
Elucidation of Teicoplanin Interactions with Drug Targets Related to COVID-19


Abstract
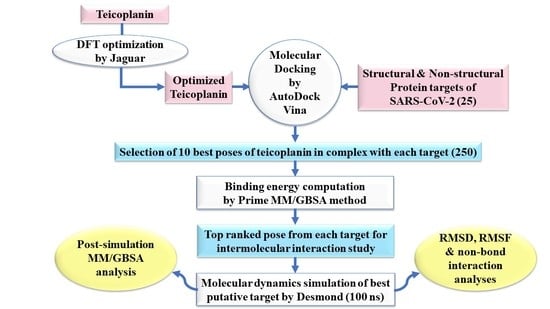
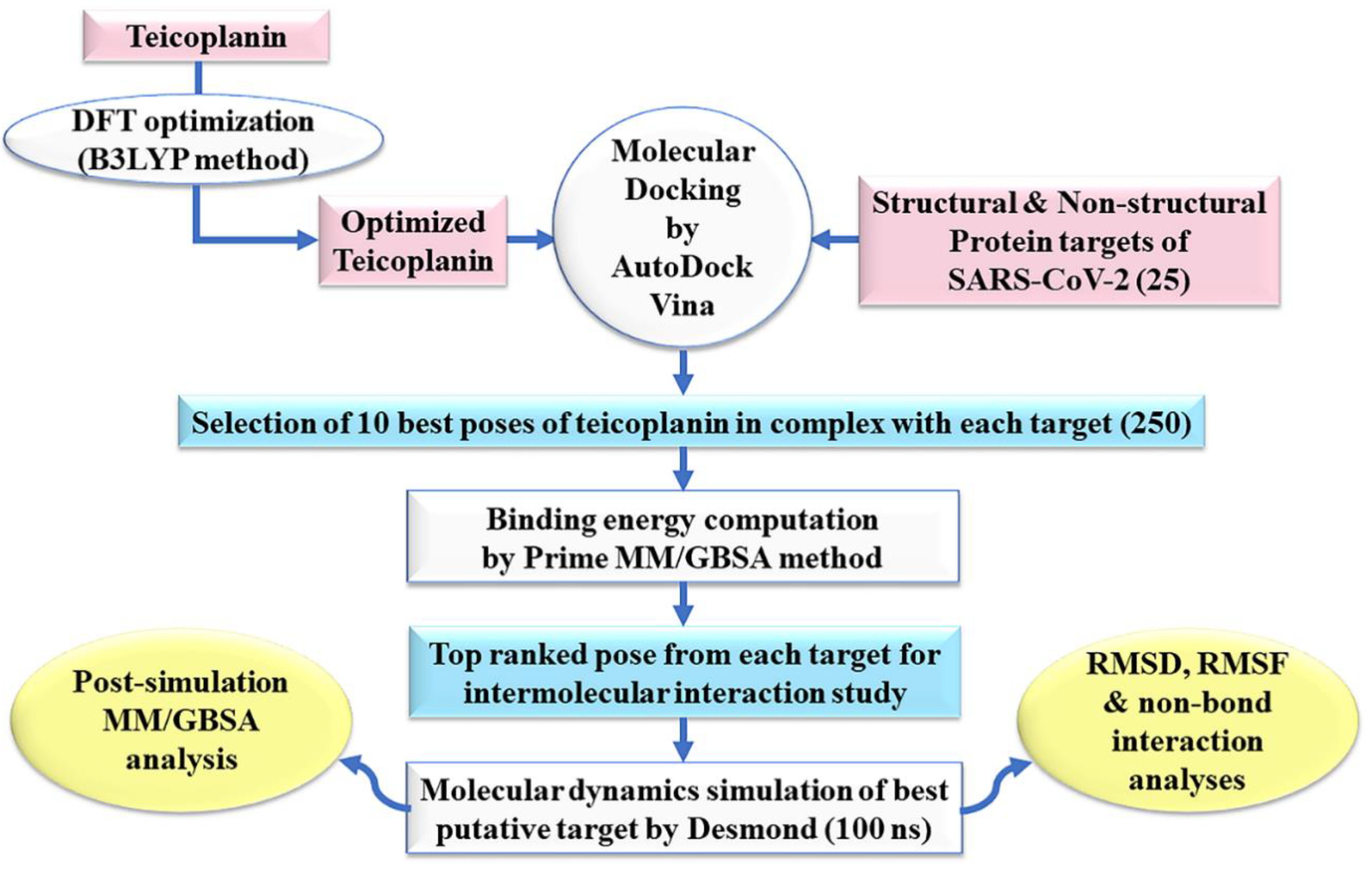
:
1. Introduction
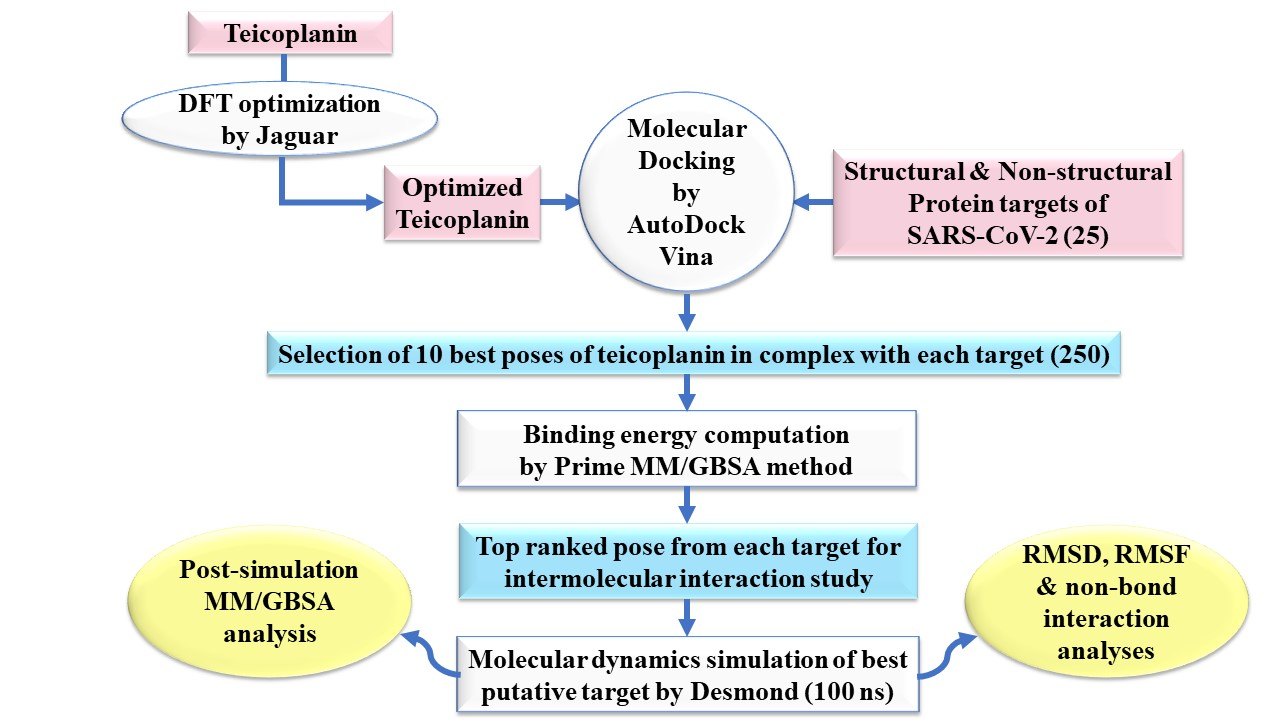
