
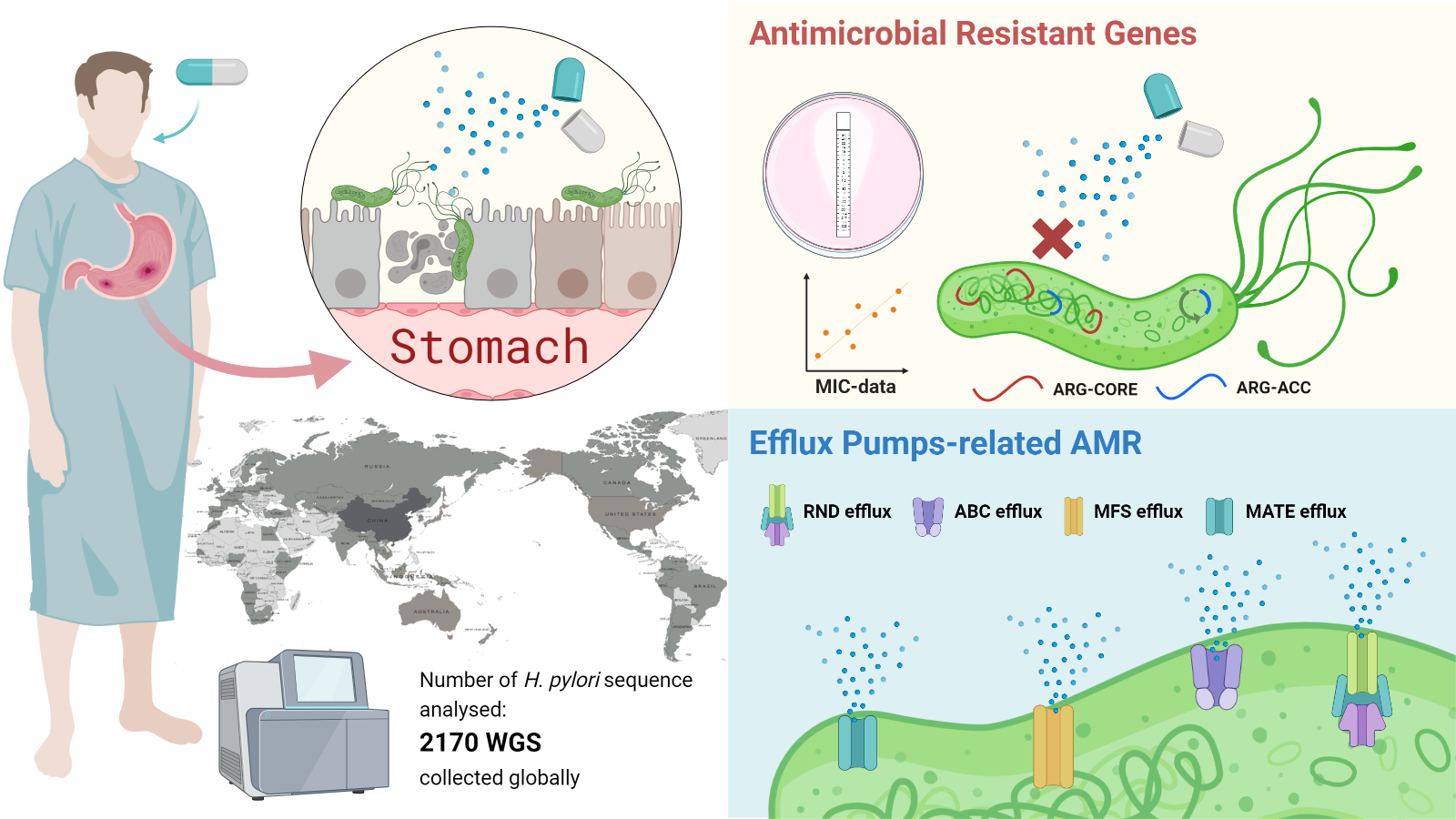
Global Antimicrobial Resistance Gene Study of Helicobacter pylori: Comparison of Detection Tools, ARG and Efflux Pump Gene Analysis, Worldwide Epidemiological Distribution, and Information Related to the Antimicrobial-Resistant Phenotype

 ,
,  , , , , ,
, , , , ,  and
and 
Abstract

1. Introduction
2. Results
2.1. ARG Detection Tools and Comparison of Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Method | Database | Provide Option for Mutation Detection | H. pylori ARG is Included | Database Version, Year Updated | Total Number of Genes Found in 2170 Strains | Total Number of Strains with ARG | Total ARG Name Found | Total ARG Class Found | Parameter Used |
|---|---|---|---|---|---|---|---|---|---|---|
| ABRICATE v1.0.1 [14] | BLAST-matches | ARG-ANNOT [15] | No | Not yet | v5, 2019 | 5 | 5 | 3 | 3 | Minimum coverage and identity of 90 |
| CARD [16,17] | No | Yes, but limited | March 2020 update | 2166 | 2161 | 4 | 4 | |||
| MEGARes [18] | No | Not yet | v2.0, 2020 | 2162 | 2159 | 3 | 3 | |||
| ResFinder [19] | No | Yes | 2019 | 5 | 5 | 3 | 3 | |||
| ResFinder v4.2.3 [19] | The aligners KMA and BLAST-matches | ResFinder and DisinFinder [19] | Yes | Yes, but limited | 2022 | 5 | 5 | 3 | 3 | Minimum coverage and identity of 90 |
| The Resistance Gene Identifier (RGI) v6.0.1 [17] | Homology and SNP models | CARD [16,17,34] | Yes | Yes, but limited | v3.2.5, 2022 | 4328 | 2170 | 7 | 4 | Default parameter and filter to obtain the ‘strict’ and ‘perfect’ results only |
| AMRFinderPlus v3.11.2 [24] | Combination of BLAST-matches, HMM screening, and other improvement methods | Combination of the following: Pathogen Detecton Reference Gene Catalog (https://www.ncbi.nlm.nih.gov/pathogens/refgene/#, accessed on 22 July 2021) Pathogen Detecton Reference HMM Catalog (https://www.ncbi.nlm.nih.gov/pathogens/hmm/, accessed on 22 July 2021) Bacterial Antimicrobial Resistance Reference Gene Database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047, accessed on 22 July 2021) NCBIfam-AMRFinder (https://ftp.ncbi.nlm.nih.gov/hmm/NCBIfam-AMRFinder/latest/, accessed on 22 July 2021) | Yes | Not yet | v2022-12-19.1, 2022 | 2176 | 2170 | 5 | 4 | Default parameter, add’—plus’ flag, and three files typed as input (.fna, .faa, and .gff) |
| HMMER v3.3.2 [25] | HMM screening | HMM ResFam Profile [26], which was trained using ARG protein sequence from CARD, the Lactamase Engineering Database (LacED), and Jacoby and Bush’s collection of curated beta-lactamase proteins. | Possible | Not yet | v1.2.2, 2018 | 15,056 | 2170 | 7 | 10 | e-value ≤ 1 × 10−10 and bit score ≥ 250 after hand curation |
2.2. Pros and Cons of ARG Detection Tools
2.3. Curation of ARG Detection Results Is Necessary
2.4. Summary of Global ARG Detection Results from All Tools and Databases
2.5. Potential Efflux Pumps (EP)-Related AMR and MDR in H. pylori
| Gene Name (According to the Databases) | ARG-CORE (Detected in ≥95% of Total Strains) or ARG-ACC | Antibiotic Target | Resistance Mechanism | Additional Information (Including Gene Description by Prokka or Protein Homologous Name or Another Alternative Name a) | AMR Gene Family (by CARD) | Prevalence in Total Genome Dataset (n = 2170) |
|---|---|---|---|---|---|---|
| ARG detected by tools after curation | ||||||
| abaF | ARG-ACC | phosphonic acid antiobiotic | MFS efflux | Major Facilitator Superfamily (MFS) antibiotic efflux pump; fosfomycin resistance protein AbaF | Major Facilitator Superfamily (MFS) antibiotic efflux pump | 1.01% (22/2170) |
| adeF | ARG-ACC | MDR (e.g., tetracycline, fluoroquinolone) | RND efflux | - | resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.05% (1/2170) |
| APH(3)-IIIa | ARG-ACC | aminoglycoside | antibiotic inactivation | - | APH(3′) | 0.09% (2/2170) |
| arlR | ARG-CORE | MDR (e.g., fluoroquinolone, disinfecting agents, and antiseptics) | MFS efflux | Response regulator ArlR | Major Facilitator Superfamily (MFS) antibiotic efflux pump | 99.91% (2168/2170) |
| baeR | ARG-ACC | MDR (e.g., aminocoumarin antibiotic, aminoglycoside antibiotic) | RND efflux | Transcriptional regulatory protein BaeR | resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.05% (1/2170) |
| bcr-1 | ARG-ACC | Bicyclomycin-like antibiotic (it is also possible as MDR) | MFS efflux | Bicyclomycin resistance protein | Major Facilitator Superfamily (MFS) antibiotic efflux pump | 0.05% (1/2170) |
| TEM-116 | ARG-ACC | MDR | antibiotic inactivation | - | TEM beta-lactamase | 0.09% (2/2170) |
