


Virtual Screening for SARS-CoV-2 Main Protease Inhibitory Peptides from the Putative Hydrolyzed Peptidome of Rice Bran

 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
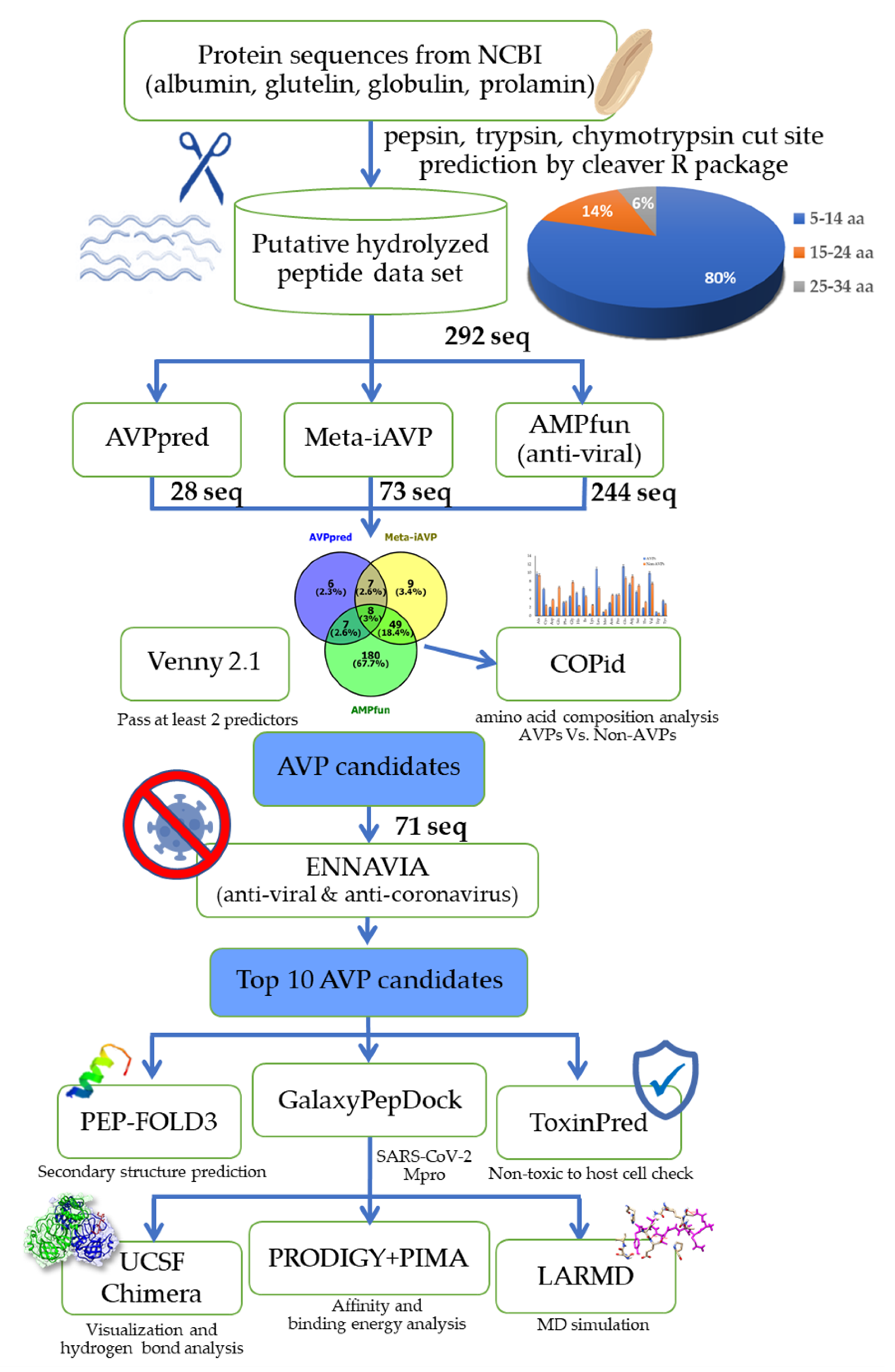
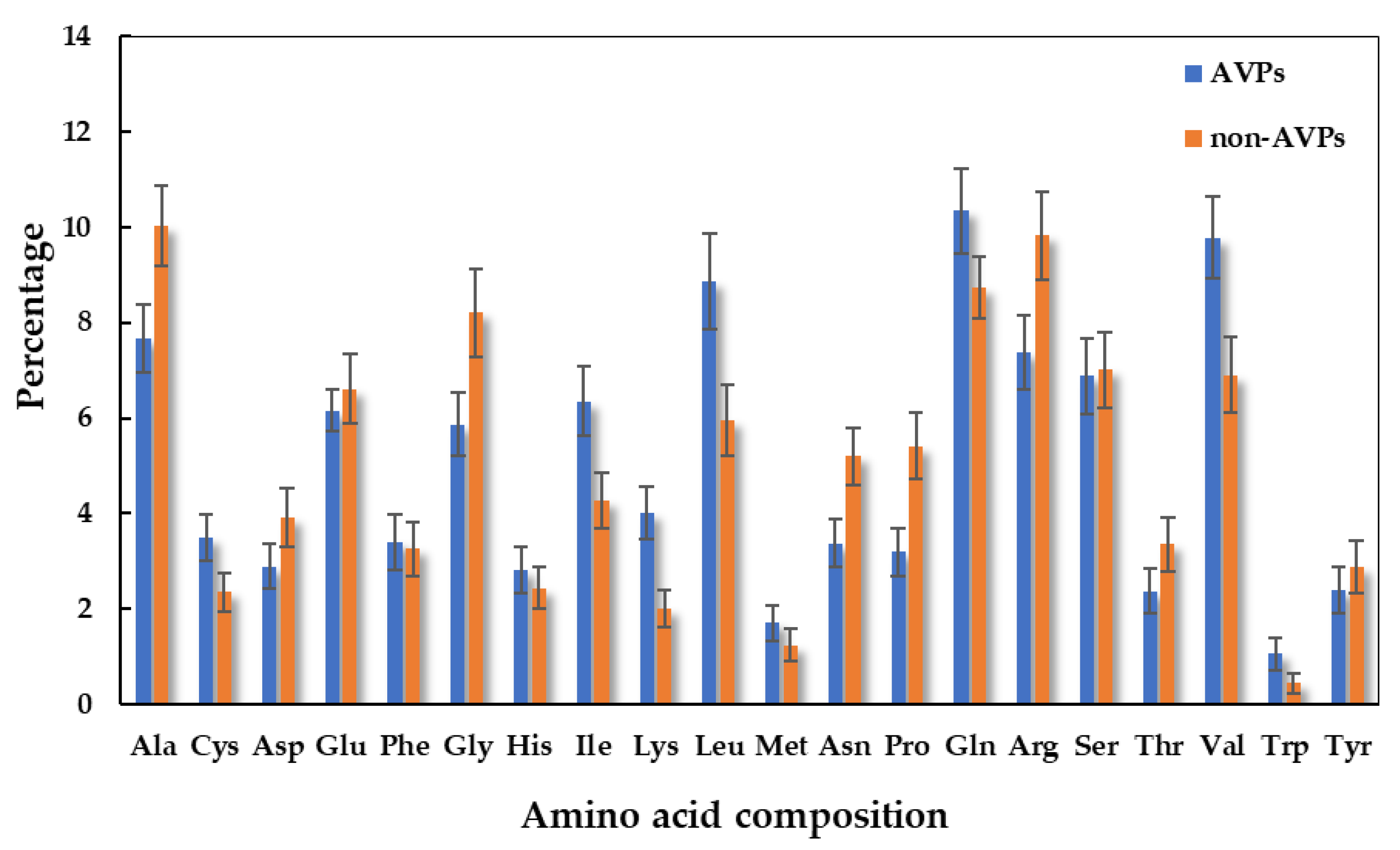
2.1. Rice Bran Putative Antiviral Peptide Screening using a Computational Method
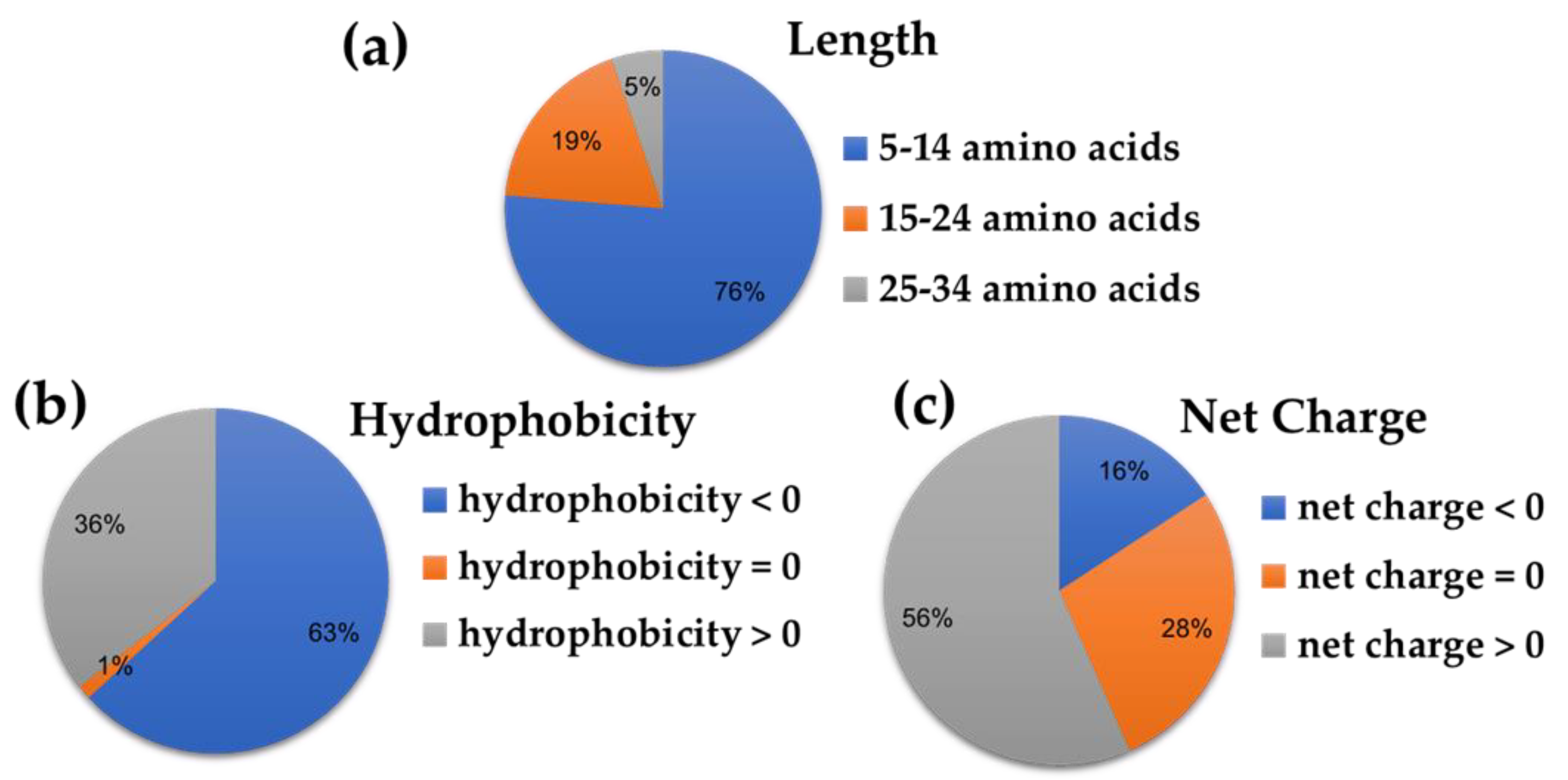
2.2. Prediction Scores and Physicochemical Properties of the Ten Top-ranked AVPs
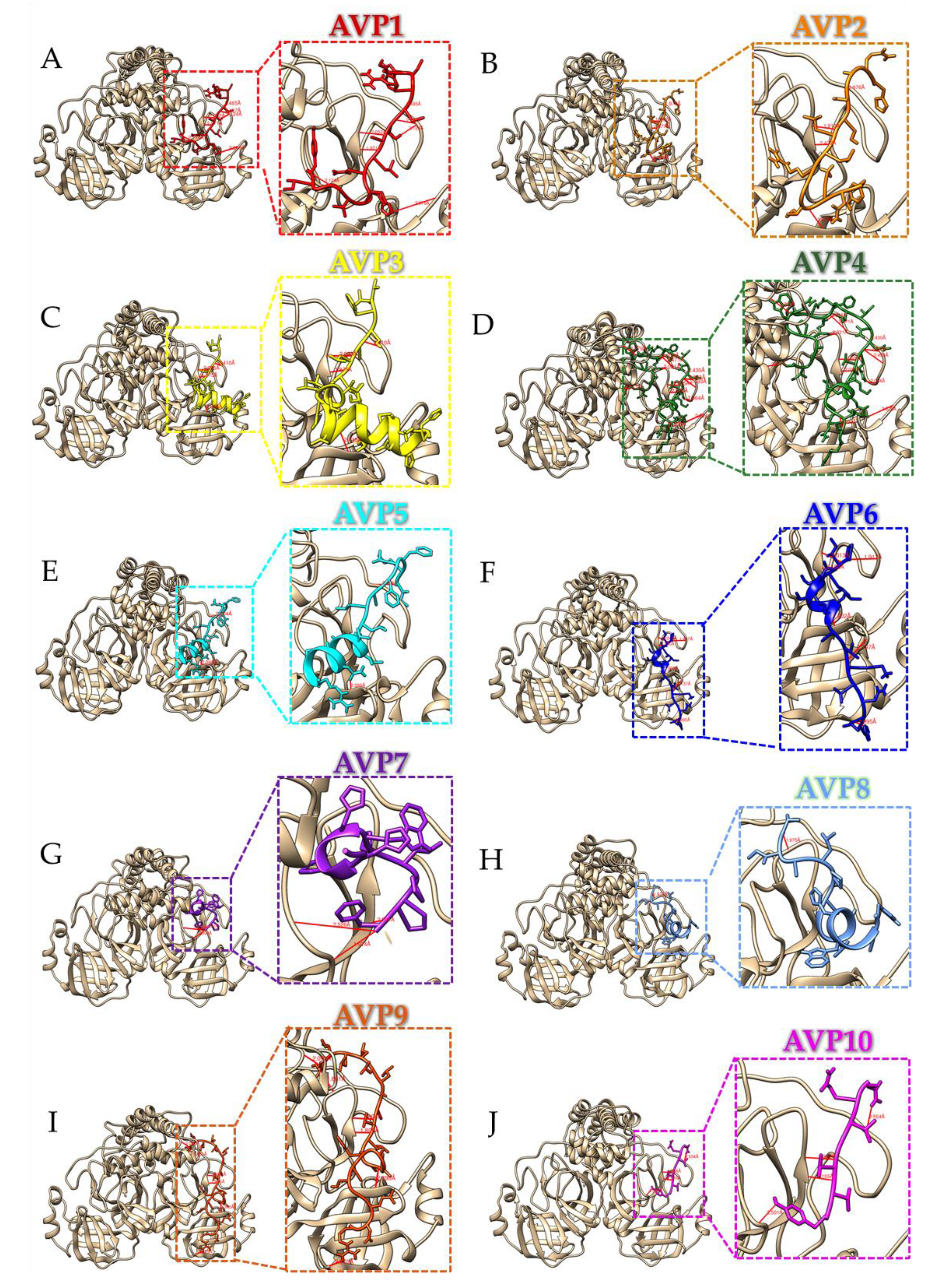
2.3. Protein–peptide Docking Simulations
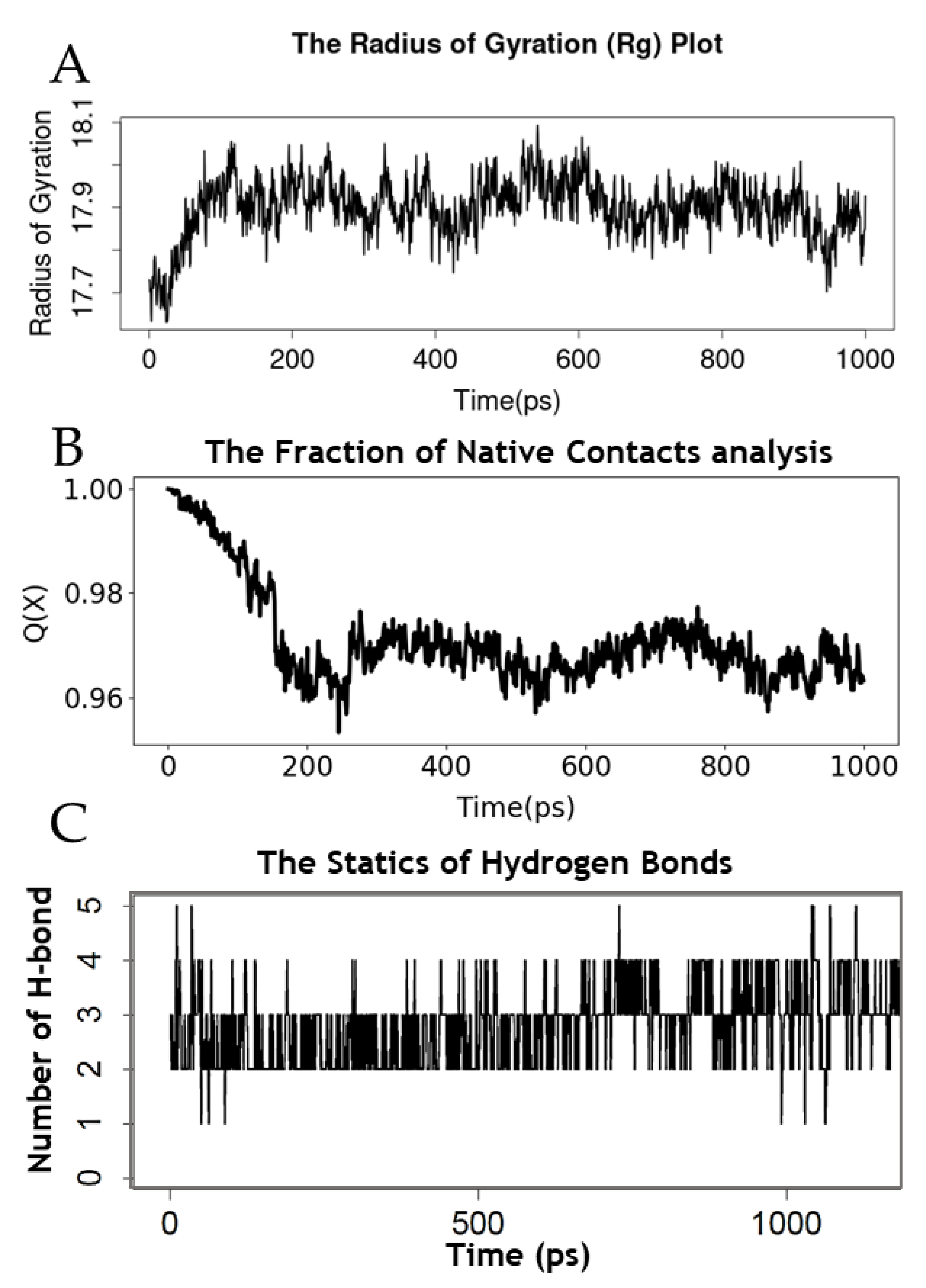
2.4. Simulation of the Molecular Dynamics of the Mpro–AVP4 Complex
2.5. In Silico Toxicity Analysis of the Selected AVPs
3. Materials and Methods
3.1. Preparation of the Rice Bran Putative Hydrolyzed Peptidome
3.2. The Bioinformatic Prediction and Screening of Antiviral Peptides
