
The Epistatic Landscape of Antibiotic Resistance of Different Clades of Mycobacterium tuberculosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
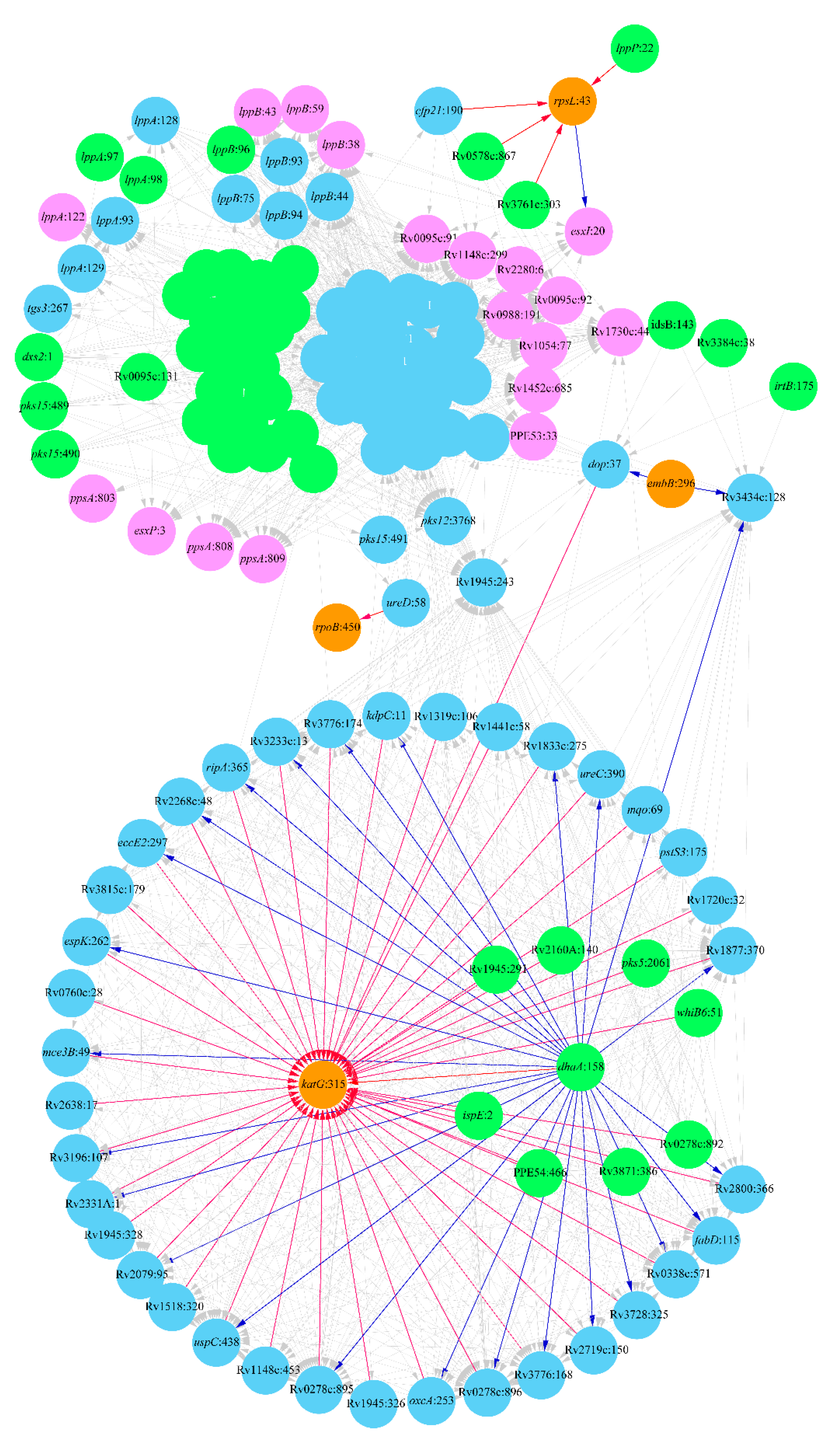
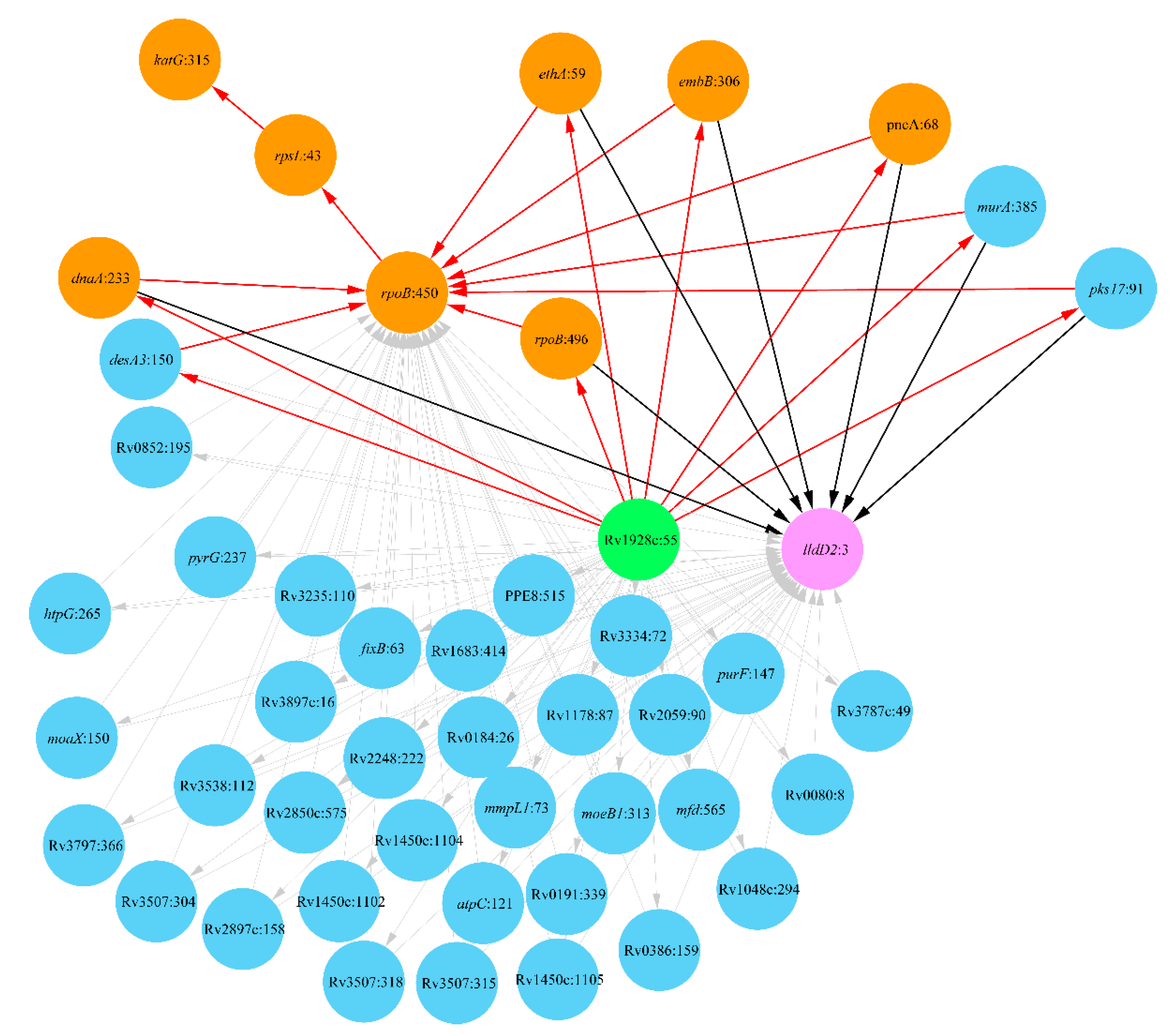
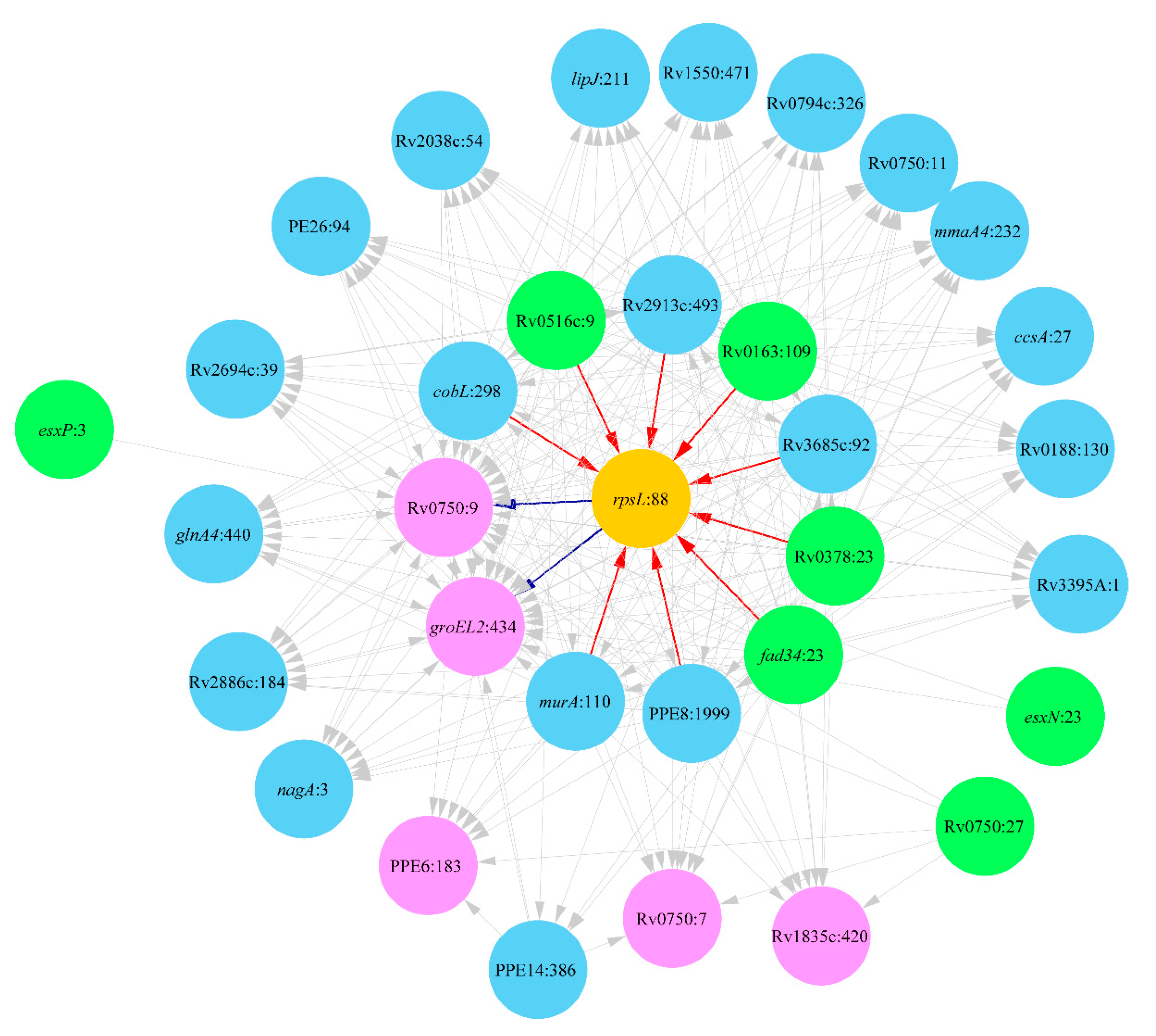
2. Results
2.1. Lineage 1: East African and Indian Clade
2.2. Lineage 2: Beijing Clade
2.3. Lineage 4: Haarlem and Ural Clades
2.3.1. Clade Haarlem
2.3.2. Clade Ural
3. Discussion
4. Materials and Methods
4.1. Data Sourcing
4.2. Functional Associations between Mtb Mutations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chakaya, J.; Khan, M.; Ntoumi, F.; Aklillu, E.; Fatima, R.; Mwaba, P.; Kapata, N.; Mfinanga, S.; Hasnain, S.E.; Katoto, P.D.; et al. Global Tuberculosis Report 2020—Reflections on the Global TB burden, treatment and prevention efforts. Int. J. Infect. Dis. in press.