| carA | ARG-CORE | macrolide | antibiotic target protection | - | Miscellaneous ABC-F subfamily ATP-binding cassette ribosomal protection proteins | 99.82% (2166/2170) |
| cnrB | ARG-ACC | metal | other | Nickel and cobalt resistance protein CnrB | 4-hydroxy-tetrahydrodipicolinate synthase | 0.18% (4/2170) |
| copA | ARG-CORE | metal | ABC efflux | Copper-exporting P-type ATPase | - | 99.95% (2169/2170) |
| czcA | ARG-CORE | metal | RND efflux | Cobalt-zinc-cadmium resistance protein CzcA | - | 99.82% (2166/2170) |
| czcB | ARG-ACC | metal | RND efflux | Cobalt-zinc-cadmium resistance protein CzcB | - | 74.65% (1620/2170) |
| ebrB | ARG-ACC | MDR (e.g., carbapenem, cephalosporin, penam) | antibiotic inactivation | - | Multidrug resistance protein EbrB | 0.05% (1/2170) |
| hp1181 | ARG-CORE | MDR (e.g., tetracycline, nitroimidazole, fluoroquinolone) | MFS efflux | in Prokka, can be detected as yfcJ; putative MFS-type transporter YfcJ | Major Facilitator Superfamily (MFS) antibiotic efflux pump | 99.95% (2169/2170) |
| lmrA | ARG-ACC | MDR (e.g., lincosamide antibiotic) | ABC efflux | Multidrug resistance ABC transporter ATP-binding and permease protein | - | 0.28% (6/2170) |
| lnuA | ARG-ACC | lincosamide | antibiotic inactivation | linA | lincosamide nucleotidyltransferase (LNU) | 0.05% (1/2170) |
| macB | ARG-ACC | macrolide | ABC efflux | pvdT | ATP-binding cassette (ABC) antibiotic efflux pump | 0.09% (2/2170) |
| mdtA | ARG-ACC | MDR (e.g., aminocoumarin) | RND efflux | yegM; Multidrug resistance protein MdtA | resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.09% (2/2170) |
| mdtB | ARG-ACC | MDR (e.g., aminocoumarin) | RND efflux | yegN; Multidrug resistance protein MdtB | resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.28% (6/2170) |
| mdtC | ARG-CORE | MDR (e.g., aminocoumarin) | RND efflux | yegO; Multidrug resistance protein MdtC | resistance-nodulation-cell division (RND) antibiotic efflux pump | 99.54% (2160/2170) |
| mdtH | ARG-ACC | MDR (e.g., fluoroquinolone antibiotic) | MFS efflux | yceL; Multidrug resistance protein MdtH | resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.32% (7/2170) |
| mdtK | ARG-ACC | MDR (e.g., aminocoumarin) | MATE efflux | norE; norM; ydhE; Multidrug resistance protein MdtK | resistance-nodulation-cell division (RND) antibiotic efflux pump | 1.15% (25/2170) |
| mdtL | ARG-ACC | MDR (e.g., aminocoumarin) | MFS efflux | yidY; Multidrug resistance protein MdtL | resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.09% (2/2170) |
| mecA | ARG-ACC | penam | antibiotic target replacement | Adapter protein MecA | Methicillin-resistant PBP2 | 0.05% (1/2170) |
| mepA | ARG-CORE | MDR (e.g., tetracycline, glycylcycline) | MATE efflux | Multidrug export protein MepA | multidrug and toxic compound extrusion (MATE) transporter | 99.63% (2162/2170) |
| MFS_efflux | ARG-CORE | MDR | ABC efflux | HP1120 (COG1131); Multidrug efflux system ATP-binding protein | - | 99.68% (2163/2170) |
| msbA | ARG-CORE | nitroimidazole | ABC efflux | Lipid A export ATP-binding/permease protein MsbA | ATP-binding cassette (ABC) antibiotic efflux pump | 99.82% (2166/2170) |
| patA | ARG-ACC | fluoroquinolone | ABC efflux | Peptidoglycan O-acetyltransferase | ATP-binding cassette (ABC) antibiotic efflux pump | 13.69% (297/2170) |
| ramA | ARG-ACC | MDR (e.g., tetracycline, rifamycin, phenicol, carbapenem, penem, penam, cephalosporin, cephamycin, glycylcycline, disinfecting agents and antiseptics, monobactam, fluoroquinolone) | Other (reduced permeability to antibiotic, based on CARD, it can actually be considered as an efflux pump complex or subunit conferring antibiotic resistance) | RamA (resistance antibiotic multiple) is a positive regulator of AcrAB-TolC. | General Bacterial Porin with reduced permeability to beta-lactams, resistance-nodulation-cell division (RND) antibiotic efflux pump | 0.09% (2/2170) |
| rsmA | ARG-CORE | MDR (e.g., diaminopyrimidine, phenicol, fluoroquinolone) | RND efflux | csrA; Ribosomal RNA small subunit methyltransferase A | resistance-nodulation-cell division (RND) antibiotic efflux pump | 99.91% (2168/2170) |
| salB | ARG-ACC | MDR (e.g., streptogramin, lincosamide, pleuromutilin, streptogramin A) | antibiotic target protection | - | sal-type ABC-F protein | 0.05% (1/2170) |
| salC | ARG-ACC | MDR (e.g., streptogramin, lincosamide, pleuromutilin, streptogramin A) | antibiotic target protection | - | sal-type ABC-F protein | 1.11% (24/2170) |