3.3. The Protein–peptide Molecular Docking Simulation
3.4. Molecular Dynamics Simulation Analysis of AVP with the Mpro Enzyme
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef] [PubMed]
- Bundó, M.; Montesinos, L.; Izquierdo, E.; Campo, S.; Mieulet, D.; Guiderdoni, E.; Rossignol, M.; Badosa, E.; Montesinos, E.; Segundo, B.S.; et al. Production of cecropin A antimicrobial peptide in rice seed endosperm. BMC Plant Biol. 2014, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Okita, T.W. Accumulation of Prolamines and Glutelins during Rice Seed Development: A Quantitative Evaluation. Plant Cell Physiol. 1993, 34, 385–390. [Google Scholar]
- Kawakatsu, T.; Yamamoto, M.P.; Hirose, S.; Yano, M.; Takaiwa, F. Characterization of a new rice glutelin gene GluD-1 expressed in the starchy endosperm. J. Exp. Bot. 2008, 59, 4233–4245. [Google Scholar]
- Cagampang, G.B.; Perdon, A.A.; Juliano, B.O. Changes in salt-soluble proteins of rice during grain development. Phytochemistry 1976, 15, 1425–1429. [Google Scholar] [CrossRef]
- Wang, M.; Hettiarachchy, N.S.; Qi, M.; Burks, W.; Siebenmorgen, T. Preparation and Functional Properties of Rice Bran Protein. Isolate. J. Agric. Food Chem. 1999, 47, 411–416. [Google Scholar] [CrossRef]
- Cicero, A.F.; Derosa, G. Rice bran and its main components: Potential role in the management of coronary risk factors. Curr. Top. Nutraceutical Res. 2005, 3, 29–46. [Google Scholar]
- Zaky, A.A.; Abd El-Aty, A.; Ma, A.; Jia, Y. An overview on antioxidant peptides from rice bran proteins: Extraction, identification, and applications. Crit. Rev. Food Sci. Nutr. 2022, 62, 1350–1362. [Google Scholar] [CrossRef]
- Phongthai, S.; Rawdkuen, S. Fractionation and characterization of antioxidant peptides from rice bran protein hydrolysates stimulated by in vitro gastrointestinal digestion. Cereal Chem. 2020, 97, 316–325. [Google Scholar] [CrossRef]
- Ngamsuk, S.; Hsu, J.-L.; Huang, T.-C.; Suwannaporn, P. Ultrasonication of Milky Stage Rice Milk with Bioactive Peptides from Rice Bran: Its Bioactivities and Absorption. Food Bioprocess Technol. 2020, 13, 462–474. [Google Scholar] [CrossRef]
- Gasymov, O.K.; Celik, S.; Agaeva, G.; Akyuz, S.; Kecel-Gunduz, S.; Qocayev, N.M.; Ozel, A.E.; Agaeva, U.; Bakhishova, M.; Aliyev, J.A. Evaluation of anti-cancer and anti-covid-19 properties of cationic pentapeptide Glu-Gln-Arg-Pro-Arg, from rice bran protein and its d-isomer analogs through molecular docking simulations. J. Mol. Graph. Model. 2021, 108, 107999. [Google Scholar] [CrossRef]
- BioMedTech. Bioinformatics. Available online: https://www.blockdit.com/posts/5db93b2bb6a0111f3f4a722b (accessed on 5 June 2020).