- Lange, C.; Chesov, D.; Heyckendorf, J.; Leung, C.C.; Udwadia, Z.; Dheda, K. Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology 2018, 23, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, K.; Pierneef, R.; Reva, O.N.; Korostetskiy, I.S.; Ilin, A.I.; Akhmetova, G.K. Clade-Specific Distribution of Antibiotic Resistance Mutations in the Population of Mycobacterium tuberculosis. Prospects for Drug Resistance Reversion. In Basic Biology and Applications of Actinobacteria; Enany, S., Ed.; IntechOpen: London, UK, 2018; pp. 79–98. [Google Scholar]
- Banaei-Esfahani, A.; Nicod, C.; Aebersold, R.; Collins, B.C. Systems proteomics approaches to study bacterial pathogens: Application to Mycobacterium tuberculosis. Curr. Opin. Microbiol. 2017, 39, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Coll, F.; Phelan, J.; Hill-Cawthorne, G.A.; Nair, M.B.; Mallard, K.; Ali, S.; Abdallah, A.M.; Alghamdi, S.; Alsomali, M.; Ahmed, A.O. Genome-wide analysis of multi-and extensively drug-resistant Mycobacterium tuberculosis. Nat. Genet. 2018, 50, 307. [Google Scholar] [CrossRef]
- Wang, F.; Shao, L.; Fan, X.; Shen, Y.; Diao, N.; Jin, J.; Sun, F.; Wu, J.; Chen, J.; Weng, X. Evolution and transmission patterns of extensively drug-resistant tuberculosis in China. Antimicrob. Agents Chemother. 2015, 59, 818–825. [Google Scholar] [CrossRef]
- Perdigão, J.; Portugal, I. Genetics and roadblocks of drug resistant tuberculosis. Infect. Genet. Evol. 2019, 72, 113–130. [Google Scholar] [CrossRef]
- Casali, N.; Nikolayevskyy, V.; Balabanova, Y.; Harris, S.R.; Ignatyeva, O.; Kontsevaya, I.; Corander, J.; Bryant, J.; Parkhill, J.; Nejentsev, S. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 2014, 46, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Gygli, S.M.; Borrell, S.; Trauner, A.; Gagneux, S. Antimicrobial resistance in Mycobacterium tuberculosis: Mechanistic and evolutionary perspectives. FEMS Microbiol. Rev. 2017, 41, 354–373. [Google Scholar] [CrossRef]
- Coll, F.R.; McNerney, M.D.; Preston, J.A.; Guerra-Assunção, A.; Warry, G.; Hill-Cawthorne, K.; Mallard, M.; Nair, A.; Miranda, A.; Alves, A. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015, 7, 51. [Google Scholar] [CrossRef]
- Cohen, K.A.; Manson, A.L.; Desjardins, C.A.; Abeel, T.; Earl, A.M. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: Progress, promise, and challenges. Genome Med. 2019, 11, 45. [Google Scholar] [CrossRef]
- Zhang, H.; Li, D.; Zhao, L.; Fleming, J.; Lin, N.; Wang, T.; Liu, Z.; Li, C.; Galwey, N.; Deng, J. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 2013, 45, 1255. [Google Scholar] [CrossRef] [PubMed]
- Comas, I.; Gagneux, S. The past and future of tuberculosis research. PLoS Pathog. 2009, 5, e1000600. [Google Scholar] [CrossRef] [PubMed]
- Farhat, M.R.; Shapiro, B.J.; Kieser, K.J.; Sultana, R.; Jacobson, K.R.; Victor, T.C.; Warren, R.M.; Streicher, E.M.; Calver, A.; Sloutsky, A. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 2013, 45, 1183. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Kohl, T.A.; Omar, S.V.; Hedge, J.; Del Ojo Elias, C.; Bradley, P.; Iqbal, Z.; Feuerriegel, S.; Niehaus, K.E.; Wilson, D.J. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 1193–1202. [Google Scholar] [CrossRef]