| srmB | ARG-ACC | macrolide | antibiotic target protection | ATP-dependent RNA helicase SrmB | Miscellaneous ABC-F subfamily ATP-binding cassette ribosomal protection proteins | 0.97% (21/2170) |
| vanT gene in vanG cluster | ARG-ACC | glycopeptide | antibiotic target alteration | - | glycopeptide resistance gene cluster, vanT | 75.62% (1641/2170) |
| vanTr gene in vanL cluster | ARG-ACC | glycopeptide | antibiotic target alteration | - | glycopeptide resistance gene cluster, vanT | 24.33% (528/2170) |
| yajC | ARG-ACC | MDR (e.g., tetracycline, disinfecting agents and antiseptics, glycylcycline, rifamycin, cephalosporin, penam, phenicol, fluoroquinolone, glycopeptide, oxazolidinone) | RND efflux | Sec translocon accessory complex subunit YajC | resistance-nodulation-cell division (RND) antibiotic efflux pump | 47.05% (1021/2170) |
| ybhF | ARG-ACC | MDR | ABC efflux | putative multidrug ABC-transporter ATP-binding protein YbhF | - | 0.05% (1/2170) |
| ybhR | ARG-ACC | MDR | ABC efflux | putative multidrug ABC transporter permease YbhR | - | 7.33% (159/2170) |
| ybhS | ARG-ACC | MDR | ABC efflux | putative multidrug ABC transporter permease YbhS | - | 57.47% (1247/2170) |
| yheH | ARG-ACC | MDR | ABC efflux | putative multidrug resistance ABC transporter ATP-binding/permease protein YheH; bmrA | - | 1.29% (28/2170) |
| MATE_efflux_yeeO | ARG-CORE | MDR | MATE efflux | Can be shown as yeeO by Prokka annotation | - | 0.05% (1/2170) |
| ACR_tran | ARG-CORE | MDR | ABC efflux | Can be shown as several different names (mdtcA, czcA, cusA, or bepA) by Prokka annotation. Should be manually curated to differ from the above genes. | - | 99.91% (2168/2170) |
| EPs related to AMR a,b (Locus tag based on H. pylori strain 26695 [NC_000915.1]. Please refer to Table S1 for more information for the reference and gene characteristics.) | ||||||
| HP0600 | ARG-ACC | MDR (e.g., metronidazole, levofloxacin) | ABC efflux | spaB | - | 62.53% (1357/2170) |
| HP0605 | ARG-CORE | MDR (bilesalt, cefotaxime, ceragenin, clindamycine, clarithromycin, erythromycin, ethidium bromide (EtBr), novobiocin, metal ion, nickel, sodium deoxycholate, tetracycline) | RND efflux | hefA; efflux RND transporter outer membane subunit HefA | - | 99.68% (2163/2170) |
| HP0606 | ARG-CORE | MDR (bilesalt, cefotaxime, ceragenin, clindamycine, clarithromycin, erythromycin, EtBr, novobiocin, metal ion, nickel, sodium deoxycholate, tetracycline) | RND efflux | hefB (alternative name: acrA or mtrC); efflux RND transporter periplasmic adaptor subunit HefB | - | 99.68% (2163/2170) |
| HP0607 | ARG-CORE | MDR (bilesalt, cefotaxime, ceragenin, clindamycine, clarithromycin, erythromycin, EtBr, novobiocin, metal ion, nickel, sodium deoxycholate, tetracycline) | RND efflux | hefC (alternative name: acrB); efflux RND transporter permease subunit HefC | - | 99.63% (2162/2170) |
| HP0759 | ARG-CORE | MDR | MATE efflux | conserved hypothetical integral membrane protein; MATE family efflux transporter | - | 99.77% (2165/2170) |
| HP0791 | ARG-CORE | metal (cadmium, zinc) | ABC efflux | cadA; heavy-metal translocating P-type ATPase | - | 99.59% (2161/2170) |
| HP0969 | ARG-CORE | MDR (e.g., cadmium, metronidazole, nickel, zinc) | RND efflux | hefF (alternative name: czcA1 or cznA) | - | 99.86% (2167/2170) |
| HP0970 | ARG-CORE | MDR (e.g., cadmium, metronidazole, nickel, zinc) | RND efflux | hefE (alternative name: czcB1 or cznB); efflux RND transporter periplasmic adaptor subunit | - | 99.77% (2165/2170) |
| HP0971 | ARG-CORE | MDR (e.g., cadmium, metronidazole, nickel, zinc) | RND efflux | hefD (alternative name: cznC) Note: HefFDE is a homolog of MexA | - | 99.77% (2165/2170) |
| HP1072 | ARG-CORE | copper | ABC efflux | copA | - | 99.72% (2164/2170) |
| HP1082 | ARG-CORE | MDR (erythromycin, etbr, novobiocin, rifampin, and lipopolysaccharide) | ABC efflux | msbA | - | 99.63% (2162/2170) |
| HP1091 | ARG-CORE | - | MFS efflux | kgtP | - | 98.57% (2139/2170) |
| HP1120 | ARG-CORE | MDR | ABC efflux | CcmA; Multidrug efflux system ATP-binding protein (NP_208012.1); ABC-type multidrug transport system, ATPase component (COG1131) | - | 99.68% (2163/2170) |
| HP1165 | ARG-CORE | tetracycline | MFS efflux | tetA | - | 99.49% (2159/2170) |
| HP1174 | ARG-CORE | D Glactose (non-drug) | MFS efflux | gluP | - | 99.68% (2163/2170) |
| HP1181 | ARG-CORE | MDR (e.g., tetracycline, nitroimidazole, fluoroquinolone) | MFS efflux | Multidrug efflux transporter | - | 99.95% (2169/2170) |
| HP1184 | ARG-CORE | norfloxacin and ethidium | MATE efflux | NorM; HP1184 family multidrug efflux MATE transporter, conserved hypothetical integral membrane protein; MatE Polysacc_synt_C | - | 99.77% (2165/2170) |
