
Short-Term Impact of Oxytetracycline Administration on the Fecal Microbiome, Resistome and Virulome of Grazing Cattle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
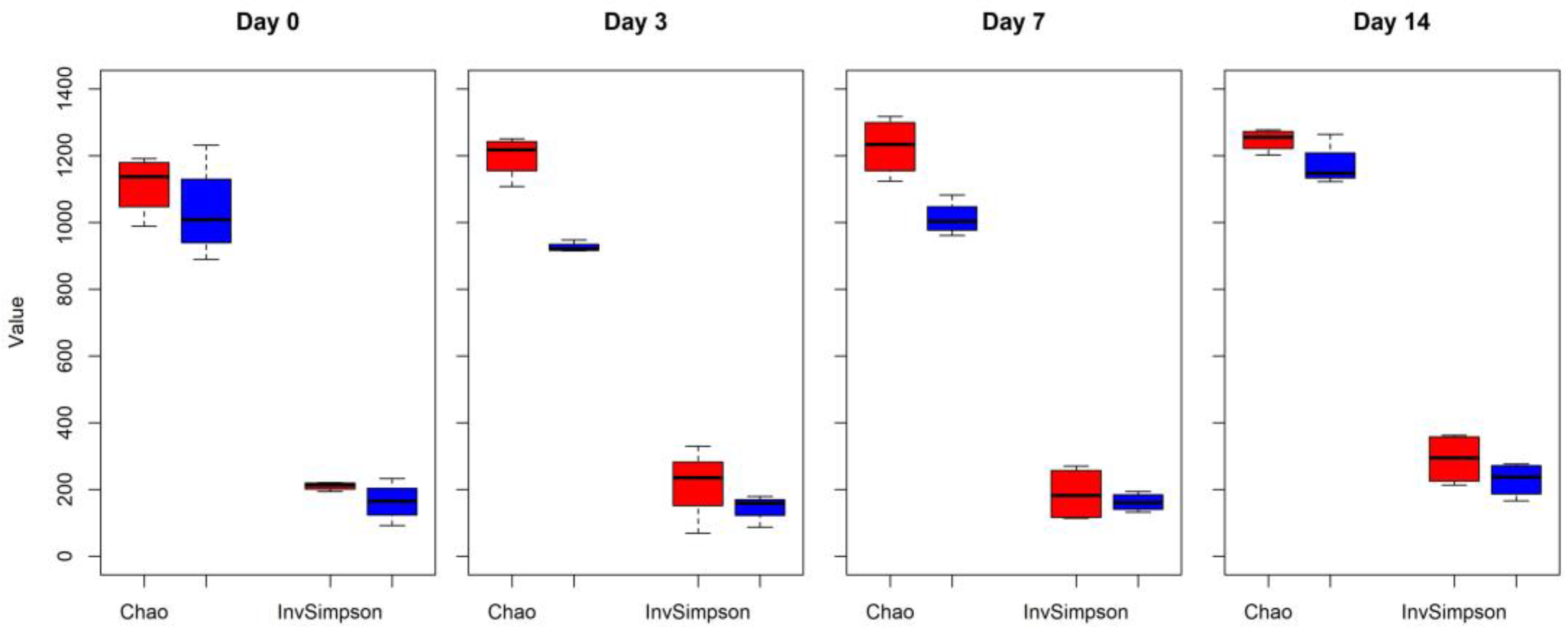
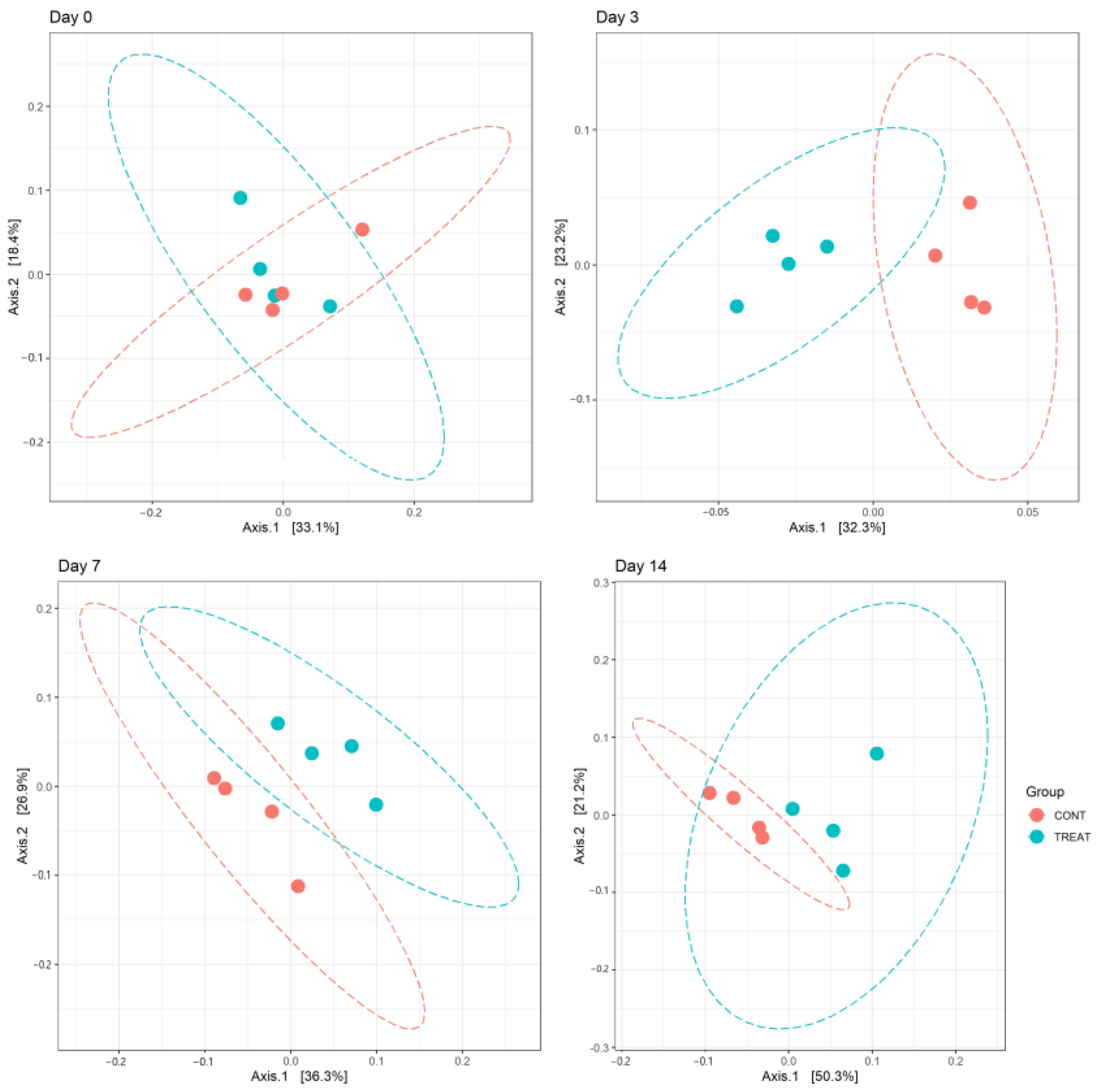
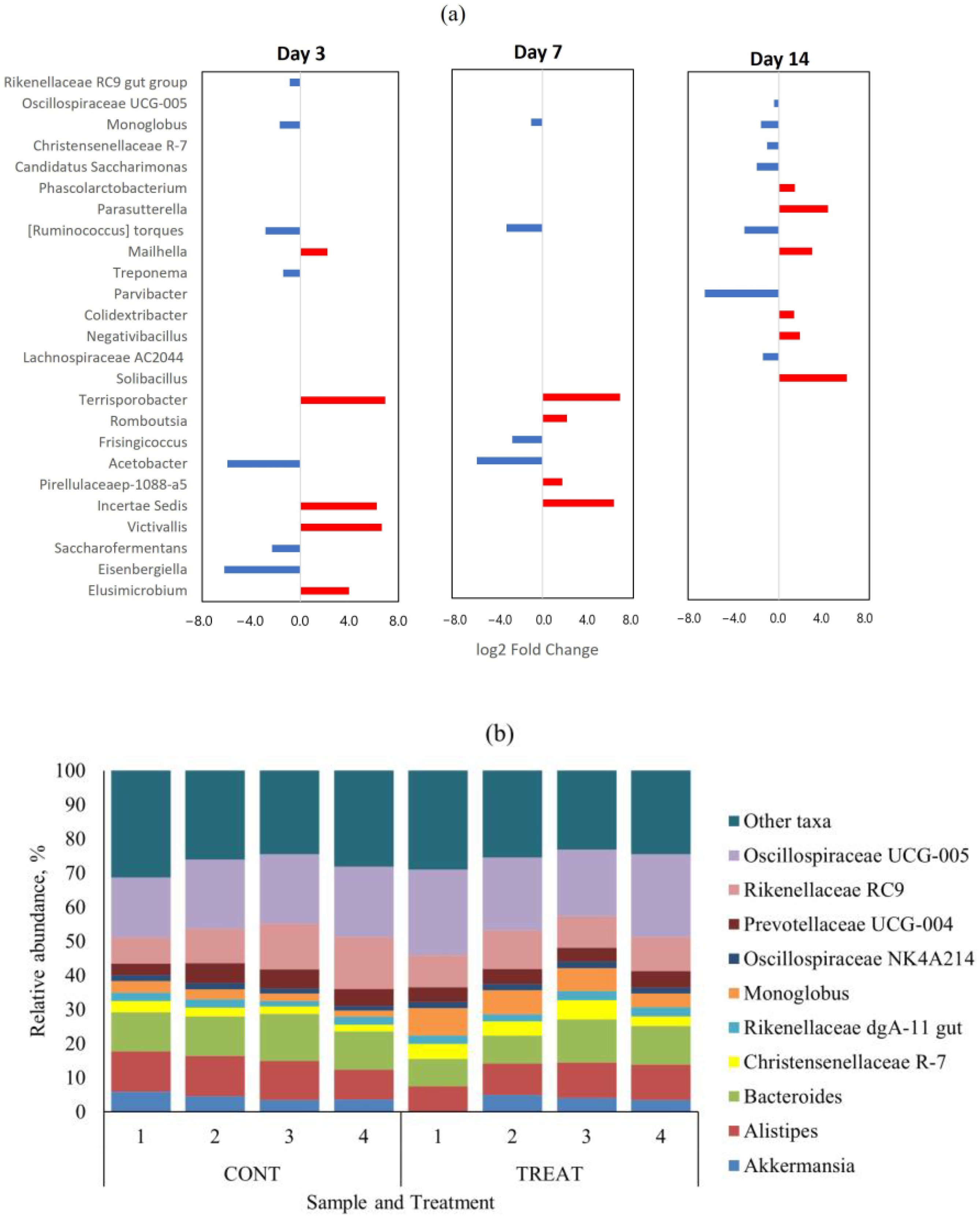
2.1. Microbiome Diversity and Composition
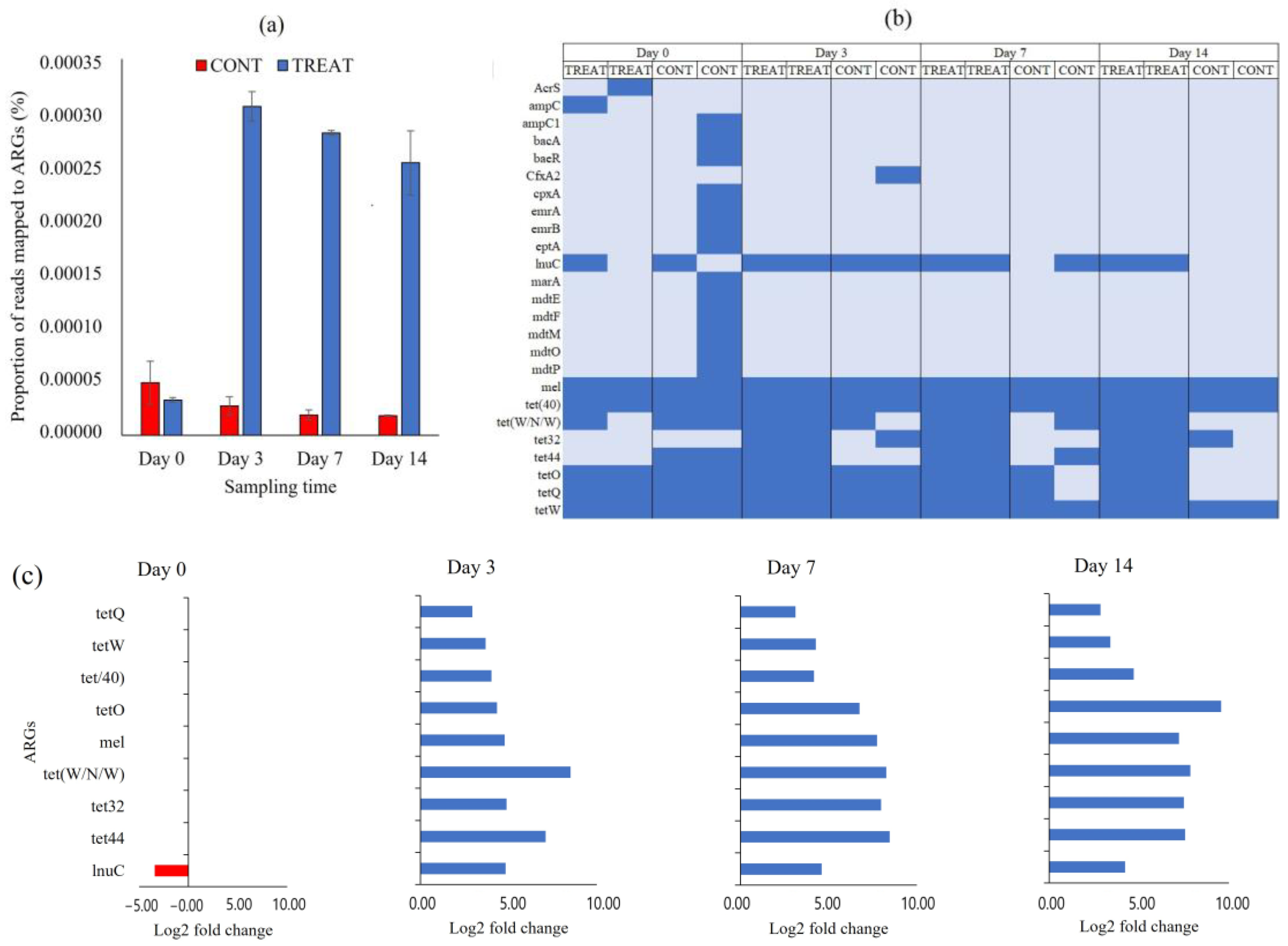
2.2. Antibiotic Resistance Genes
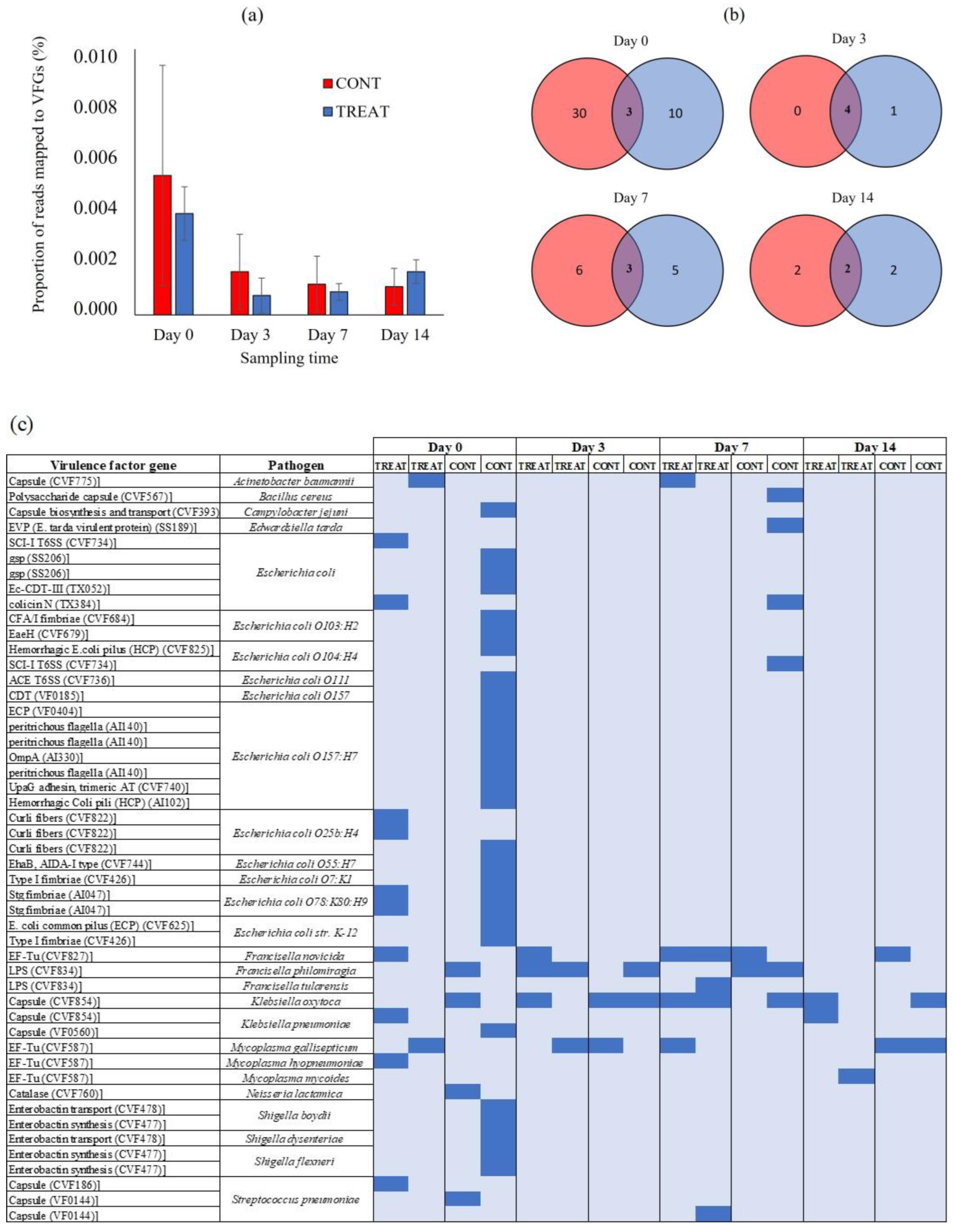
2.3. Virulence Factor Genes
3. Discussion
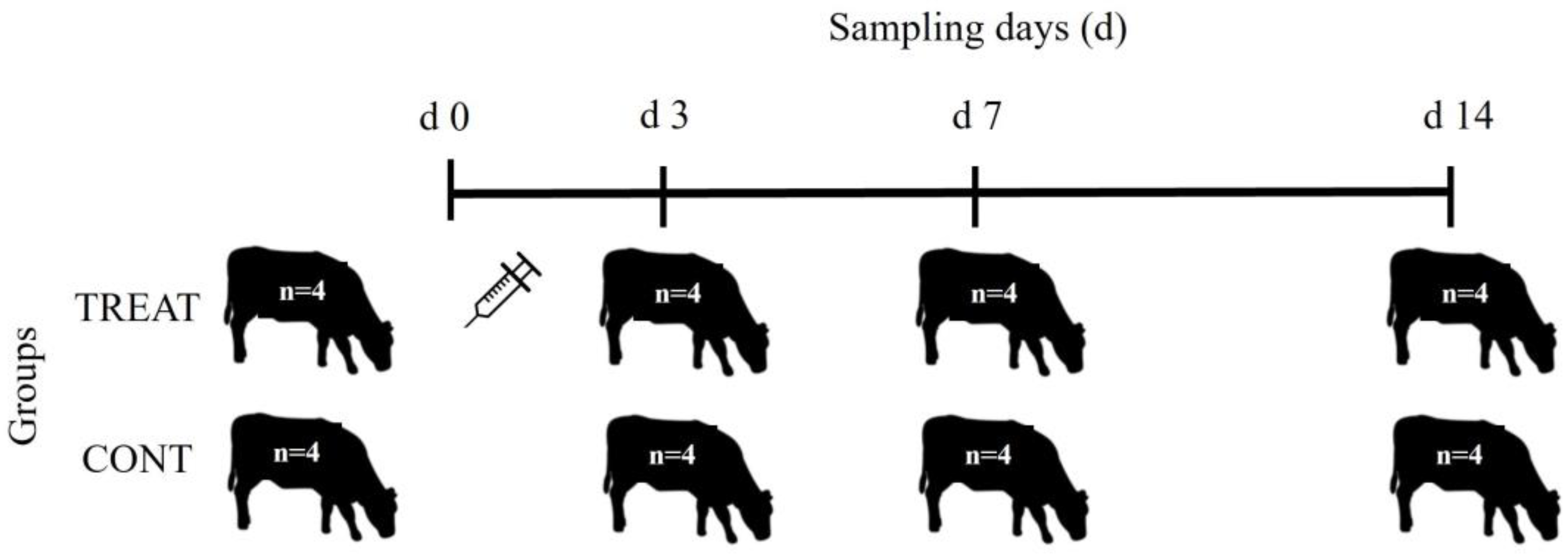
4. Materials and Methods
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Penders, J.; Stobberingh, E.E.; Savelkoul, P.H.M.; Wolffs, F.G. The human microbiome as a reservoir of antimicrobial resistance. Front. Microbiol. 2013, 4, 87. [Google Scholar] [CrossRef]
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Chambers, L.; Yang, Y.; Littier, H.; Ray, P.; Zhang, T.; Pruden, A.; Strickland, M.; Knowlton, K. Metagenomic analysis of antibiotic resistance genes in dairy cow feces following therapeutic administration of third generation cephalosporin. PLoS ONE 2015, 10, e0133764. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.W. Antimicrobial resistance issues in beef production. In Proceedings of the 68th Reciprocal Meat Conference, Lincoln, NE, USA, 13–17 June 2015; pp. 55–58. [Google Scholar]
- Thanner, S.; Drissner, D.; Walsh, F. Antimicrobial resistance in agriculture. mBio 2016, 7, e02227-15. [Google Scholar] [CrossRef]
- Doster, E.; Rovira, P.; Noyes, N.R.; Burgess, B.A.; Yang, X.; Weinroth, M.D.; Lakin, S.M.; Dean, C.J.; Linke, L.; Magnuson, R.; et al. Investigating effects of tulathromycin metaphylaxis on the fecal resistome and microbiome of commercial feedlot cattle early in the feeding period. Front. Microbiol. 2018, 9, 1715. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Yang, W.; Alexander, T.W. Antibiotic treatment in feedlot cattle: A longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharingeal microbiota. Microbiome 2019, 7, 86. [Google Scholar] [CrossRef]
- Rovira, P.; McAllister, T.; Lakin, S.M.; Cook, S.R.; Doster, E.; Noyes, N.R.; Weinroth, M.D.; Yang, X.; Parker, J.K.; Boucher, C.; et al. Characterization of the microbial resistome in conventional and “raised without antibiotics” beef and dairy production systems. Front. Microbiol. 2019, 10, 1980. [Google Scholar] [CrossRef]