- Chang, K.Y.; Yang, J.-R. Analysis and Prediction of Highly Effective Antiviral Peptides Based on Random Forests. PLoS ONE 2013, 8, e70166. [Google Scholar] [CrossRef]
- Skalickova, S.; Heger, Z.; Krejcova, L.; Pekarik, V.; Bastl, K.; Janda, J.; Kostolansky, F.; Vareckova, E.; Zitka, O.; Adam, V.; et al. Perspective of Use of Antiviral Peptides against Influenza Virus. Viruses 2015, 7, 5428–5442. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- Nyanguile, O. Peptide Antiviral Strategies as an Alternative to Treat Lower Respiratory Viral Infections. Front. Immunol. 2019, 10, 1366. [Google Scholar] [CrossRef]
- Sala, A.; Ardizzoni, A.; Ciociola, T.; Magliani, W.; Conti, S.; Blasi, E.; Cermelli, C. Antiviral Activity of Synthetic Peptides Derived from Physiological Proteins. Intervirology 2018, 61, 166–173. [Google Scholar] [CrossRef]
- Vilas Boas, L.C.P.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Unraveling the bioactivity of anticancer peptides as deduced from machine learning. EXCLI J. 2018, 17, 734–752. [Google Scholar]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. Meta-iAVP: A sequence-based meta-predictor for improving the prediction of antiviral peptides using effective feature representation. Int. J. Mol. Sci. 2019, 20, 5743. [Google Scholar]
- Bulet, P.; Stocklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic α-helical antimicrobial peptides. Pept. Sci. 1998, 47, 451–463. [Google Scholar]
- Li, S.; Guo, S.; Li, F.; Xiang, J. Functional Diversity of Anti-Lipopolysaccharide Factor Isoforms in Shrimp and Their Characters Related to Antiviral Activity. Mar. Drugs 2015, 13, 2602–2616. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Hancock, R.E.W. Cationic antimicrobial peptides and their multifunctional role in the immune system. Crit. Rev. Immunol. 2000, 20, 24. [Google Scholar] [CrossRef]
- Wang, C.-K.; Shih, L.-Y.; Chang, K.Y. Large-Scale Analysis of Antimicrobial Activities in Relation to Amphipathicity and Charge Reveals Novel Characterization of Antimicrobial Peptides. Molecules 2017, 22, 2037. [Google Scholar]
- Chowdhury, A.S.; Reehl, S.M.; Kehn-Hall, K.; Bishop, B.; Webb-Robertson, B.-J.M. Better understanding and prediction of antiviral peptides through primary and secondary structure feature importance. Sci. Rep. 2020, 10, 1–8. [Google Scholar]
- Timmons, P.B.; Hewage, C.M. ENNAVIA is a novel method which employs neural networks for antiviral and anti-coronavirus activity prediction for therapeutic peptides. Brief. Bioinform. 2021, 22, bbab258. [Google Scholar]
- Sterri, S.H.; Fonnum, F. CHAPTER 68—Role of Carboxylesterases in Therapeutic Intervention of Nerve Gas Poisoning. In Handbook of Toxicology of Chemical Warfare Agents; Gupta, R.C., Ed.; Academic Press: San Diego, CA, USA, 2009; pp. 1033–1040. [Google Scholar]
- Fakih, T.M. Dermaseptin-based antiviral peptides to prevent COVID-19 through in silico molecular docking studies against SARS-Cov-2 spike protein. Pharm. Sci. Res. 2020, 7, 8. [Google Scholar]
- Jeffrey, G.A.; Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- El-Demerdash, A.; Al-Karmalawy, A.A.; Abdel-Aziz, T.M.; Elhady, S.S.; Darwish, K.M.; Hassan, A.H. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021, 11, 31339–31363. [Google Scholar] [CrossRef]
- Kumar, N.; Sood, D.; van der Spek, P.J.; Sharma, H.S.; Chandra, R. Molecular binding mechanism and pharmacology comparative analysis of noscapine for repurposing against SARS-CoV-2 protease. J. Proteome Res. 2020, 19, 4678–4689. [Google Scholar] [CrossRef] [PubMed]
- Robles-Loaiza, A.A.; Pinos-Tamayo, E.A.; Mendes, B.; Ortega-Pila, J.A.; Proaño-Bolaños, C.; Plisson, F.; Teixeira, C.; Gomes, P.; Almeida, J.R. Traditional and Computational Screening of Non-Toxic Peptides and Approaches to Improving Selectivity. Pharmaceuticals 2022, 15, 323. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Maione, F.; Iqbal, A.J.; Mascolo, N.; De Feo, V. The potential therapeutic application of peptides and peptidomimetics in cardiovascular disease. Front. Pharmacol. 2017, 7, 526. [Google Scholar] [CrossRef] [PubMed]
