. Dis. 2011, 204, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Pendini, N.R.; Bailey, L.M.; Booker, G.W.; Wilce, M.; Wallace, J.C.; Polyak, S.W. Microbial biotin protein ligases aid in understanding holocarboxylase synthetase deficiency. Biochim. Biophys. Acta Prot. Proteom. 2008, 784, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Bagautdinov, B.; Kuroishi, C.; Sugahara, M.; Kunishima, N. Crystal structures of biotin protein ligase from Pyrococcus horikoshii OT3 and its complexes: Structural basis of biotin activation. J. Mol. Biol. 2005, 353, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.W.; Abell, A.D.; Wilce, M.C.J.; Zhang, L.; Booker, G.W. Structure, function and selective inhibition of bacterial acetyl CoA carboxylase. Appl. Microbiol. Biotechnol. 2012, 93, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Arpornsuwan, T.; Carey, K.J.; Stojkoski, C.; Booker, G.W.; Polyak, S.W.; Wallace, J.C. Localization of inhibitory antibodies to the biotin domain of human pyruvate carboxylase. Hybridoma 2012, 31, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Salaemae, W.; Azhar, A.; Booker, G.W.; Polyak, S.W. Biotin biosynthesis in Mycobacterium tuberculosis: Physiology, biochemistry and molecular intervention. Protein Cell 2011, 2, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.C.; Jitrapakdee, S.; Chapman-Smith, A. Pyruvate carboxylase. Int. J. Biochem. Cell Biol. 1998, 30, 1–5. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, R.; Haselbeck, R.J.; Ohlsen, K.L.; Yamamoto, R.T.; Xu, H.; Trawick, J.D.; Wall, D.; Wang, L.; Brown-Driver, V.; Froelich, J.M. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 2002, 43, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.; Beckett, D. Cooperative binding of the Escherichia coli repressor of biotin biosynthesis to the biotin operator sequence. Biochemistry 1993, 32, 9649–9656. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; Mironov, A.A.; Gelfand, M.S. Conservation of the biotin regulon and the BirA regulatory signal in eubacteria and archaea. Genome Res. 2002, 12, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Beckett, D. Biotin sensing at the molecular level. J. Nutr. 2009, 139, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Weaver, L.H.; Brown, P.H.; Beckett, D.; Matthews, B.W. Co-repressor induced order and biotin repressor dimerization: A case for divergent followed by convergent evolution. J. Mol. Biol. 2006, 357, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Smith, A.; Cronan, J.E., Jr. In vivo enzymatic protein biotinylation. Biomol. Eng. 1999, 16, 119–125. [Google Scholar] [CrossRef]
- Bagautdinov, B.; Matsuura, Y.; Bagautdinova, S.; Kunishima, N. Protein biotinylation visualized by a complex structure of biotin protein ligase with a substrate. J. Biol. Chem. 2008, 283, 14739–14750. [Google Scholar] [CrossRef] [PubMed]
- Paparella, A.S.; Soares da Costa, T.P.; Yap, M.Y.; Tieu, W.; Wilce, M.C.; Booker, G.W.; Abell, A.D.; Polyak, S.W. Structure guided design of biotin protein ligase inhibitors for antibiotic discovery. Curr. Top. Med. Chem. 2014, 14, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Mayende, L.; Swift, R.D.; Bailey, L.M.; da Costa, T.P.S.; Wallace, J.C.; Booker, G.W.; Polyak, S.W. A novel molecular mechanism to explain biotin-unresponsive holocarboxylase synthetase deficiency. J. Mol. Med. 2012, 90, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.W.; Chapman-Smith, A.; Brautigan, P.J.; Wallace, J.C. Biotin protein ligase from Saccharomyces cerevisiae: The N-terminal domain is required for complete activity. J. Biol. Chem. 1999, 274, 32847–32854. [Google Scholar] [CrossRef] [PubMed]