- Nagaraja, T.; Chengappa, M. Liver abscesses in feedlot cattle: A review. J. Anim. Sci. 1998, 76, 287–298. [Google Scholar] [CrossRef]
- Cameron, A.; McAllister, T.A. Antimicrobial usage and resistance in beef production. J. Anim. Sci. Biotechnol. 2016, 7, 68. [Google Scholar] [CrossRef]
- Santamaria, J.; López, L.; Soto, C.Y. Detection and diversity evaluation of tetracycline resistance genes in grassland-based production systems in Colombia, South America. Front. Microbiol. 2011, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Markland, S.; Weppelmann, T.A.; Ma, Z.; Lee, S.; Mir, R.A.; Teng, L.; Ginn, A.; Lee, C.; Ukhanova, M.; Galindo, S.; et al. High prevalence of cefotaxime resistant bacteria in grazing beef cattle: A cross sectional study. Front. Microbiol. 2019, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Michalova, E.; Novotna, P.; Schlegelova, J. Tetracyclines in veterinary medicine and bacterial resistance to them. Vet. Med.-Czech 2004, 49, 79–100. [Google Scholar] [CrossRef]
- Granados-Chinchilla, F.; Rodríguez, C. Tetracyclines in food and feeding stuffs: From regulation to analytical methods, bacterial resistance, and environmental and health implications. J. Anal. Methods Chem. 2017, 1315497. [Google Scholar] [CrossRef]
- Shin, S.W.; Shin, M.K.; Jung, M.; Belaynehe, K.M.; Yoo, H.S. Prevalence of antimicrobial resistance and transfer of tetracycline resistance genes in Escherichia coli isolates from beef cattle. Appl. Environ. Microbiol. 2015, 81, 5560–5566. [Google Scholar] [CrossRef]
- Otto, S.; Ponich, K.L.; Cassis, R.; Goertz, C.; Peters, D.; Checkley, S.L. Antimicrobial resistance of bovine Salmonella enterica ssp. enterica isolates from the Alberta Agriculture and Forestry Disease Investigation Program (2006–2014). Can. Vet. J. 2018, 59, 1195–1201. [Google Scholar]
- Premarathne, J.; Anuar, A.S.; Thung, T.Y.; Satharasinghe, D.A.; Jambari, N.N.; Abdul-Mutalib, N.A.; Huat, J.; Basri, D.F.; Rukayadi, Y.; Nakaguchi, Y.; et al. Prevalence and antibiotic resistance against tetracycline in Campylobacter jejuni and C. coli in cattle and beef meat from Selangor, Malaysia. Front. Microbiol. 2017, 8, 2254. [Google Scholar] [CrossRef]