| HP1206 | ARG-ACC | MDR (possibly related to metronidazole and levofloxacin resistance) | ABC efflux | hetA; multidrug resistance protein (HetA) | - | 94.70% (2050/2170) |
| HP1250 | ARG-CORE | - | ABC efflux | Csd5 | - | 96.96% (2104/2170) |
| HP1251 | ARG-CORE | - | ABC efflux | oligopeptide ABC transporter, permease protein (OppB); microcin C transport system permease protein | - | 99.72% (2164/2170) |
| HP1252 | ARG-CORE | - | ABC efflux | OppA | - | 99.59% (2161/2170) |
| HP1327 | ARG-CORE | metal (copper, cobalt, zinc cadmium ion) | RND efflux | hefG (alternative name: crdB); copper resistance outer membrane protein CrdB | - | 99.40% (2157/2170) |
| HP1328 | ARG-CORE | metal (copper, cobalt, zinc cadmium ion) | RND efflux | hefH (alternative name: czcB2); efflux RND transporter periplasmic adaptor subunit | - | 99.63% (2162/2170) |
| HP1329 | ARG-CORE | metal (copper, cobalt, zinc cadmium ion) | RND efflux | hefI (alternative name: czcA2, cusA) | - | 99.31% (2155/2170) |
| HP1487 | ARG-CORE | MDR (e.g., novobiocin, deoxycholate, EtBr resistance) | RND efflux (or ABC efflux) | ABC-2 type transport system permease protein | - | 99.59% (2161/2170) |
| HP1488 | ARG-CORE | MDR (e.g., novobiocin, deoxycholate, EtBr resistance) | RND efflux (or ABC efflux) | Membrane-fusion protein HlyD family secretion protein | - | 99.59% (2161/2170) |
| HP1489 | ARG-CORE | MDR (e.g., novobiocin, deoxycholate, EtBr resistance) | RND efflux (or ABC efflux) | TolC-like outer membrane efflux protein | - | 99.59% (2161/2170) |
| HP1503 | ARG-CORE | metal | ABC efflux | cation-transporting ATPase, P-type (copA), P-type Cu+ transporter | - | 99.77% (2165/2170) |
| HP1561 | ARG-CORE | metal (nickel, copper) | ABC efflux | Iron(III) ABC transporter, periplasmic iron-binding protein (ceuE), iron complex transport system substrate-binding protein | - | 98.02% (2127/2170) |
2.6. Global Geographic and Population Distribution of the ‘Set of ARG Commonly Found in the Accessory Genome of H. pylori’ (ARG-ACC)
2.7. Association between ARG and Antimicrobial-Resistant Phenotype in H. pylori
3. Discussion
4. Materials and Methods
4.1. WGS Dataset Collection
4.2. WGS Annotation and H. pylori Population Construction
4.3. ARG and EP Detection
- ABRICATE v1.0.1 (BLAST-matches-based method that applies BLASTN) [14], against several ARG databases:Parameter settings: minimum coverage and identity, 90.
- ResFinder v4.2.3 (BLAST-matches-based method) [19], against the ResFinder database (last update October 2022).Parameter settings: minimum coverage and identity, 90.
- The Resistance Gene Identifier (RGI) v6.0.1 (homology and SNP models) [17], against the CARD database (v3.2.5, last update 2022) [34].Parameter settings: default, including ‘strict’ and ‘perfect’ only.
- AMRFinderPlus v3.11.2 (combination of BLAST-matches, HMM screening, and other improvements) [24], against a set of databases that combine:
- (a)
- Pathogen Detection Reference Gene Catalog (https://www.ncbi.nlm.nih.gov/pathogens/refgene/#, accessed on 22 July 2021)
- (b)
- Pathogen Detection Reference HMM Catalog (https://www.ncbi.nlm.nih.gov/pathogens/hmm/, accessed on 22 July 2021)
- (c)
- Bacterial Antimicrobial Resistance Reference Gene Database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA313047, accessed on 22 July 2021)
- (d)
- NCBIfam-AMRFinder (https://ftp.ncbi.nlm.nih.gov/hmm/NCBIfam-AMRFinder/latest/, accessed on 22 July 2021)
The aforementioned information regarding databases is derived automatically from ‘AMRFinderFinderPlus’ as the default source. Our access to these database was not manually one by one; rather, we obtained it by querying the ‘AMRFinderFinderPlus’ database as a unified resource (for more information please visit: https://github.com/ncbi/amr/wiki, accessed on 21 December 2022). Parameter settings: default parameter, add ‘—plus’ flag, and three file types as input (.fna, .faa, and .gff). - HMMER v3.3.2 (http://hmmer.org/ (accessed on 21 December 2022) Howard Hughes Medical Institute) (The Hidden Markov model (HMM)-based method), against the ResFam Profile Database [25,26] (v1.2.2, last update 2018).Parameter settings: --incE, 1 × 10−10; -E, 1 × 10−10; bit score ≥100, ≥200, ≥250, ≥300, ≥400, and ≥500. We used the Prokka (.faa) output file to run HMMER. We excluded the results if they contained ‘antibiotic target alteration’ or unclear information. After hand curation against the Prokka annotation results, we excluded genes that should not belong to ARG.