- Pendini, N.R.; Bailey, L.M.; Booker, G.W.; Wilce, M.C.; Wallace, J.C.; Polyak, S.W. Biotin protein ligase from Candida albicans: Expression, purification and development of a novel assay. Arch. Biochem. Biophys. 2008, 479, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Campeau, E.; Gravel, R.A. Expression in Escherichia coli of N- and C-terminally deleted human holocarboxylase synthetase influence of the N-terminus on biotinylation and identification of a minimum functional protein. J. Biol. Chem. 2001, 276, 12310–12316. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.P.; Geders, T.W.; Tiwari, D.; Boshoff, H.I.; Sibbald, P.A.; Barry, C.E., 3rd; Schnappinger, D.; Finzel, B.C.; Aldrich, C.C. Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem. Biol. 2011, 18, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Tron, C.M.; McNae, I.W.; Nutley, M.; Clarke, D.J.; Cooper, A.; Walkinshaw, M.D.; Baxter, R.L.; Campopiano, D.J. Structural and functional studies of the biotin protein ligase from Aquifex aeolicus reveal a critical role for a conserved residue in target specificity. J. Mol. Biol. 2009, 387, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Pendini, N.R.; Yap, M.Y.; Polyak, S.W.; Cowieson, N.P.; Abell, A.; Booker, G.W.; Wallace, J.C.; Wilce, J.A.; Wilce, M.C. Structural characterization of Staphylococcus aureus biotin protein ligase and interaction partners: An antibiotic target. Protein Sci. 2013, 22, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Soares da Costa, T.P.; Tieu, W.; Yap, M.Y.; Zvarec, O.; Bell, J.M.; Turnidge, J.D.; Wallace, J.C.; Booker, G.W.; Wilce, M.C.; Abell, A.D. Biotin analogues with antibacterial activity are potent inhibitors of biotin protein ligase. ACS Med. Chem. Lett. 2012, 3, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.; Beckett, D. Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 2000, 9, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Tieu, W.; Soares da Costa, T.P.; Yap, M.Y.; Keeling, K.L.; Wilce, M.C.; Wallace, J.C.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Optimising in situ click chemistry: The screening and identification of biotin protein ligase inhibitors. Chem. Sci. 2013, 4, 3533–3537. [Google Scholar] [CrossRef]
- Naganathan, S.; Beckett, D. Nucleation of an allosteric response via ligand-induced loop folding. J. Mol. Biol. 2007, 373, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Rozwarski, D.A.; Vilchèze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Smith, A.; Cronan, J.E. The enzymatic biotinylation of proteins: A post-translational modification of exceptional specificity. Trends Biochem. Sci. 1999, 24, 359–363. [Google Scholar] [CrossRef]
- Brown, P.H.; Cronan, J.E.; Grøtli, M.; Beckett, D. The biotin repressor: Modulation of allostery by corepressor analogs. J. Mol. Biol. 2004, 337, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Tieu, W.; Polyak, S.W.; Paparella, A.S.; Yap, M.Y.; Soares da Costa, T.P.; Ng, B.; Wang, G.; Lumb, R.; Bell, J.M.; Turnidge, J.D. Improved synthesis of biotinol-5′-AMP: Implications for antibacterial discovery. ACS Med. Chem. Lett. 2014, 6, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Sittiwong, W.; Cordonier, E.L.; Zempleni, J.; Dussault, P.H. Β-keto and β-hydroxyphosphonate analogs of biotin-5′-AMP are inhibitors of holocarboxylase synthetase. Bioorg. Med. Chem. Lett. 2014, 24, 5568–5571. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.H.; Beckett, D. Use of binding enthalpy to drive an allosteric transition. Biochemistry 2005, 44, 3112–3121. [Google Scholar] [CrossRef] [PubMed]
- Soares da Costa, T.P.; Tieu, W.; Yap, M.Y.; Pendini, N.R.; Polyak, S.W.; Sejer Pedersen, D.; Morona, R.; Turnidge, J.D.; Wallace, J.C.; Wilce, M.C.; et al. Selective inhibition of biotin protein ligase from Staphylococcus aureus. J. Biol. Chem. 2012, 287, 17823–17832. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002, 114, 2708–2711. [Google Scholar] [CrossRef]