- Yang, X.; Noyes, N.R.; Doster, E.; Martin, J.N.; Magnuson, R.J.; Yang, H.; Geornaras, I.; Woerner, D.R.; Jones, K.L.; Ruiz, J.; et al. Use of metagenomic shotgun sequencing technology to detect foodborne pathogens within their microbiome in beef production chain. Appl. Environ. Microbiol. 2016, 82, 2433–2443. [Google Scholar] [CrossRef]
- Auffret, M.D.; Dewhurst, R.J.; Duthie, C.A.; Rooke, J.A.; Wallace, R.J.; Freeman, T.C.; Stewart, R.; Watson, M.; Roehe, R. The rumen microbiome as a reservoir of antimicrobial resistance and pathogenicity genes is directly affected by diet in beef cattle. Microbiome 2017, 5, 159. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Metagenomics and its connection to microbial community organization. F1000 Biol. Rep. 2012, 4, 15. [Google Scholar] [CrossRef]
- Noyes, N.R.; Yang, X.; Linke, L.M.; Magnuson, R.J.; Dettenwanger, A.; Cook, S.; Geornaras, I.; Woerner, D.E.; Gow, S.P.; McAllister, T.A.; et al. Resistome diversity in cattle and the environment decreases during beef production. eLife 2016, 5, e13195. [Google Scholar] [CrossRef] [PubMed]
- Weinroth, M.D.; Martin, J.N.; Doster, E.; Geornaras, I.; Parker, J.K.; Carlson, C.R.; Metcalf, J.L.; Morley, P.S.; Belk, K.E. Investigation of tylosin in feed of feedlot cattle and effects on liver abscess prevalence, and fecal and soil microbiomes and resistomes1. J. Anim. Sci. 2019, 97, 4567–4578. [Google Scholar] [CrossRef] [PubMed]
- Beyi, A.F.; Brito-Goulart, D.; Hawbecker, T.; Slagel, C.; Ruddell, B.; Hassall, A.; Dewell, R.; Dewell, G.; Sahin, O.; Zhang, Q.; et al. Danofloxacin treatment alters the diversity and resistome profile of gut microbiota in calves. Microorganisms 2020, 9, 2023. [Google Scholar] [CrossRef] [PubMed]
- Rochegüe, T.; Haenni, M.; Mondot, S.; Astruc, C.; Cazeau, G.; Ferry, T.; Madec, J.Y.; Lupo, A. Impact of antibiotic therapies on resistance genes dynamic and composition of the animal gut microbiota. Animals 2021, 11, 3280. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Meng, L.; Liu, H.; Wu, H.; Schroyen, M.; Zheng, N.; Wang, J. Effect of cephalosporin treatment on the microbiota and antibiotic resistance genes in feces of dairy cows with clinical mastitis. Antibiotics 2022, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Nielsen, P.; Gyrd-Hanse, N. Oxytetracyclines in cattle: A comparison between a conventional and a long-acting preparation. Acta Vet. Scand. 1983, 24, 120–128. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Webb, M.; Ghimire, S.; Blair, A.; Olson, K.; Fenske, G.J.; Fonder, A.T.; Christopher-Hennings, J.; Brake, D.; Scaria, J. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci. Rep. 2017, 7, 12257. [Google Scholar] [CrossRef]
- Mosca, A.; Leclerc, M.; Hugot, J.P. Gut microbiota diversity and human diseases: Should we reintroduce key predators in our ecosystem? Front. Microbiol. 2016, 7, 455. [Google Scholar] [CrossRef]
- Jenkins, T.P.; Formenti, F.; Castro, C.; Piubelli, C.; Perandin, F.; Buonfrate, D.; Otranto, D.; Griffin, J.L.; Krause, L.; Bisoffi, Z.; et al. A comprehensive analysis of the faecal microbiome and metabolome of Strongyloides stercoralis infected volunteers from a non-endemic area. Sci. Rep. 2018, 8, 15651. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kellingray, L.; Zhai, Q.; Le Gall, G.; Narbad, A.; Zhao, J.; Zhang, H.; Chen, W. Structural and functional alterations in the microbial community and immunological consequences in a mouse model of antibiotic-induced dysbiosis. Front. Microbiol. 2018, 9, 1948. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Anthony, W.E.; Wang, B.; Sukhum, K.V.; D’Souza, A.W.; Hink, T.; Cas, C.; Seiler, S.; Reske, K.A.; Coon, C.; Dubberke, E.R.; et al. Acute and persistent effects of commonly used antibiotics on the gut microbiome and resistome in healthy adults. Cell. Rep. 2022, 39, 110649. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, S.; Fox, J.T.; Shi, X.; Alam, M.J.; Renter, D.G.; Nagaraja, T.G. Prevalence of Escherichia coli O157:H7 in organically and naturally raised beef cattle. Environ. Microbiol. 2009, 75, 5421–5423. [Google Scholar] [CrossRef] [PubMed]
- Morley, P.S.; Dargatz, D.A.; Hyatt, D.R.; Dewell, G.A.; Gage Patterson, J.; Burgess, B.A.; Wittum, T.E. Effects of restricted antimicrobial exposure on antimicrobial resistance in fecal Escherichia coli from feedlot cattle. Foodborne Pathog. Dis. 2011, 8, 87–98. [Google Scholar] [CrossRef]