4.4. Curation of ARG Detection Results
4.5. Finding Additional ARG from Prokka
4.6. Protein Model Analysis
4.7. Analysis of the ARG Presence Status with the AMR Phenotype and Detection of New ARG Candidates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander, S.M.; Retnakumar, R.J.; Chouhan, D.; Devi, T.N.B.; Dharmaseelan, S.; Devadas, K.; Thapa, N.; Tamang, J.P.; Lamtha, S.C.; Chattopadhyay, S. Helicobacter pylori in Human Stomach: The Inconsistencies in Clinical Outcomes and the Probable Causes. Front. Microbiol. 2021, 12, 713955. [Google Scholar] [CrossRef] [PubMed]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Alfaray, R.I.; Saruuljavkhlan, B.; Ansari, S.; Fauzia, K.A.; Yamaoka, Y. Review: Epidemiology of Helicobacter pylori Infection. Microbiota Health Dis. 2022, 4, e733. [Google Scholar] [CrossRef]
- Yamaoka, Y. How to eliminate gastric cancer-related death worldwide? Nat. Rev. Clin. Oncol. 2018, 15, 407–408. [Google Scholar] [CrossRef]
- Tshibangu-Kabamba, E.; Yamaoka, Y. Helicobacter pylori infection and antibiotic resistance—From biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Megraud, F.; O’Morain, C.A.; Gisbert, J.P.; Kuipers, E.J.; Axon, A.T.; Bazzoli, F.; Gasbarrini, A.; Atherton, J.; Graham, D.Y.; et al. Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut 2017, 66, 6–30. [Google Scholar] [CrossRef]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of Antibiotic Resistance in Helicobacter pylori: A Systematic Review and Meta-analysis in World Health Organization Regions. Gastroenterology 2018, 155, 1372–1382.e17. [Google Scholar] [CrossRef]
- Fallone, C.A.; Moss, S.F.; Malfertheiner, P. Reconciliation of Recent Helicobacter pylori Treatment Guidelines in a Time of Increasing Resistance to Antibiotics. Gastroenterology 2019, 157, 44–53. [Google Scholar] [CrossRef]
- Gong, Y.; Yuan, Y. Resistance mechanisms of Helicobacter pylori and its dual target precise therapy. Crit. Rev. Microbiol. 2018, 44, 371–392. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Yang, F.; Chi, W.; Ding, L.; Liu, T.; Zhu, F.; Ji, D.; Zhou, J.; Fang, Y.; et al. Antimicrobial resistance patterns and genetic elements associated with the antibiotic resistance of Helicobacter pylori strains from Shanghai. Gut Pathog. 2022, 14, 14. [Google Scholar] [CrossRef]
- Tshibangu-Kabamba, E.; Ngoma-Kisoko, P.J.; Tuan, V.P.; Matsumoto, T.; Akada, J.; Kido, Y.; Tshimpi-Wola, A.; Tshiamala-Kashala, P.; Ahuka-Mundeke, S.; Ngoy, D.M.; et al. Next-Generation Sequencing of the Whole Bacterial Genome for Tracking Molecular Insight into the Broad-Spectrum Antimicrobial Resistance of Helicobacter pylori Clinical Isolates from the Democratic Republic of Congo. Microorganisms 2020, 8, 887. [Google Scholar] [CrossRef]
- Mehrotra, T.; Devi, T.B.; Kumar, S.; Talukdar, D.; Karmakar, S.P.; Kothidar, A.; Verma, J.; Kumari, S.; Alexander, S.M.; Retnakumar, R.J.; et al. Antimicrobial resistance and virulence in Helicobacter pylori: Genomic insights. Genomics 2021, 113, 3951–3966. [Google Scholar] [CrossRef]
- Sukri, A.; Hanafiah, A.; Yusoff, H.; Shamsul Nizam, N.A.; Nameyrra, Z.; Wong, Z.; Raja Ali, R.A. Multidrug-Resistant Helicobacter pylori Strains: A Five-Year Surveillance Study and Its Genome Characteristics. Antibiotics 2022, 11, 1391. [Google Scholar] [CrossRef]
- Seemann, T. ABRIcate. Available online: https://github.com/tseemann/abricate (accessed on 22 July 2021).