- Georghiou, S.B.; Magana, M.; Garfein, R.S.; Catanzaro, D.G.; Catanzaro, A.; Rodwell, T.C. Evaluation of genetic mutations associated with Mycobacterium tuberculosis resistance to amikacin, kanamycin and capreomycin: A systematic review. PLoS ONE 2021, 7, e33275. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Contamin, L.; Nguyen, T.V.A.; Bañuls, A.L. Insights into the processes that drive the evolution of drug resistance in Mycobacterium tuberculosis. Evol. Appl. 2018, 11, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Trauner, A.; Borrell, S.; Reither, K.; Gagneux, S. Evolution of drug resistance in tuberculosis: Recent progress and implications for diagnosis and therapy. Drugs 2014, 74, 1063–1072. [Google Scholar] [CrossRef]
- Spies, F.S.; von Groll, A.; Ribeiro, A.W.; Ramos, D.F.; Ribeiro, M.O.; Dalla Costa, E.R.; Martin, A.; Palomino, J.C.; Rossetti, M.L.; Zaha, A. Biological cost in Mycobacterium tuberculosis with mutations in the rpsL, rrs, rpoB, and katG genes. Tuberculosis 2013, 93, 150–154. [Google Scholar] [CrossRef]
- Salvatore, P.P.; Becerra, M.C.; Abel zur Wiesch, P.; Hinkley, T.; Kaur, D.; Sloutsky, A.; Cohen, T. Fitness costs of drug resistance mutations in multidrug-resistant Mycobacterium tuberculosis: A household-based case-control study. J. Infect. Dis. 2016, 213, 149–155. [Google Scholar] [CrossRef]
- Huo, F.; Luo, J.; Shi, J.; Zong, Z.; Jing, W.; Dong, W.; Dong, L.; Ma, Y.; Liang, Q.; Shang, Y. A 10-year comparative analysis shows that increasing prevalence of rifampin-resistant Mycobacterium tuberculosis in China is associated with the transmission of strains harboring compensatory mutations. Antimicrob. Agents Chemother. 2018, 62, e02303–e02317. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; Abeel, T.; McGuire, A.M.; Desjardins, C.A.; Munsamy, V.; Shea, T.P.; Walker, B.J.; Bantubani, N.; Almeida, D.V.; Alvarado, L. Evolution of extensively drug-resistant tuberculosis over four decades: Whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med. 2015, 12, e1001880. [Google Scholar] [CrossRef] [PubMed]
- Zimic, M.; Vargas, A.; Kirwan, D.; Rios, A.; Gilman, R.; Sheen, P.; Grandjean, L. Determination of potentially novel compensatory mutations in rpoc associated with rifampin resistance and rpob mutations in Mycobacterium tuberculosis clinical isolates from peru. Int. J. Mycobacteriol. 2020, 9, 121–137. [Google Scholar]
- Al-Saeedi, M.; Al-Hajoj, S. Diversity and evolution of drug resistance mechanisms in Mycobacterium tuberculosis. Infect. Drug Resist. 2017, 10, 333–342. [Google Scholar] [CrossRef]
- Merker, M.; Barbier, M.; Cox, H.; Rasigade, J.-P.; Feuerriegel, S.; Kohl, T.A.; Diel, R.; Borrell, S.; Gagneux, S.; Nikolayevskyy, V. Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. eLife 2018, 7, e38200. [Google Scholar] [CrossRef]
- Safi, H.; Lingaraju, S.; Amin, A.; Kim, S.; Jones, M.; Holmes, M.; McNeil, M.; Peterson, S.N.; Chatterjee, D.; Fleischmann, R. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-D-arabinose biosynthetic and utilization pathway genes. Nat. Gen. 2013, 45, 1190–1197. [Google Scholar] [CrossRef]
- Shea, J.; Halse, T.A.; Kohlerschmidt, D.; Lapierre, P.; Modestil, H.A.; Kearns, C.H.; Dworkin, F.F.; Rakeman, J.L.; Escuyer, V.; Musser, K.A. Low-level rifampin resistance and rpoB mutations in Mycobacterium tuberculosis: An analysis of whole-genome sequencing and drug susceptibility test data in New York. J. Clin. Microbiol. 2021, 59, e01885-20. [Google Scholar] [CrossRef]
- Gagneux, S. Strain Variation in the Mycobacterium Tuberculosis Complex: Its Role in Biology, Epidemiology and Control; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1019. [Google Scholar]