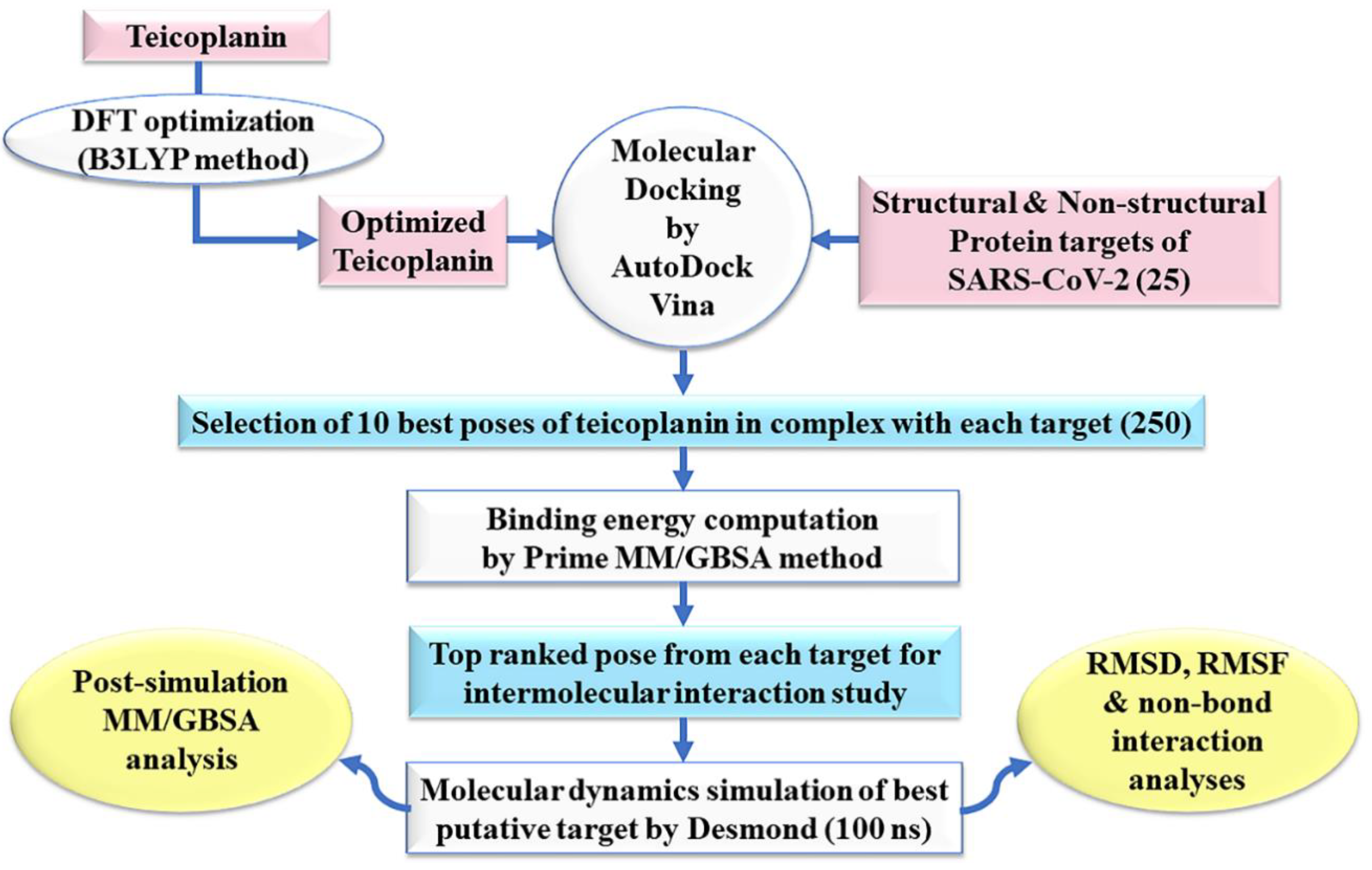
2. Experimental Section
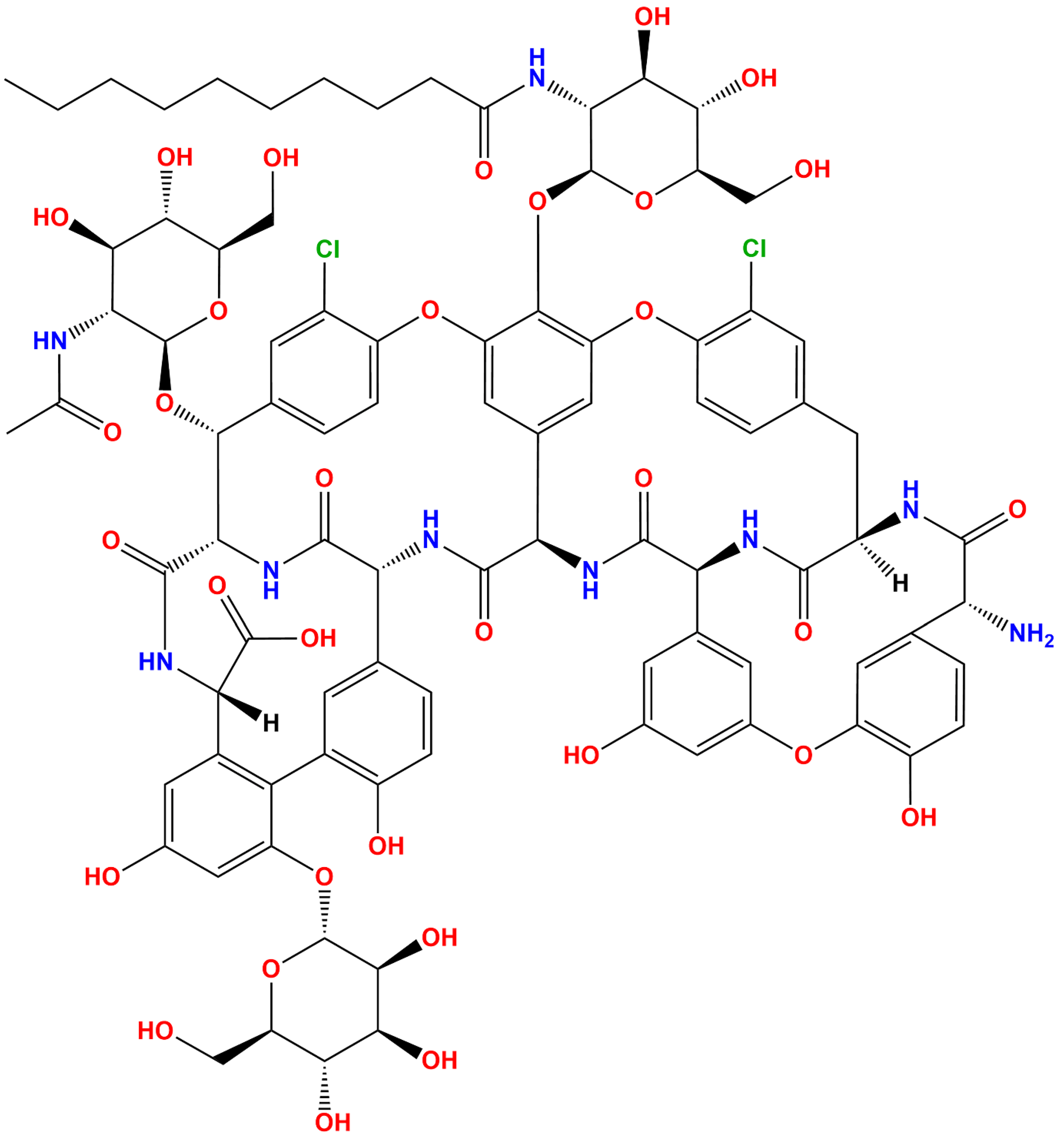