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.; Solymosi, N. Review and Comparison of Antimicrobial Resistance Gene Databases. Antibiotics 2022, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Gibson, M.K.; Forsberg, K.J.; Dantas, G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015, 9, 207–216. [Google Scholar] [CrossRef]
- Falsafi, T.; Ehsani, A.; Attaran, B.; Niknam, V. Association of hp1181 and hp1184 Genes With the Active Efflux Phenotype in Multidrug-Resistant Isolates of Helicobacter pylori. Jundishapur. J. Microbiol. 2016, 9, e30726. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Y.; Wang, X.; Fan, J.; Heng, J.; Liu, X.; Feng, W.; Kang, X.; Huang, B.; Liu, J.; et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. USA 2013, 110, 14664–14669. [Google Scholar] [CrossRef]
- Nishino, K.; Yamaguchi, A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J. Bacteriol. 2001, 183, 5803–5812. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Dror, O.; Benyamini, H.; Nussinov, R.; Wolfson, H. MASS: Multiple structural alignment by secondary structures. Bioinformatics 2003, 19 (Suppl. S1), i95–i104. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2022, 51, D690–D699. [Google Scholar] [CrossRef]
- van Amsterdam, K.; Bart, A.; van der Ende, A. A Helicobacter pylori TolC efflux pump confers resistance to metronidazole. Antimicrob. Agents Chemother. 2005, 49, 1477–1482. [Google Scholar] [CrossRef]
- Megraud, F.; Lehours, P. Helicobacter pylori detection and antimicrobial susceptibility testing. Clin. Microbiol. Rev. 2007, 20, 280–322. [Google Scholar] [CrossRef]
- Nielsen, D.; Skovsgaard, T. P-glycoprotein as multidrug transporter: A critical review of current multidrug resistant cell lines. Biochim. Biophys. Acta 1992, 1139, 169–183. [Google Scholar] [CrossRef]
- Bina, J.E.; Alm, R.A.; Uria-Nickelsen, M.; Thomas, S.R.; Trust, T.J.; Hancock, R.E. Helicobacter pylori uptake and efflux: Basis for intrinsic susceptibility to antibiotics in vitro. Antimicrob. Agents Chemother. 2000, 44, 248–254. [Google Scholar] [CrossRef]
- Miyamae, S.; Ueda, O.; Yoshimura, F.; Hwang, J.; Tanaka, Y.; Nikaido, H. A MATE family multidrug efflux transporter pumps out fluoroquinolones in Bacteroides thetaiotaomicron. Antimicrob. Agents Chemother. 2001, 45, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Waidner, B.; Melchers, K.; Ivanov, I.; Loferer, H.; Bensch, K.W.; Kist, M.; Bereswill, S. Identification by RNA profiling and mutational analysis of the novel copper resistance determinants CrdA (HP1326), CrdB (HP1327), and CzcB (HP1328) in Helicobacter pylori. J. Bacteriol. 2002, 184, 6700–6708. [Google Scholar] [CrossRef]
- Yang, S.; Clayton, S.R.; Zechiedrich, E.L. Relative contributions of the AcrAB, MdfA and NorE efflux pumps to quinolone resistance in Escherichia coli. J. Antimicrob. Chemother. 2003, 51, 545–556. [Google Scholar] [CrossRef]
- Cagliero, C.; Mouline, C.; Cloeckaert, A.; Payot, S. Synergy between efflux pump CmeABC and modifications in ribosomal proteins L4 and L22 in conferring macrolide resistance in Campylobacter jejuni and Campylobacter coli. Antimicrob. Agents Chemother. 2006, 50, 3893–3896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dannelly, H.K. Inactivation of the putative tetracycline resistance gene HP1165 in Helicobacter pylori led to loss of inducible tetracycline resistance. Arch. Microbiol. 2006, 185, 255–262. [Google Scholar] [CrossRef]
- Stahler, F.N.; Odenbreit, S.; Haas, R.; Wilrich, J.; Van Vliet, A.H.; Kusters, J.G.; Kist, M.; Bereswill, S. The novel Helicobacter pylori CznABC metal efflux pump is required for cadmium, zinc, and nickel resistance, urease modulation, and gastric colonization. Infect. Immun. 2006, 74, 3845–3852. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Suzuki, H.; Nishizawa, T.; Tsugawa, H.; Muraoka, H.; Saito, Y.; Matsuzaki, J.; Hibi, T. Contribution of efflux pumps to clarithromycin resistance in Helicobacter pylori. J. Gastroenterol. Hepatol. 2010, 25 (Suppl. S1), S75–S79. [Google Scholar] [CrossRef]
- Ge, X.; Cai, Y.; Chen, Z.; Gao, S.; Geng, X.; Li, Y.; Li, Y.; Jia, J.; Sun, Y. Bifunctional Enzyme SpoT Is Involved in Biofilm Formation of Helicobacter pylori with Multidrug Resistance by Upregulating Efflux Pump Hp1174 (gluP). Antimicrob. Agents Chemother. 2018, 62, e00957-18. [Google Scholar] [CrossRef]
- Raj, D.S.; Kumar Kesavan, D.; Muthusamy, N.; Umamaheswari, S. Efflux pumps potential drug targets to circumvent drug Resistance—Multi drug efflux pumps of Helicobacter pylori. Mater. Today Proc. 2021, 45, 2976–2981. [Google Scholar] [CrossRef]
- Trainor, E.A.; Horton, K.E.; Savage, P.B.; Testerman, T.L.; McGee, D.J. Role of the HefC efflux pump in Helicobacter pylori cholesterol-dependent resistance to ceragenins and bile salts. Infect. Immun. 2011, 79, 88–97. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, J.; Im, H.N.; An, D.R.; Lee, M.; Hesek, D.; Mobashery, S.; Kim, J.Y.; Cho, K.; Yoon, H.J.; et al. Structural basis for the recognition of muramyltripeptide by Helicobacter pylori Csd4, a D,L-carboxypeptidase controlling the helical cell shape. Acta Cryst. D Biol. Cryst. 2014, 70, 2800–2812. [Google Scholar] [CrossRef]
- Sycuro, L.K.; Wyckoff, T.J.; Biboy, J.; Born, P.; Pincus, Z.; Vollmer, W.; Salama, N.R. Multiple peptidoglycan modification networks modulate Helicobacter pylori’s cell shape, motility, and colonization potential. PLoS Pathog. 2012, 8, e1002603. [Google Scholar] [CrossRef]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef]