- Li, Q.-j.; Jiao, W.-W.; Yin, Q.-Q.; Li, Y.-J.; Li, J.-Q.; Xu, F.; Sun, L.; Xiao, J.; Qi, H.; Wang, T. Positive epistasis of major low-cost drug resistance mutations rpoB531-TTG and katG315-ACC depends on the phylogenetic background of Mycobacterium tuberculosis strains. Int. J. Antimicrob. Agents 2017, 49, 757–762. [Google Scholar] [CrossRef]
- Fenner, L.; Egger, M.; Bodmer, T.; Altpeter, E.; Zwahlen, M.; Jaton, K.; Pfyffer, G.E.; Borrell, S.; Dubuis, O.; Bruderer, T. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3047–3053. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; El-Hay, T.; Wyres, K.L.; Weissbrod, O.; Munsamy, V.; Yanover, C.; Aharonov, R.; Shaham, O.; Conway, T.C.; Goldschmidt, Y.; et al. Paradoxical Hypersusceptibility of Drug-resistant Mycobacteriumtuberculosis to β-lactam Antibiotics. EBioMedicine 2016, 9, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, M.I.; Walker, T.M.; van der Werf, T.S.; Lange, C.; Niemann, S.; Merker, M. Pathogen-based precision medicine for drug-resistant tuberculosis. PLoS Pathog. 2018, 14, e1007297. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.D.; Knight, G.M.; McHugh, T.D. The complex evolution of antibiotic resistance in Mycobacterium tuberculosis. Int. J. Infect. Dis. 2015, 32, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Muzondiwa, D.; Mutshembele, A.; Pierneef, R.E.; Reva, O.N. Resistance Sniffer: An online tool for prediction of drug resistance patterns of Mycobacterium tuberculosis isolates using next generation sequencing data. Int. J. Med. Microbiol. 2020, 310, 151399. [Google Scholar] [CrossRef] [PubMed]
- Chernyaeva, E.N.; Shulgina, M.V.; Rotkevich, M.S.; Dobrynin, P.V.; Simonov, S.A.; Shitikov, E.A.; Ischenko, D.S.; Karpova, I.Y.; Kostryukova, E.S.; Ilina, E.N. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: A new tool for integrating sequence variations and epidemiology. BMC Genom. 2014, 15, 308. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, J.L.; Levin, B.; Paik, M.C. Statistical Methods for Rates and Proportions; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med. 2009, 6, e2. [Google Scholar] [CrossRef]
- Wiens, K.E.; Woyczynski, L.P.; Ledesma, J.R.; Ross, J.M.; Zenteno-Cuevas, R.; Goodridge, A.; Ullah, I.; Mathema, B.; Siawaya, J.F.D.; Biehl, M.H. Global variation in bacterial strains that cause tuberculosis disease: A systematic review and meta-analysis. BMC Med. 2018, 16, 196. [Google Scholar] [CrossRef]
- Avalos, E.; Catanzaro, D.; Catanzaro, A.; Ganiats, T.; Brodine, S.; Alcaraz, J.; Rodwell, T. Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: A systematic review. PLoS ONE 2015, 10, e0120470. [Google Scholar] [CrossRef]
- Dash, P.; Divya, M.B.; Guruprasad, L.; Guruprasad, K. Three-dimensional models of Mycobacterium tuberculosis proteins Rv1555, Rv1554 and their docking analyses with sildenafil, tadalafil, vardenafil drugs, suggest interference with quinol binding likely to affect protein’s function. BMC Struct. Biol. 2018, 18, 5. [Google Scholar] [CrossRef]
- White, M.J.; He, H.; Penoske, R.M.; Twining, S.S.; Zahrt, T.C. PepD participates in the mycobacterial stress response mediated through MprAB and SigE. J. Bacteriol. 2010, 192, 1498–1510. [Google Scholar] [CrossRef]
- Ramón-García, S.; Martín, C.; De Rossi, E.; Aínsa, J.A. Contribution of the Rv2333c efflux pump (the Stp protein) from Mycobacterium tuberculosis to intrinsic antibiotic resistance in Mycobacterium bovis BCG. J. Antimicrob. Chemother. 2007, 59, 544–547. [Google Scholar] [CrossRef]