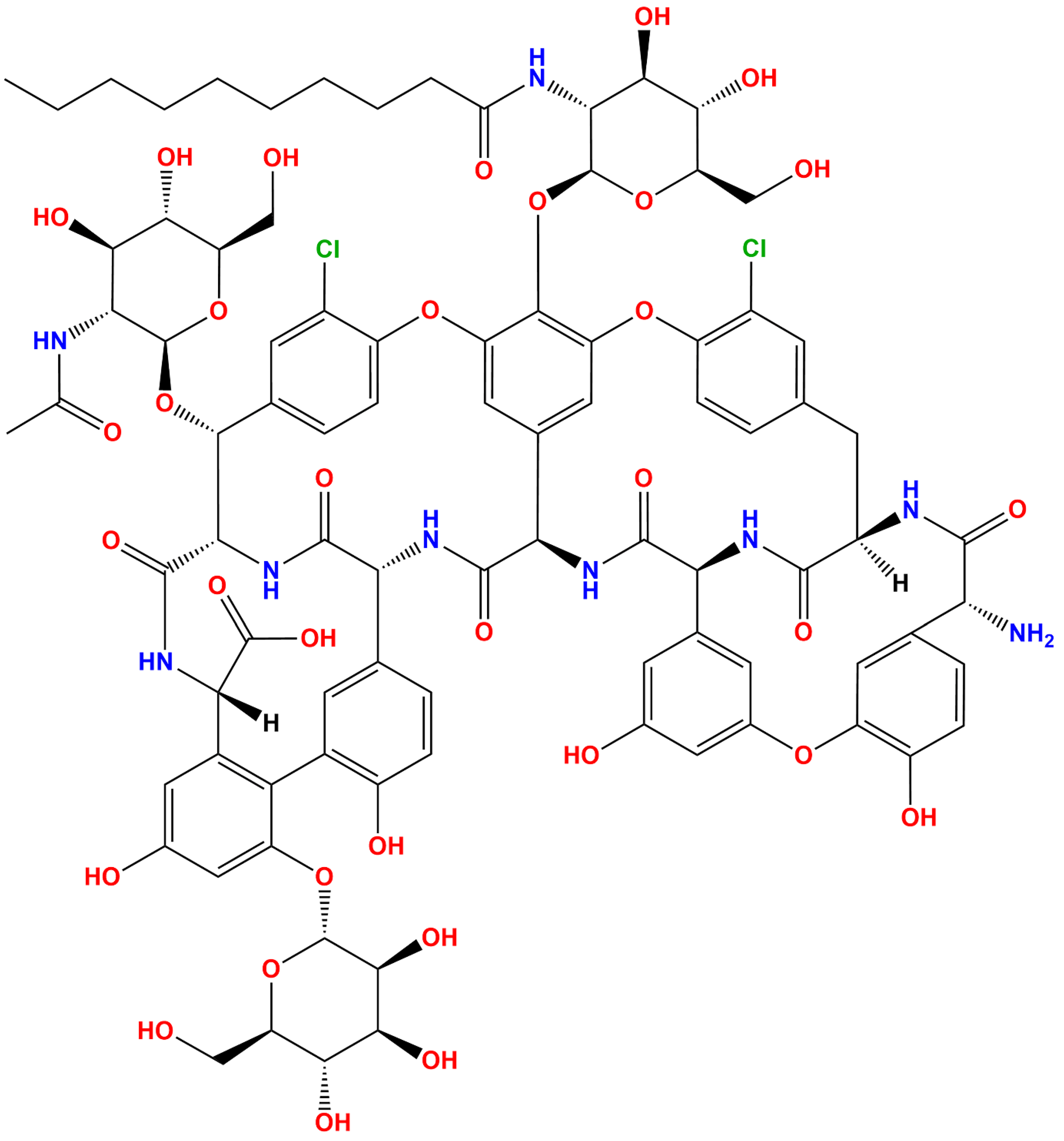
2.1. Ligand Preparation
2.2. Protein Preparation