- Mamidyala, S.K.; Finn, M.G. In situ click chemistry: Probing the binding landscapes of biological molecules. Chem. Soc. Rev. 2010, 39, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, K.B.; Manetsch, R. In situ click chemistry: A powerful means for lead discovery. Expert Opin. Drug Discov. 2006, 1, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical-biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Krasiński, A.; Radić, Z.; Manetsch, R.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In situ selection of lead compounds by click chemistry: Target-guided optimization of acetylcholinesterase inhibitors. J. Am. Chem. Soc. 2005, 127, 6686–6692. [Google Scholar] [CrossRef] [PubMed]
- Mocharla, V.P.; Colasson, B.; Lee, L.V.; Röper, S.; Sharpless, K.B.; Wong, C.-H.; Kolb, H.C. In situ click chemistry: Enzyme-generated inhibitors of carbonic anhydrase II. Angew. Chem. Int. Ed. 2005, 44, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Sunazuka, T.; Sugawara, A.; Endo, A.; Iguchi, K.; Yamamoto, T.; Ui, H.; Shiomi, K.; Watanabe, T.; Sharpless, K.B.; et al. Chitinase inhibitors: Extraction of the active framework from natural argifin and use of in situ click chemistry. J. Antibiot. 2009, 62, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.B.; Mensah, L.M.; Doyle, M.L.; Broom, N.J.P.; Osbourne, N.; Forrest, A.K.; Richardson, C.M.; O'Hanlon, P.J.; Pope, A.J. Rational design of femtomolar inhibitors of isoleucyl tRNA synthetase from a binding model for pseudomonic acid-A. Biochemistry 2000, 39, 6003–6011. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Richardson, C.M.; Mensah, L.M.; O’Hanlon, P.J.; Osborne, N.F.; Pope, A.J.; Walker, G. Molecular recognition of tyrosinyl adenylate analogues by prokaryotic tyrosyl tRNA synthetases. Bioorg. Med. Chem. 1999, 7, 2473–2485. [Google Scholar] [CrossRef]
- Somu, R.V.; Boshoff, H.; Qiao, C.; Bennett, E.M.; Barry, C.E.; Aldrich, C.C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2005, 49, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Ferreras, J.A.; Ryu, J.-S.; di Lello, F.; Tan, D.S.; Quadri, L.E.N. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005, 1, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Tuck, K.L.; Saldanha, S.A.; Birch, L.M.; Smith, A.G.; Abell, C. The design and synthesis of inhibitors of pantothenate synthetase. Org. Biomol. Chem. 2006, 4, 3598–3610. [Google Scholar] [CrossRef] [PubMed]
- Ciulli, A.; Scott, D.E.; Ando, M.; Reyes, F.; Saldanha, S.A.; Tuck, K.L.; Chirgadze, D.Y.; Blundell, T.L.; Abell, C. Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. Chem. Bio. Chem. 2008, 9, 2606–2611. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, C.; Dennis, E.G.; Booker, G.W.; Polyak, S.W.; Boss, P.K.; Davies, C. A novel tool for studying auxin-metabolism: The inhibition of grapevine indole-3-acetic acid-amido synthetases by a reaction intermediate analogue. PLoS ONE 2012, 7, e37632. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
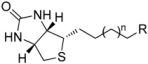 | n | R | SaBPL Ki (μM) | EcBPL Ki (μM) | HsBPL Ki (μM) |
|---|---|---|---|---|---|
| 7 | 2 | OH | 3.4 | 4.0 | 9.0 |
| 8 | 3 | OH | >20 | >20 | >20 |
| 9 | 1 | CH3 | 0.05 | 1.1 | 0.1 |
| 10 | 2 | CH3 | 0.5 | 7.3 | 6.4 |
| 11 | 1 | C≡C | 0.08 | 0.9 | 0.2 |
| 12 | 2 | C≡C | 0.3 | 7.3 | 3.5 |
| 13 | 3 | C≡C | 2.4 | 20 | 12 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, J.; Paparella, A.S.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Biotin Protein Ligase Is a Target for New Antibacterials. Antibiotics 2016, 5, 26. https://doi.org/10.3390/antibiotics5030026
Feng J, Paparella AS, Booker GW, Polyak SW, Abell AD. Biotin Protein Ligase Is a Target for New Antibacterials. Antibiotics. 2016; 5(3):26. https://doi.org/10.3390/antibiotics5030026
Chicago/Turabian StyleFeng, Jiage, Ashleigh S. Paparella, Grant W. Booker, Steven W. Polyak, and Andrew D. Abell. 2016. "Biotin Protein Ligase Is a Target for New Antibacterials" Antibiotics 5, no. 3: 26. https://doi.org/10.3390/antibiotics5030026
APA StyleFeng, J., Paparella, A. S., Booker, G. W., Polyak, S. W., & Abell, A. D. (2016). Biotin Protein Ligase Is a Target for New Antibacterials. Antibiotics, 5(3), 26. https://doi.org/10.3390/antibiotics5030026