- Ryoo, S.W.; Park, Y.K.; Park, S.N.; Shim, Y.S.; Liew, H.; Kang, S.; Bai, G.H. Comparative proteomic analysis of virulent Korean Mycobacterium tuberculosis K-strain with other mycobacteria strain following infection of U-937 macrophage. J. Microbiol. 2007, l45, 268–271. [Google Scholar]
- Vandal, O.H.; Pierini, L.M.; Schnappinger, D.; Nathan, C.F.; Ehrt, S. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 2008, 14, 849–854. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.; Yan, B.; Lyu, L.; Takiff, H.E.; Gao, Q. The potassium transporter KdpA affects persister formation by regulating ATP levels in Mycobacterium marinum. Emerg. Microbes Infect. 2020, 9, 129–139. [Google Scholar] [CrossRef]
- Kremer, L.; Nampoothiri, K.M.; Lesjean, S.; Dover, L.G.; Graham, S.; Betts, J.; Brennan, P.J.; Minnikin, D.E.; Locht, C.; Besra, G.S. Biochemical characterization of acyl carrier protein (AcpM) and malonyl-CoA:AcpM transacylase (mtFabD), two major components of Mycobacterium tuberculosis fatty acid synthase II. J. Biol. Chem. 2001, 276, 27967–27974. [Google Scholar] [CrossRef]
- Kavvas, E.S.; Catoiu, E.; Mih, N.; Yurkovich, J.T.; Seif, Y.; Dillon, N.; Heckmann, D.; Anand, A.; Yang, L.; Nizet, V. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat. Commun. 2018, 9, 4306. [Google Scholar] [CrossRef] [PubMed]
- Healy, C.; Gouzy, A.; Ehrt, S. Peptidoglycan hydrolases RipA and Ami1 are critical for replication and persistence of Mycobacterium tuberculosis in the host. MBio 2020, 11, e03315–e03319. [Google Scholar] [CrossRef]
- Özcelik, D.; Barandun, J.; Schmitz, N.; Sutter, M.; Guth, E.; Damberger, F.F.; Allain, F.H.; Ban, N.; Weber-Ban, E. Structures of Pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat. Commun. 2012, 3, 1014. [Google Scholar] [CrossRef]
- Sutcliffe, I.C.; Harrington, D.J. Lipoproteins of Mycobacterium tuberculosis: An abundant and functionally diverse class of cell envelope components. FEMS Microbiol. Rev. 2004, 28, 645–659. [Google Scholar] [CrossRef]
- Torrey, H.L.; Keren, I.; Via, L.E.; Lee, J.S.; Lewis, K. High persister mutants in Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0155127. [Google Scholar] [CrossRef] [PubMed]
- Bisson, G.P.; Mehaffy, C.; Broeckling, C.; Prenni, J.; Rifat, D.; Lun, D.S.; Burgos, M.; Weissman, D.; Karakousis, P.C.; Dobos, K. Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 6441–6452. [Google Scholar] [CrossRef] [PubMed]
- Brynildsrud, O.B.; Pepperell, C.S.; Suffys, P.; Grandjean, L.; Monteserin, J.; Debech, N.; Bohlin, J.; Alfsnes, K.; Pettersson, J.O.; Kirkeleite, I. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci. Adv. 2018, 4, eaat5869. [Google Scholar] [CrossRef]
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 2013, 45, 1176. [Google Scholar] [CrossRef] [PubMed]
- Furió, V.; Moreno-Molina, M.; Chiner-Oms, Á.; Villamayor, L.M.; Torres-Puente, M.; Comas, I. An evolutionary functional genomics approach identifies novel candidate regions involved in isoniazid resistance in Mycobacterium tuberculosis. BioRxiv 2020. [Google Scholar] [CrossRef]
- Eddabra, R.; Neffa, M. Mutations associated with rifampicin resistance in Mycobacterium tuberculosis isolates from Moroccan patients: Systematic review. Interdiscip. Perspect. Infect. Dis. 2020, 2020, 5185896. [Google Scholar] [CrossRef] [PubMed]