2.3. Molecular Docking Simulation
2.4. Prime MM-GBSA Calculations
2.5. Molecular Dynamics Simulation
2.6. Post-Simulation MM-GBSA Analysis
3. Results and Discussion
3.1. Validation of Docking Protocol
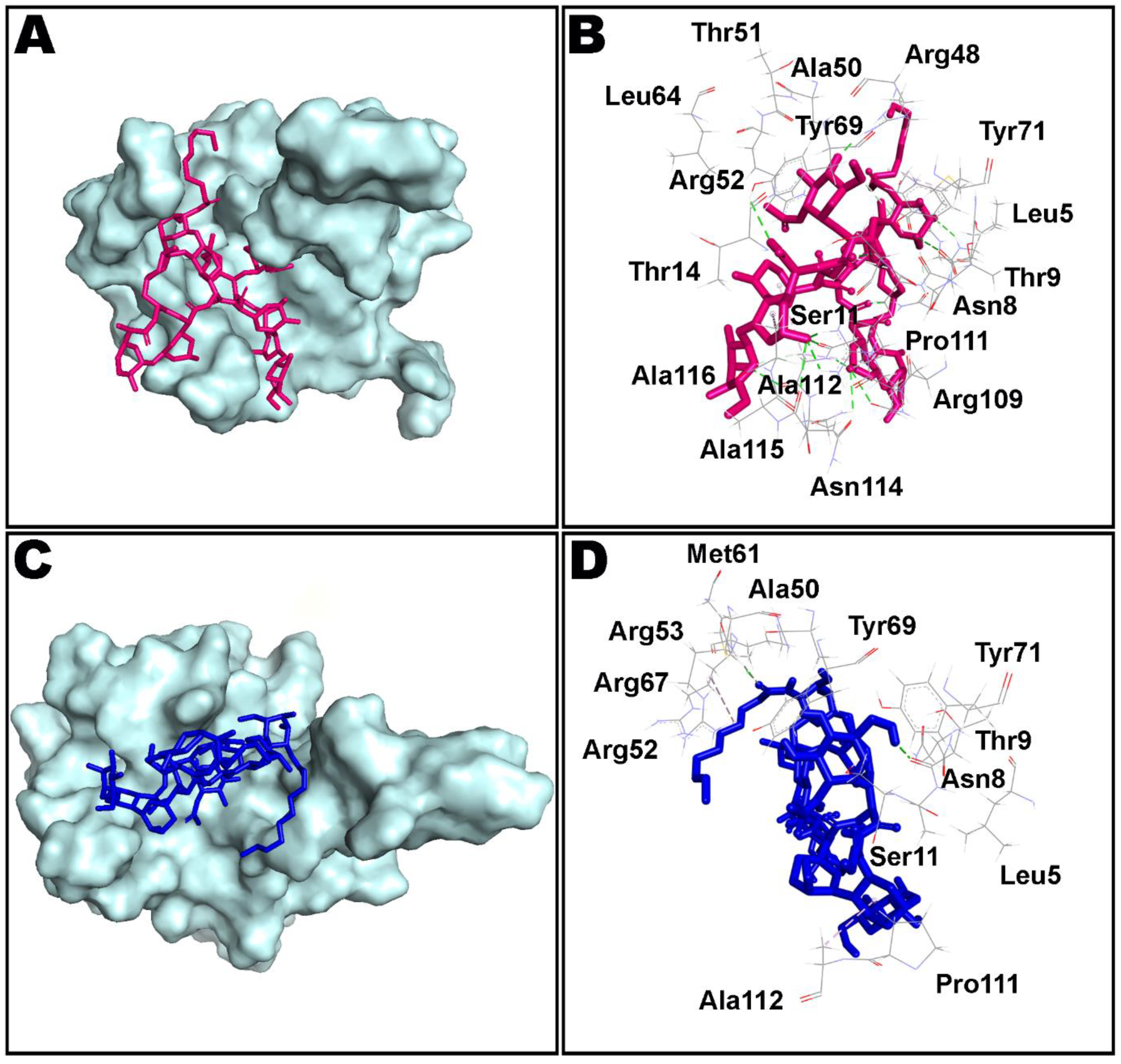