- Cook, Q.S.; Burks, A.W. Peptide and Recombinant Allergen Vaccines for Food Allergy. Clin. Rev. Allergy Immunol. 2018, 55, 162–171. [Google Scholar] [CrossRef]
- Silva, A.F.; Bastos, E.L.; Torres, M.D.T.; Costa-Da-Silva, A.L.; Ioshino, R.S.; Capurro, M.L.; Alves, F.L.; Miranda, A.; Vieira, R.D.F.F.; Oliveira, V.X. Antiplasmodial activity study of angiotensin II via Ala scan analogs. J. Pept. Sci. 2014, 20, 640–648. [Google Scholar] [CrossRef]
- Osorio, D.; Rondón-Villarreal, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. Small 2015, 12, 44–444. [Google Scholar] [CrossRef]
- IDimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2013, 30, 846–851. [Google Scholar] [CrossRef]
- Dimitrov, I.; Doytchinova, I. An alignment-independent platform for allergenicity prediction. In Immunoinformatics; Springer: New York, NY, USA, 2020; pp. 147–153. [Google Scholar]
- Bhattacharya, M.; Sharma, A.R.; Patra, P.; Ghosh, P.; Sharma, G.; Patra, B.C.; Ghosh, P.; Sharma, G.; Patra, B.C.; Saha, R.P.; et al. A SARS-CoV-2 vaccine candidate: In-silico cloning and validation. Inform. Med. Unlocked 2020, 20, 100394. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Strappe, P.; Shang, W.T.; Zhou, Z.K. Functional peptides derived from rice bran proteins. Crit. Rev. Food Sci. Nutr. 2017, 59, 349–356. [Google Scholar] [CrossRef]
- Wattanasiritham, L.; Theerakulkait, C.; Wickramasekara, S.; Maier, C.S.; Stevens, J.F. Isolation and identification of antioxidant peptides from enzymatically hydrolyzed rice bran protein. Food Chem. 2016, 192, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Wang JHu, J.; Cui, J.; Bai, X.; Du, Y.; Miyaguchi, Y.; Lin, B. Purification and identification of a ACE inhibitory peptide from oyster proteins hydrolysate and the antihypertensive effect of hydrolysate in spontaneously hypertensive rats. Food Chem. 2008, 111, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Kameda, M.; Namae, T.; Ochiai, A.; Saitoh, E.; Tanaka, T. Identification and characterization of multifunctional cationic peptides derived from peptic hydrolysates of rice bran protein. J. Funct. Foods 2017, 34, 287–296. [Google Scholar] [CrossRef]
- Ochiai, A.; Tanaka, S.; Tanaka, T.; Taniguchi, M. Rice Bran Protein as a Potent Source of Antimelanogenic Peptides with Tyrosinase Inhibitory Activity. J. Nat. Prod. 2016, 79, 2545–2551. [Google Scholar] [CrossRef] [PubMed]
- Gibb, S. Cleaver: Cleavage of Polypeptide Sequences, R package version 1.34.1; 2022. Available online: https://www.bioconductor.org/packages/release/bioc/html/cleaver.html (accessed on 12 January 2022).
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef]
- Chung, C.-R.; Kuo, T.-R.; Wu, L.-C.; Lee, T.-Y.; Horng, J.-T. Characterization and identification of antimicrobial peptides with different functional activities. Briefings Bioinform. 2019, 21, 1098–1114. [Google Scholar] [CrossRef]
- Oliveros, J.; Venny, C. An Interactive Tool for Comparing Lists with Venn’s Diagrams; BioinfoGP, CNB-CSIC; Spanish National Biotechnology Centre: Madrid, Spain, 2007; Available online: https://bioinfogp.cnb.csic.es/tools/venny/ (accessed on 12 January 2022).
- Zhou, X.; Zhong, F.; Lin, C.; Hu, X.; Zhang, Y.; Xiong, B.; Yin, X.; Fu, J.; He, W.; Duan, J.; et al. Structure of SARS-CoV-2 main protease in the apo state. Sci. China Life Sci. 2021, 64, 656–659. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Mathew, O.K.; Sowdhamini, R. PIMA: Protein-Protein interactions in Macromolecular Assembly—A web server for its Analysis and Visualization. Bioinformation 2016, 12, 9–11. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar]