- Hicks, N.D.; Giffen, S.R.; Culviner, P.H.; Chao, M.C.; Dulberger, C.L.; Liu, Q.; Stanley, S.; Brown, J.; Sixsmith, J.; Wolf, I.D. Mutations in dnaA and a cryptic interaction site increase drug resistance in Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1009063. [Google Scholar] [CrossRef] [PubMed]
- Vilchèze, C.; Jacobs, W.R., Jr. Resistance to isoniazid and ethionamide in Mycobacterium tuberculosis: Genes, mutations, and causalities. Microbiol. Spectr. 2014, 2, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Plinke, C.; Walter, K.; Aly, S.; Ehlers, S.; Niemann, S. Mycobacterium tuberculosis embB codon 306 mutations confer moderately increased resistance to ethambutol in vitro and in vivo. Antimicrob. Agents Chemother. 2011, 55, 2891–2896. [Google Scholar] [CrossRef]
- Li, K.; Yang, Z.; Gu, J.; Luo, M.; Deng, J.; Chen, Y. Characterization of pncA mutations and prediction of PZA resistance in Mycobacterium tuberculosis clinical isolates from Chongqing, China. Front. Microbiol. 2021, 11, 594171. [Google Scholar] [CrossRef]
- Sinkov, V.; Ogarkov, O.; Mokrousov, I.; Bukin, Y.; Zhdanova, S.; Heysell, S.K. New epidemic cluster of pre-extensively drug resistant isolates of Mycobacterium tuberculosis Ural family emerging in Eastern Europe. BMC Genom. 2018, 19, 762. [Google Scholar] [CrossRef]
- Hatzios, S.K.; Baer, C.E.; Rustad, T.R.; Siegrist, M.S.; Pang, J.M.; Ortega, C.; Alber, T.; Grundner, C.; Sherman, D.R.; Bertozzi, C.R. Osmosensory signaling in Mycobacterium tuberculosis mediated by a eukaryotic-like Ser/Thr protein kinase. Proc. Natl. Acad. Sci. USA 2013, 110, E5069–E5077. [Google Scholar] [CrossRef]
- Goltermann, L.; Sarusie, M.V.; Bentin, T. Chaperonin GroEL/GroES over-expression promotes aminoglycoside resistance and reduces drug susceptibilities in Escherichia coli following exposure to sublethal aminoglycoside doses. Front. Microbiol. 2016, 6, 1572. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.; Anand, M.; Bhatt, A.; Kremer, L.; Jacobs, W.R., Jr.; Hatfull, G.F. GroEL1: A dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell 2005, 123, 861–873. [Google Scholar] [CrossRef]
- Goltermann, L.; Good, L.; Bentin, T. Chaperonins fight aminoglycoside-induced protein misfolding and promote short-term tolerance in Escherichia coli. J. Biol. Chem. 2013, 288, 10483–10489. [Google Scholar] [CrossRef] [PubMed]
- Borrell, S.; Gagneux, S. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2009, 13, 1456–1466. [Google Scholar] [PubMed]
- Javid, B.; Sorrentino, F.; Toosky, M.; Zheng, W.; Pinkham, J.T.; Jain, N.; Pan, M.; Deighan, P.; Rubin, E.J. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 1132–1137. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzondiwa, D.; Hlanze, H.; Reva, O.N. The Epistatic Landscape of Antibiotic Resistance of Different Clades of Mycobacterium tuberculosis. Antibiotics 2021, 10, 857. https://doi.org/10.3390/antibiotics10070857
Muzondiwa D, Hlanze H, Reva ON. The Epistatic Landscape of Antibiotic Resistance of Different Clades of Mycobacterium tuberculosis. Antibiotics. 2021; 10(7):857. https://doi.org/10.3390/antibiotics10070857
Chicago/Turabian StyleMuzondiwa, Dillon, Hleliwe Hlanze, and Oleg N. Reva. 2021. "The Epistatic Landscape of Antibiotic Resistance of Different Clades of Mycobacterium tuberculosis" Antibiotics 10, no. 7: 857. https://doi.org/10.3390/antibiotics10070857
APA StyleMuzondiwa, D., Hlanze, H., & Reva, O. N. (2021). The Epistatic Landscape of Antibiotic Resistance of Different Clades of Mycobacterium tuberculosis. Antibiotics, 10(7), 857. https://doi.org/10.3390/antibiotics10070857