- Wright, G.D. Q&A: Antibiotic resistance: Where does it come from and what can we do about it? BMC Biol. 2010, 8, 123. [Google Scholar]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Rahman, H.M.; Sakamoto, K.Q.; Nonaka, L.; Suzuki, S. Occurrence and diversity of tetracycline resistance gene tet(M) in enteric bacteria of Antarctic Adélie penguin. J. Antimicrob. Chemother. 2008, 62, 627–628. [Google Scholar] [CrossRef]
- Yang, H.; Byelashov, O.A.; Geornaras, I.; Goodridge, L.D.; Nightingale, K.K.; Belk, K.E.; Smith, G.C.; Sofos, J.N. Presence of antibiotic resistant commensal bacteria in samples from agricultural, city, and national park environments evaluated by standard culture and real time PCR methods. Can. J. Microbiol. 2010, 56, 761–770. [Google Scholar] [CrossRef]
- Looft, T.; Allen, H.K. Collateral effects of antibiotics on mammalian gut microbiomes. Gut Microbes 2012, 3, 463–467. [Google Scholar] [CrossRef]
- Perry, J.A.; Wright, G.D. The antibiotic resistance “mobilome”: Searching for the link between environment and clinic. Front. Microbiol. 2013, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Rovira, P.; Agga, G.E.; Arthur, T.M.; Bosilevac, J.M.; Wheeler, T.L.; Morley, P.S.; Belk, K.E.; Smith, J.W. Impact of “raised without antibiotics” beef cattle production practices on occurrences of antimicrobial resistance. Appl. Environ. Microbiol. 2017, 83, e01682-17. [Google Scholar] [CrossRef]
- Roberts, M.C. Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 2005, 245, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.A.; Narasimha, V.H.; Straley, B.A.; Donaldson, S.C.; Love, B.; Knabel, S.J.; Jayarao, B.M. Antimicrobial resistant enteric bacteria from dairy cattle. Appl. Environ. Microbiol. 2007, 73, 156–163. [Google Scholar] [CrossRef]
- Kobashi, Y.; Hasebe, A.; Nishi, M.; Uchiyama, H. Diversity of tetracycline resistance genes in bacteria isolated from various agricultural environments. Microbes Environ. 2007, 22, 44–51. [Google Scholar] [CrossRef]
- Iwu, C.D.; Korsten, L.; Okoh, A.I. The incidence of antibiotic resistance within and beyond the agricultural ecosystem: A concern for public health. MicrobiologyOpen 2020, 9, e1035. [Google Scholar] [CrossRef] [PubMed]