- Yang, J.-F.; Wang, F.; Chen, Y.-Z.; Hao, G.-F.; Yang, G.-F. LARMD: Integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Brief. Bioinform. 2020, 21, 2206–2218. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide ID | Secondary Structures | Sequences (from Protein/Cut by) | Length | AVPpred | Meta-iAVP | AMP Fun | ENNAVIA | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M3* | M4* | A* | B* | C* | D* | ||||||
| AVP1 |  | SWCRCSALNHMVGGIY (albumin/pepsin) | 16 | 55.56 | 72.37 | 0.51 | 0.39 | 0.47 | 1.00 | 0.79 | 1.00 |
| AVP2 |  | HQASSLLRGIKNY (globulin/pepsin) | 13 | 54.82 | 24.41 | 0.98 | 0.49 | 0.97 | 0.74 | 0.57 | 0.23 |
| AVP3 |  | VVFSALLLIIVSVLAATATMADHHK (albumin/trypsin) | 25 | 52.40 | 25.59 | 0.54 | 0.59 | 1.00 | 0.00 | 1.00 | 0.01 |
| AVP4 |  | QQHSIVATPFWQPATFQLINNQVMQQQCCQQLR (prolamin/trypsin) | 33 | 37.06 | 64.03 | 0.06 | 0.65 | 0.68 | 0.00 | 0.99 | 0.16 |
| AVP5 |  | IIFVFALLAIVACNASAR (prolamin/pepsin/trypsin) | 18 | 56.12 | 33.78 | 0.99 | 0.43 | 1.00 | 0.00 | 0.75 | 0.00 |
| AVP6 |  | PILSLVQMSAVKVNLY (glutelin/pepsin) | 16 | 46.62 | 55.77 | 0.32 | 0.53 | 0.10 | 0.00 | 0.80 | 0.32 |
| AVP7 |  | GGHGPHWPLPPF (globulin/pepsin) | 12 | 42.04 | 25.90 | 1.00 | 0.51 | 0.74 | 0.91 | 0.00 | 0.08 |
| AVP8 |  | VIALPAGVAHWCY (glutelin/pepsin) | 13 | 41.67 | 62.94 | 0.54 | 0.46 | 0.50 | 0.31 | 0.35 | 0.28 |
| AVP9 |  | ATILLLLAAVLFAAAAAASGEDR (globulin/trypsin) | 23 | 47.39 | 62.95 | 0.74 | 0.43 | 0.96 | 0.00 | 0.40 | 0.00 |
| AVP10 |  | QQVGVVY (prolamin/chymotrypsin) | 7 | 50.46 | 43.09 | 0.99 | 0.66 | 0.05 | 0.20 | 0.20 | 0.45 |
| Peptide ID | Hydrophobicity | Steric Hindrance | Sidebulk | Hydropathicity | Amphipathicity | Hydrophilicity | Net Hydrogen | Charge | pI | Mol wt |
|---|---|---|---|---|---|---|---|---|---|---|
| AVP1 | −0.01 | 0.6 | 0.6 | 0.34 | 0.24 | −0.71 | 0.69 | 1.5 | 8.38 | 1797.33 |
| AVP2 | −0.22 | 0.58 | 0.58 | −0.62 | 0.68 | −0.13 | 1.08 | 2.5 | 10.01 | 1486.89 |
| AVP3 | 0.17 | 0.56 | 0.56 | 1.52 | 0.26 | −0.73 | 0.36 | 1 | 7.26 | 2621.56 |
| AVP4 | −0.15 | 0.61 | 0.61 | −0.45 | 0.5 | −0.56 | 1 | 1.5 | 8.4 | 3916.01 |
| AVP5 | 0.23 | 0.62 | 0.62 | 2.1 | 0.14 | −0.94 | 0.39 | 1 | 8.6 | 1892.59 |
| AVP6 | 0.09 | 0.62 | 0.62 | 1.05 | 0.31 | −0.74 | 0.56 | 1 | 8.94 | 1775.43 |
| AVP7 | 0.08 | 0.43 | 0.43 | −0.69 | 0.24 | −0.72 | 0.25 | 1 | 7.26 | 1298.65 |
| AVP8 | 0.25 | 0.54 | 0.54 | 1.32 | 0.11 | −1.18 | 0.23 | 0.5 | 7.06 | 1399.86 |
| AVP9 | 0.15 | 0.58 | 0.58 | 1.45 | 0.16 | −0.45 | 0.35 | −1 | 4.38 | 2228.94 |
| AVP10 | 0.06 | 0.69 | 0.69 | 0.56 | 0.36 | −0.91 | 0.71 | 0 | 5.88 | 792.01 |
| Peptides | Peptide Residues | Protease Residues | Distance (Å) | Peptides | Peptide Residues | Protease Residues | Distance (Å) |
|---|---|---|---|---|---|---|---|
| AVP1 | Arg4 | Gln180 | 2.485 | AVP4 | Arg33 | Thr19 | 2.285 |
| Ser6 | Thr178 | 2.124 | (continue) | Arg33 | Thr24 | 2.335 | |
| Ala7 | Glu154 | 1.971 | Arg33 | Gln61 | 2.041 | ||
| His10 | Cys42 | 2.978 | Arg33 | Thr19 | 2.018 | ||
| Val12 | Gly131 | 2.124 | AVP5 | Ile1 | Ala181 | 2.404 | |
| AVP2 | Ser4 | Gln180 | 1.976 | Asn14 | Thr24 | 2.085 | |
| Leu6 | Glu154 | 1.912 | AVP6 | Pro1 | Gln177 | 1.917 | |
| Arg8 | His151 | 2.036 | Ile2 | Glu154 | 2.013 | ||
| Arg8 | His160 | 2.047 | Val6 | Asn111 | 2.428 | ||
| Ile10 | Thr24 | 2.078 | Gln7 | Thr24 | 1.902 | ||
| Tyr13 | His39 | 2.778 | Ala10 | Thr24 | 2.463 | ||
| AVP3 | Ala5 | Thr178 | 2.410 | Lys12 | Gly21 | 2.137 | |
| Leu6 | Glu154 | 1.875 | Lys12 | Asn57 | 1.939 | ||
| Ser12 | Asn111 | 2.330 | Tyr16 | Asn55 | 2.536 | ||
| Ala15 | Thr24 | 2.045 | Tyr16 | Val69 | 1.995 | ||
| Thr19 | Thr22 | 2.017 | AVP7 | Phe12 | Gly131 | 2.526 | |
| AVP4 | Ser4 | Val286 | 2.047 | Phe12 | Cys133 | 1.924 | |
| Thr8 | Pro156 | 2.031 | Phe12 | His152 | 2.091 | ||
| Trp11 | Ser274 | 2.279 | AVP8 | Val1 | Asp209 | 1.898 | |
| Gln12 | Asp209 | 2.021 | Val1 | Thr157 | 1.975 | ||
| Pro13 | Arg123 | 2.196 | Cys12 | Thr23 | 3.071 | ||