- Zanotti, G.; Cendron, L. Structural and functional aspects of the Helicobacter pylori secretome. World J. Gastroenterol. 2014, 20, 1402–1423. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Gama-Castro, S.; Mackie, A.; Billington, R.; Bonavides-Martinez, C.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Muniz-Rascado, L.; et al. The EcoCyc Database in 2021. Front. Microbiol. 2021, 12, 711077. [Google Scholar] [CrossRef] [PubMed]
- von Wintersdorff, C.J.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.; Wolffs, P.F. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef]
- Phuc, B.H.; Tuan, V.P.; Dung, H.D.Q.; Binh, T.T.; Tung, P.H.; Tri, T.D.; Thuan, N.P.M.; Van Khien, V.; Trang, T.T.H.; Akada, J.; et al. Helicobacter pylori type 4 secretion systems as gastroduodenal disease markers. Sci. Rep. 2021, 11, 4584. [Google Scholar] [CrossRef]
- Delahay, R.M.; Croxall, N.J.; Stephens, A.D. Phylogeographic diversity and mosaicism of the Helicobacter pylori tfs integrative and conjugative elements. Mob. DNA 2018, 9, 5. [Google Scholar] [CrossRef]
- McClain, M.S.; Bryant, K.N.; McDonald, W.H.; Algood, H.M.S.; Cover, T.L. Identification of an Essential LolD-Like Protein in Helicobacter pylori. J. Bacteriol. 2023, 205, e0005223. [Google Scholar] [CrossRef]
- Castledine, M.; Newbury, A.; Lewis, R.; Hacker, C.; Meaden, S.; Buckling, A. Antagonistic Mobile Genetic Elements Can Counteract Each Other’s Effects on Microbial Community Composition. mBio 2023, 14, e00460-23. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.S.; Dwibedy, S.K.; Padhy, I. Polymyxins, the last-resort antibiotics: Mode of action, resistance emergence, and potential solutions. J. Biosci. 2021, 46, 85. [Google Scholar] [CrossRef]
- Tamayo, R.; Choudhury, B.; Septer, A.; Merighi, M.; Carlson, R.; Gunn, J.S. Identification of cptA, a PmrA-regulated locus required for phosphoethanolamine modification of the Salmonella enterica serovar typhimurium lipopolysaccharide core. J. Bacteriol. 2005, 187, 3391–3399. [Google Scholar] [CrossRef]
- Gupta, N.; Behera, D.K.; Prajapati, V.K.; Verma, V.K. A comprehensive approach to discover Toxin-Antitoxin systems from human pathogen Helicobacter pylori: A poison and its antidote encapsulated in the genome. Life Sci. 2022, 288, 120149. [Google Scholar] [CrossRef]
- Yang, Q.E.; Walsh, T.R. Toxin-antitoxin systems and their role in disseminating and maintaining antimicrobial resistance. FEMS Microbiol. Rev. 2017, 41, 343–353. [Google Scholar] [CrossRef]
- Wielders, C.L.; Fluit, A.C.; Brisse, S.; Verhoef, J.; Schmitz, F.J. mecA gene is widely disseminated in Staphylococcus aureus population. J. Clin. Microbiol. 2002, 40, 3970–3975. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Durrant, M.G.; Li, M.M.; Siranosian, B.A.; Montgomery, S.B.; Bhatt, A.S. A Bioinformatic Analysis of Integrative Mobile Genetic Elements Highlights Their Role in Bacterial Adaptation. Cell Host Microbe 2020, 27, 140–153.e149. [Google Scholar] [CrossRef]
- Fauzia, K.A.; Aftab, H.; Tshibangu-Kabamba, E.; Alfaray, R.I.; Saruuljavkhlan, B.; Cimuanga-Mukanya, A.; Matsumoto, T.; Subsomwong, P.; Akada, J.; Miftahussurur, M.; et al. Mutations Related to Antibiotics Resistance in Helicobacter pylori Clinical Isolates from Bangladesh. Antibiotics 2023, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Alba, C.; Blanco, A.; Alarcon, T. Antibiotic resistance in Helicobacter pylori. Curr. Opin. Infect. Dis. 2017, 30, 489–497. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R.; International Nucleotide Sequence Database Collaboration. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.F.; Mohamed, K.; Fan, Y.; Agama Study, G.; Achtman, M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Kanz, C.; Aldebert, P.; Althorpe, N.; Baker, W.; Baldwin, A.; Bates, K.; Browne, P.; van den Broek, A.; Castro, M.; Cochrane, G.; et al. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 2005, 33, D29–D33. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- RefSeq. Assembly Anomalies and Other Reasons a Genome Assembly may be Excluded from RefSeq. Available online: https://www.ncbi.nlm.nih.gov/assembly/help/anomnotrefseq/ (accessed on 18 December 2020).
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Saranathan, R.; Levi, M.H.; Wattam, A.R.; Malek, A.; Asare, E.; Behin, D.S.; Pan, D.H.; Jacobs, W.R., Jr.; Szymczak, W.A. Helicobacter pylori Infections in the Bronx, New York: Surveying Antibiotic Susceptibility and Strain Lineage by Whole-Genome Sequencing. J. Clin. Microbiol. 2020, 58, e01591-19. [Google Scholar] [CrossRef]
- King, Z. Genomic Characterisation and Comparison of Antibiotic Resistant and Sensitive Helicobacter Pylori in New Zealand; Auckland University of Technology: Auckland, New Zealand, 2021. [Google Scholar]
- Tuan, V.P.; Narith, D.; Tshibangu-Kabamba, E.; Dung, H.D.Q.; Viet, P.T.; Sokomoth, S.; Binh, T.T.; Sokhem, S.; Tri, T.D.; Ngov, S.; et al. A Next-Generation Sequencing-Based Approach to Identify Genetic Determinants of Antibiotic Resistance in Cambodian Helicobacter pylori Clinical Isolates. J. Clin. Med. 2019, 8, 858. [Google Scholar] [CrossRef]
- Miftahussurur, M.; Syam, A.F.; Nusi, I.A.; Makmun, D.; Waskito, L.A.; Zein, L.H.; Akil, F.; Uwan, W.B.; Simanjuntak, D.; Wibawa, I.D.; et al. Surveillance of Helicobacter pylori Antibiotic Susceptibility in Indonesia: Different Resistance Types among Regions and with Novel Genetic Mutations. PLoS ONE 2016, 11, e0166199. [Google Scholar] [CrossRef]