- Nogrado, K.; Unno, T.; Hur, H.G.; Lee, J.H. Tetracycline-resistant bacteria and ribosomal protection protein genes in soils from selected agricultural fields and livestock farms. Appl. Biol. Chem. 2021, 64, 42. [Google Scholar] [CrossRef]
- Jacob, M.E.; Fox, J.T.; Narayanan, S.K.; Drouillard, J.S.; Renter, D.G.; Nagaraja, T.G. Effects of feeding wet corn distillers grains with soluble with or without monensin and tylosin on the prevalence and antimicrobial susceptibilities of fecal foodborne pathogenic and commensal bacteria in feedlot cattle. J. Anim. Sci. 2008, 86, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Bosman, A.B.; Wagenaar, J.A.; Stegemn, J.A.; Vernooij, J.C.M.; Mevius, D.J. Antimicrobial resistance in commensal Escherichia coli in veal calves is associated with antimicrobial drug use. Epidemiol. Infect. 2014, 142, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Benedict, K.M.; Gow, S.P.; McAllister, T.A.; Booker, C.W.; Hannon, S.J.; Checkley, S.L.; Noyes, N.R.; Morley, P.S. Antimicrobial resistance in Escherichia coli recovered from feedlot cattle and associations with antimicrobial use. PLoS ONE 2015, 10, e143995. [Google Scholar] [CrossRef]
- Ambrose, K.D.; Nisbet, R.; Stephens, D.S. Macrolide efflux in Streptococcus pneumoniae is mediated by a dual efflux pump (mel and mef) and is erythromycin inducible. Antimicrob. Agents Chemother. 2005, 49, 4203–4209. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.O.; Young, K.; Leng, Z.; Roberts, M.C. Mobile elements carrying ermF and tetQ genes in Gram-positive and Gram-negative bacteria. J. Antimicrob. Chemother. 1999, 44, 329–335. [Google Scholar] [CrossRef]
- Achard, A.; Villers, C.; Pichereau, V.; Leclercq, R. New lnu(C) gene conferring resistance to lincomycin by nucleotidylation in Streptococcus agalactiae UCN36. Antimicrob. Agents Chemother. 2005, 49, 2716–2719. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Stedfteld, R.D.; Wang, Q.; Cole, J.R.; Hashsham, S.A.; Looft, T.; Zhu, Y.G.; Tiedje, J.M. Clusters of antibiotic resistance genes enriched together stay together in swine agriculture. mBio 2015, 7, e02214-15. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genom. 2015, 16, 964. [Google Scholar] [CrossRef]