3.2. Molecular Docking of Teicoplanin with Potential Targets of SARS-CoV-2
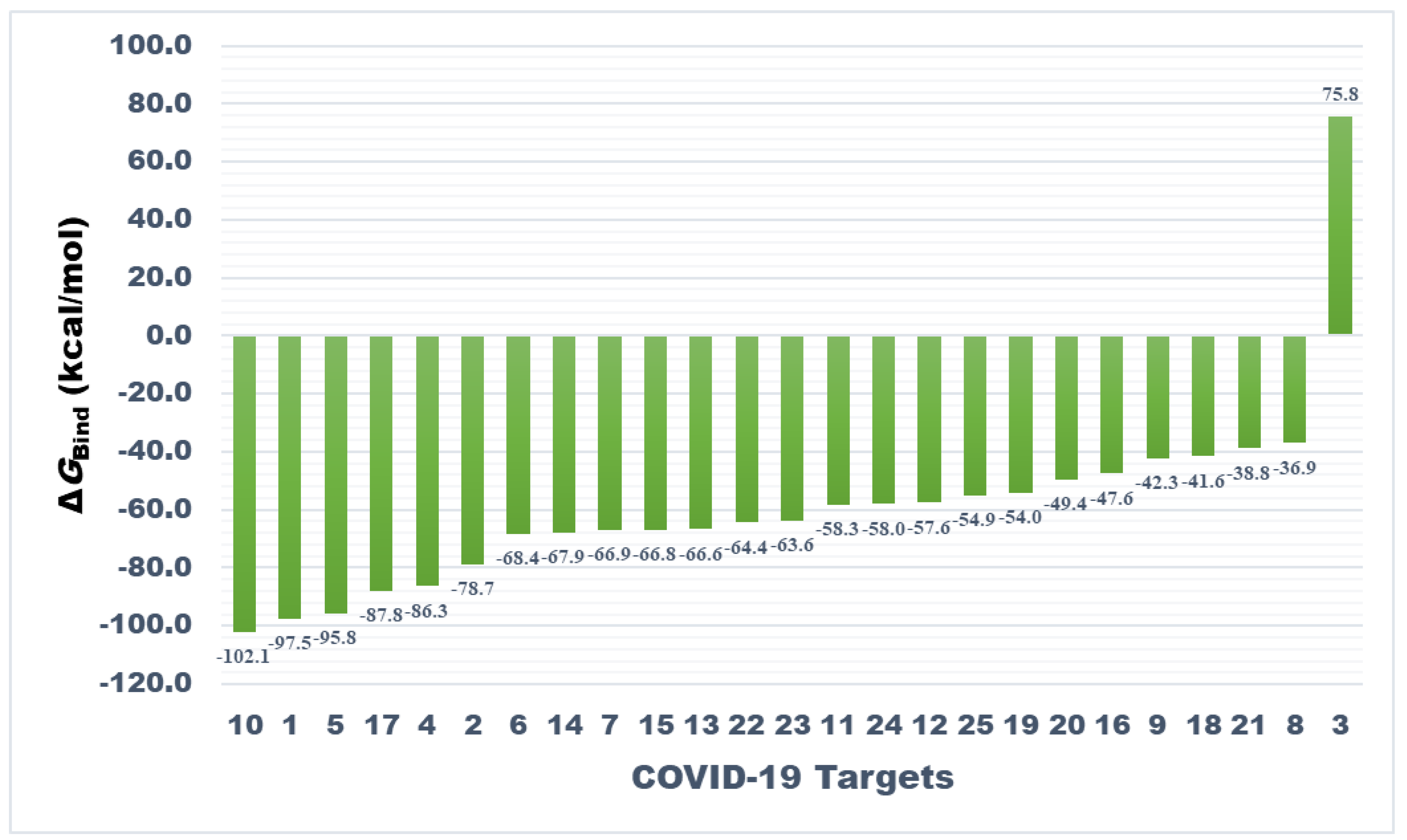