| Thr15 | Ala182 | 2.471 | AVP9 | Ala1 | Arp209 | 2.000 | |
| Thr15 | Gly183 | 1.938 | Ala1 | Thr157 | 1.861 | ||
| Asn20 | Gln180 | 2.430 | Thr2 | Gly271 | 2.093 | ||
| Gln22 | Gln180 | 2.057 | Thr2 | Arp185 | 1.972 | ||
| Gln22 | Gln177 | 2.493 | Val10 | Glu154 | 1.894 | ||
| Gln22 | Thr178 | 2.523 | Ala13 | Thr24 | 2.390 | ||
| Gln22 | Asp175 | 2.064 | Ser19 | Asn111 | 2.178 | ||
| Val23 | Glu154 | 1.935 | Glu21 | Thr24 | 2.242 | ||
| Gln25 | Ser132 | 2.372 | Arp22 | Gln61 | 2.349 | ||
| Gln25 | Glu154 | 2.035 | Arg23 | Gln17 | 2.163 | ||
| Gln26 | Cys42 | 2.026 | Arg23 | Asn64 | 2.379 | ||
| Cys28 | Glu154 | 3.828 | AVP10 | Gln1 | Ala179 | 2.554 | |
| Gln30 | Thr24 | 1.993 | Val5 | Glu154 | 1.868 | ||
| Gln31 | Thr22 | 2.426 | Tyr7 | Ser132 | 2.501 | ||
| Gln31 | Thr23 | 2.093 | Tyr7 | Glu154 | 2.111 |
| Protein-Peptide Complex | ΔG (kcal/mol) | Kd (M) at 25.0 °C | H-Bond Ener. (kJ/mol) | Elec. Ener. (kJ/mol) | VDW. Ener. (kJ/mol) | Molecular Docking Score (kJ/mol) |
|---|---|---|---|---|---|---|
| Mpro–AVP1 | −11.2 | 5.8E−09 | −23.62 | 4.48 | −206.06 | −225.20 |
| Mpro–AVP2 | −10.5 | 1.9E−08 | −31.67 | 2.43 | −196.55 | −225.79 |
| Mpro–AVP3 | −11.0 | 9.3E−09 | −27.37 | 0.00 | −153.91 | −181.28 |
| Mpro–AVP4 | −14.5 | 2.2E−11 | −37.56 | 0.00 | −325.48 | −363.04 |
| Mpro–AVP5 | −9.9 | 5.7E−08 | −3.19 | 0.00 | −160.49 | −163.68 |
| Mpro–AVP6 | −11.0 | 9.1E−09 | −42.27 | 4.91 | −187.82 | −225.19 |
| Mpro–AVP7 | −8.2 | 9.3E−07 | −23.68 | 0.00 | −133.58 | −157.26 |
| Mpro–AVP8 | −10.0 | 4.5E−08 | −6.39 | −13.14 | −107.89 | −127.42 |
| Mpro–AVP9 | −12.6 | 5.9E−10 | −23.33 | 0.00 | −219.64 | −242.97 |
| Mpro–AVP10 | −9.4 | 1.3E−07 | −25.32 | 0.00 | −109.52 | −134.84 |
| Peptide ID | SVM Method | Quantitative Matrix (QM) Method | ||||||
|---|---|---|---|---|---|---|---|---|
| Model A* | Model B* | Model C* | Model D* | Model E* | Model F* | Model G* | Model H* | |
| AVP1 | −0.22 | −0.22 | −0.28 | −0.28 | 19.70 | 9.06 | 0.33 | −0.91 |
| AVP2 | −0.94 | −0.94 | −1.16 | −1.16 | −14.50 | −12.95 | −3.82 | −2.13 |
| AVP3 | −1.40 | −1.40 | −1.77 | −1.77 | −27.60 | −46.75 | −4.69 | −7.79 |
| AVP4 | −1.07 | −1.07 | −1.14 | −1.14 | −1.90 | −17.13 | −1.64 | −1.91 |
| AVP5 | −0.68 | −0.68 | −0.68 | −0.68 | −8.50 | −13.44 | −2.30 | −2.32 |
| AVP6 | −1.31 | −1.31 | −2.03 | −2.03 | −15.70 | −23.15 | −0.32 | −1.35 |
| AVP7 | −0.75 | −0.75 | −1.30 | −1.30 | 12.50 | 22.19 | 1.28 | 1.85 |
| AVP8 | −0.19 | −0.19 | −0.61 | −0.61 | 7.70 | 9.28 | −1.36 | 0.43 |
| AVP9 | −1.69 | −1.69 | −1.38 | −1.38 | −42.50 | −52.14 | −9.10 | −10.11 |
| AVP10 | −1.45 | −1.45 | −1.18 | −1.18 | −16.00 | −7.16 | −0.91 | −0.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harnkit, N.; Khongsonthi, T.; Masuwan, N.; Prasartkul, P.; Noikaew, T.; Chumnanpuen, P. Virtual Screening for SARS-CoV-2 Main Protease Inhibitory Peptides from the Putative Hydrolyzed Peptidome of Rice Bran. Antibiotics 2022, 11, 1318. https://doi.org/10.3390/antibiotics11101318
Harnkit N, Khongsonthi T, Masuwan N, Prasartkul P, Noikaew T, Chumnanpuen P. Virtual Screening for SARS-CoV-2 Main Protease Inhibitory Peptides from the Putative Hydrolyzed Peptidome of Rice Bran. Antibiotics. 2022; 11(10):1318. https://doi.org/10.3390/antibiotics11101318
Chicago/Turabian StyleHarnkit, Nathaphat, Thanakamol Khongsonthi, Noprada Masuwan, Pornpinit Prasartkul, Tipanart Noikaew, and Pramote Chumnanpuen. 2022. "Virtual Screening for SARS-CoV-2 Main Protease Inhibitory Peptides from the Putative Hydrolyzed Peptidome of Rice Bran" Antibiotics 11, no. 10: 1318. https://doi.org/10.3390/antibiotics11101318
APA StyleHarnkit, N., Khongsonthi, T., Masuwan, N., Prasartkul, P., Noikaew, T., & Chumnanpuen, P. (2022). Virtual Screening for SARS-CoV-2 Main Protease Inhibitory Peptides from the Putative Hydrolyzed Peptidome of Rice Bran. Antibiotics, 11(10), 1318. https://doi.org/10.3390/antibiotics11101318