- Pickering, H.; Hart, J.D.; Burr, S.; Stabler, R.; Maleta, K.; Kalua, K.; Bailey, R.L.; Holland, M.J. Impact of azithromycin mass drug administration on the antibiotic-resistant gut microbiome in children: A randomized, controlled trial. Gut Pathog. 2022, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C.; Schwarz, S. Tetracycline and phenicol resistance genes and mechanisms: Importance for agriculture, the environment, and humans. J. Environ. Qual. 2016, 45, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Bok, E.; Mazurek, J.; Stosik, M.; Wojciech, M.; Baldy-Chudzik, K. Prevalence and virulence determinants and antimicrobial resistance among commensal Escherichia coli derived from dairy and beef cattle. Int. J. Environ. Public Health 2015, 12, 970–985. [Google Scholar] [CrossRef]
- Lambertini, E.; Karns, J.S.; Van Kessel, J.A.S.; Cao, H.; Schukken, Y.H.; Wolfgang, D.R.; Smith, J.M.; Pradhan, A.K. Dynamics of Escherichia coli virulence factors in dairy herds and farm environments in a longitudinal study in the United States. Appl. Environ. Microbiol. 2015, 81, 4477–4488. [Google Scholar] [CrossRef] [PubMed]
- Darmancier, H.; Domingues, C.P.F.; Rebelo, J.S.; Amaro, A.; Dionisio, F.; Pothier, J.; Serra, O.; Nogueira, T. Are virulence and antibiotic resistance genes linked? A Comprehensive Analysis of Bacterial Chromosomes and Plasmids. Antibiotics 2022, 11, 706. [Google Scholar] [CrossRef]
- Forster, S.C.; Liu, J.; Kumar, N.; Gulliver, E.L.; Gould, J.A.; Escobar-Zepeda, A.; Mkandawire, T.; Pike, L.J.; Shao, Y.; Stares, M.D.; et al. Strain-level characterization of broad host range mobile genetic elements transferring antibiotic resistance from the human microbiome. Nat. Commun. 2022, 13, 1445. [Google Scholar] [CrossRef] [PubMed]
- Podder, M.P.; Rogers, L.; Daley, P.K.; Keefe, G.P.; Whitney, H.G.; Tahlan, K. Klebsiella species associated with bovine mastitis in Newfoundland. PLoS ONE 2014, 9, e106518. [Google Scholar] [CrossRef]
- Ahangari Cohan, H.; Jamshidian, M.; Rohani, M.; Moravedji, M.; Mostafavi, E. Francisella tularensis survey among ranchers and livestock in western Iran. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101598. [Google Scholar] [CrossRef] [PubMed]