- Miftahussurur, M.; Waskito, L.A.; Syam, A.F.; Nusi, I.A.; Siregar, G.; Richardo, M.; Bakry, A.F.; Rezkitha, Y.A.A.; Wibawa, I.D.N.; Yamaoka, Y. Alternative eradication regimens for Helicobacter pylori infection in Indonesian regions with high metronidazole and levofloxacin resistance. Infect. Drug Resist. 2019, 12, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Aftab, H.; Miftahussurur, M.; Subsomwong, P.; Ahmed, F.; Khan, A.K.; Yamaoka, Y. Helicobacter pylori antibiotic susceptibility patterns in Bangladesh: Emerging levofloxacin resistance. J. Infect. Dev. Ctries. 2016, 10, 245–253. [Google Scholar] [CrossRef] [PubMed]




| Gene Annotation (Gene Name or Non-Unique Gene Name) | Total Present in Resistant Strains, pr (pr/r; %) | Total Present in Susceptible Strains, ps (ps/s; %) | Naive p-Value |
|---|---|---|---|
| Clarithromycin (n = 61, r = 35, s = 26) | |||
| hypothetical protein | 10 (10/35; 28.57) | 0 (0/26; 0.00) | 0.003 |
| phosphoethanolamine transferase CptA (cptA_1) | 18 (18/35; 51.43) | 5 (5/26; 19.23) | 0.016 |
| hypothetical protein | 8 (8/35; 22.86) | 0 (0/26; 0.00) | 0.016 |
| hypothetical protein | 7 (7/35; 20.00) | 0 (0/26; 0.00) | 0.017 |
| hypothetical protein | 19 (19/35; 54.29) | 6 (6/26; 23.08) | 0.019 |
| phosphoethanolamine transferase CptA (cptA_2) | 17 (17/35; 48.57) | 5 (5/26; 19.23) | 0.030 |
| hypothetical protein | 11 (11/35; 31.43) | 2 (2/26; 7.69) | 0.030 |
| DNA topoisomerase 1 (topA_2) | 11 (11/35; 31.43) | 2 (2/26; 7.69) | 0.030 |
| hypothetical protein | 9 (9/35; 25.71) | 1 (1/26; 3.85) | 0.034 |
| hypothetical protein | 13 (13/35; 37.14) | 3 (3/26; 11.54) | 0.038 |
| hypothetical protein (recF_1) | 14 (14/35; 40.00) | 4 (4/26; 15.38) | 0.049 |
| Metronidazole (n = 61, r = 43, s = 18) | |||
| lipoprotein-releasing system ATP-binding protein LolD | 37 (37/43; 86.05) | 9 (9/18; 50.00) | 0.007 |
| hypothetical protein | 17 (17/43; 39.53) | 1 (1/18; 5.56) | 0.012 |
| hypothetical protein | 16 (16/43; 37.21) | 1 (1/18; 5.56) | 0.013 |
| trifunctional nucleotide phosphoesterase protein YfkN (yfkN) | 41 (41/43; 95.35) | 13 (13/18; 72.22) | 0.020 |
| hypothetical protein (SIRT5_2) | 10 (10/43; 23.26) | 0 (0/18; 0.00) | 0.026 |
| hypothetical protein | 10 (10/43; 23.26) | 0 (0/18; 0.00) | 0.026 |
| apolipoprotein N-acyltransferase (Int) | 34 (34/43; 79.07) | 9 (9/18; 50.00) | 0.033 |
| hypothetical protein | 20 (20/43; 46.51) | 3 (3/18; 16.67) | 0.042 |
| hypothetical protein (gspA) | 40 (40/43; 93.02) | 13 (13/18; 72.22) | 0.042 |
| hypothetical protein | 9 (9/43; 20.93) | 0 (0/18; 00) | 0.047 |
| hypothetical protein | 9 (9/43; 20.93) | 0 (0/18; 00) | 0.047 |
| hypothetical protein (hsdM) | 9 (9/43; 20.93) | 0 (0/18; 00) | 0.047 |
| Levofloxacin (n = 42, r = 12, s = 30) | |||
| chromosome partition protein Smc (smc) | 4 (4/12; 33.33) | 1 (1/30; 3.33) | 0.018 |
| hypothetical protein | 4 (4/12; 33.33) | 1 (1/30; 3.33) | 0.018 |
| hypothetical protein | 4 (4/12; 33.33) | 1 (1/30; 3.33) | 0.018 |
| hypothetical protein | 4 (4/12; 33.33) | 1 (1/30; 3.33) | 0.018 |
| hypothetical protein | 4 (4/12; 33.33) | 1 (1/30; 3.33) | 0.018 |
| hypothetical protein | 3 (3/12; 25.00) | 0 (0/30; 0.00) | 0.019 |
| transcription-repair-coupling factor (mfd) | 3 (3/12; 25.00) | 0 (0/30; 0.00) | 0.019 |
| peptide deformylase 1 (def) | 3 (3/12; 25.00) | 0 (0/30; 0.00) | 0.019 |
| competence protein ComM (comM) | 3 (3/12; 25.00) | 0 (0/30; 0.00) | 0.019 |
| hypothetical protein | 3 (3/12; 25.00) | 0 (0/30; 0.00) | 0.019 |
| flagellar basal-body rod protein FlgG (flgG_2) | 3 (3/12; 25.00) | 0 (0/30; 0.00) | 0.019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfaray, R.I.; Saruuljavkhlan, B.; Fauzia, K.A.; Torres, R.C.; Thorell, K.; Dewi, S.R.; Kryukov, K.A.; Matsumoto, T.; Akada, J.; Vilaichone, R.-k.; et al. Global Antimicrobial Resistance Gene Study of Helicobacter pylori: Comparison of Detection Tools, ARG and Efflux Pump Gene Analysis, Worldwide Epidemiological Distribution, and Information Related to the Antimicrobial-Resistant Phenotype. Antibiotics 2023, 12, 1118. https://doi.org/10.3390/antibiotics12071118
Alfaray RI, Saruuljavkhlan B, Fauzia KA, Torres RC, Thorell K, Dewi SR, Kryukov KA, Matsumoto T, Akada J, Vilaichone R-k, et al. Global Antimicrobial Resistance Gene Study of Helicobacter pylori: Comparison of Detection Tools, ARG and Efflux Pump Gene Analysis, Worldwide Epidemiological Distribution, and Information Related to the Antimicrobial-Resistant Phenotype. Antibiotics. 2023; 12(7):1118. https://doi.org/10.3390/antibiotics12071118
Chicago/Turabian StyleAlfaray, Ricky Indra, Batsaikhan Saruuljavkhlan, Kartika Afrida Fauzia, Roberto C. Torres, Kaisa Thorell, Selva Rosyta Dewi, Kirill A. Kryukov, Takashi Matsumoto, Junko Akada, Ratha-korn Vilaichone, and et al. 2023. "Global Antimicrobial Resistance Gene Study of Helicobacter pylori: Comparison of Detection Tools, ARG and Efflux Pump Gene Analysis, Worldwide Epidemiological Distribution, and Information Related to the Antimicrobial-Resistant Phenotype" Antibiotics 12, no. 7: 1118. https://doi.org/10.3390/antibiotics12071118
APA StyleAlfaray, R. I., Saruuljavkhlan, B., Fauzia, K. A., Torres, R. C., Thorell, K., Dewi, S. R., Kryukov, K. A., Matsumoto, T., Akada, J., Vilaichone, R.-k., Miftahussurur, M., & Yamaoka, Y. (2023). Global Antimicrobial Resistance Gene Study of Helicobacter pylori: Comparison of Detection Tools, ARG and Efflux Pump Gene Analysis, Worldwide Epidemiological Distribution, and Information Related to the Antimicrobial-Resistant Phenotype. Antibiotics, 12(7), 1118. https://doi.org/10.3390/antibiotics12071118