- Valeris-Chacin, R.; Powledge, S.; McAtee, T.; Morley, P.S.; Richeson, J. Mycoplasma bovis is associated with Mannheimia haemolytica during acute bovine respiratory disease in feedlot cattle. Front. Microbiol. 2022, 13, 946792. [Google Scholar] [CrossRef] [PubMed]
- Thames, C.H.; Pruden, A.; James, R.E.; Ray, P.P.; Knowlton, K.F. Excretion of antibiotic resistance genes by dairy calves fed milk replacers with varying doses of antibiotics. Front. Microbiol. 2012, 3, 139. [Google Scholar] [CrossRef]
- Forslund, K.; Sunagawa, S.; Kultima, J.R.; Mende, D.R.; Arumugam, M.; Typas, A.; Bork, P. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013, 23, 1163–1169. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Wright, E.S. Using DECIPHER v2.0 to analyze big biological sequence data in R. R Journal 2016, 8, 352–359. [Google Scholar] [CrossRef]
- Schliep, K. phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef]
- McMurdie, P.; Holmes, S. phyloseq: An R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Amolegang, R.R.; Tammy, T.Y.L.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the Comprehensive. Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Yang, J.; Yu, J.; Yao, Z.J.; Sun, L.L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. 2014. Version 2.2-0. Available online: http://CRAN.R-project.org/package=vegan (accessed on 14 February 2023).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovira, P. Short-Term Impact of Oxytetracycline Administration on the Fecal Microbiome, Resistome and Virulome of Grazing Cattle. Antibiotics 2023, 12, 470. https://doi.org/10.3390/antibiotics12030470
Rovira P. Short-Term Impact of Oxytetracycline Administration on the Fecal Microbiome, Resistome and Virulome of Grazing Cattle. Antibiotics. 2023; 12(3):470. https://doi.org/10.3390/antibiotics12030470
Chicago/Turabian StyleRovira, Pablo. 2023. "Short-Term Impact of Oxytetracycline Administration on the Fecal Microbiome, Resistome and Virulome of Grazing Cattle" Antibiotics 12, no. 3: 470. https://doi.org/10.3390/antibiotics12030470
APA StyleRovira, P. (2023). Short-Term Impact of Oxytetracycline Administration on the Fecal Microbiome, Resistome and Virulome of Grazing Cattle. Antibiotics, 12(3), 470. https://doi.org/10.3390/antibiotics12030470