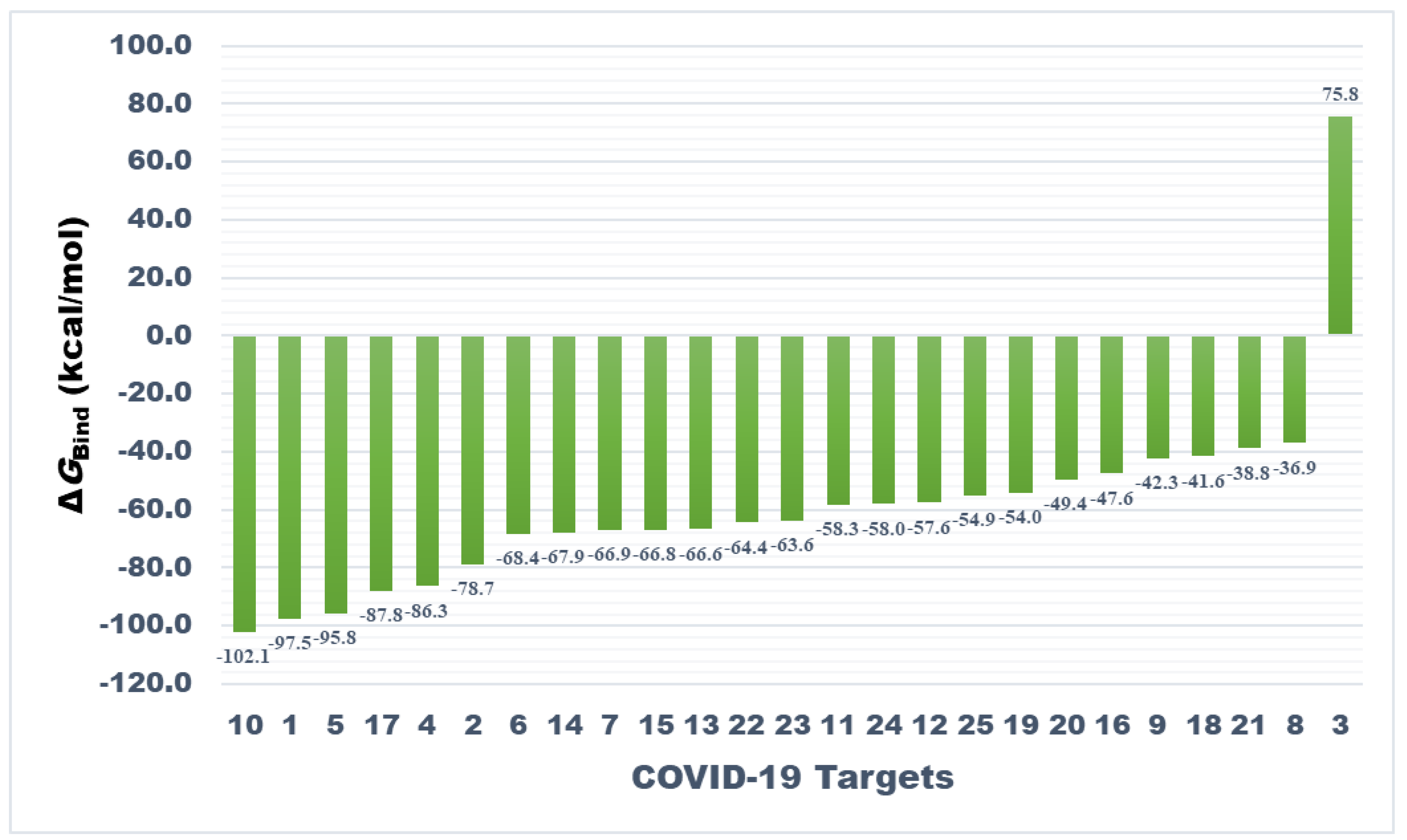
3.3. Prime MM-GBSA Calculations of Docked Complexes
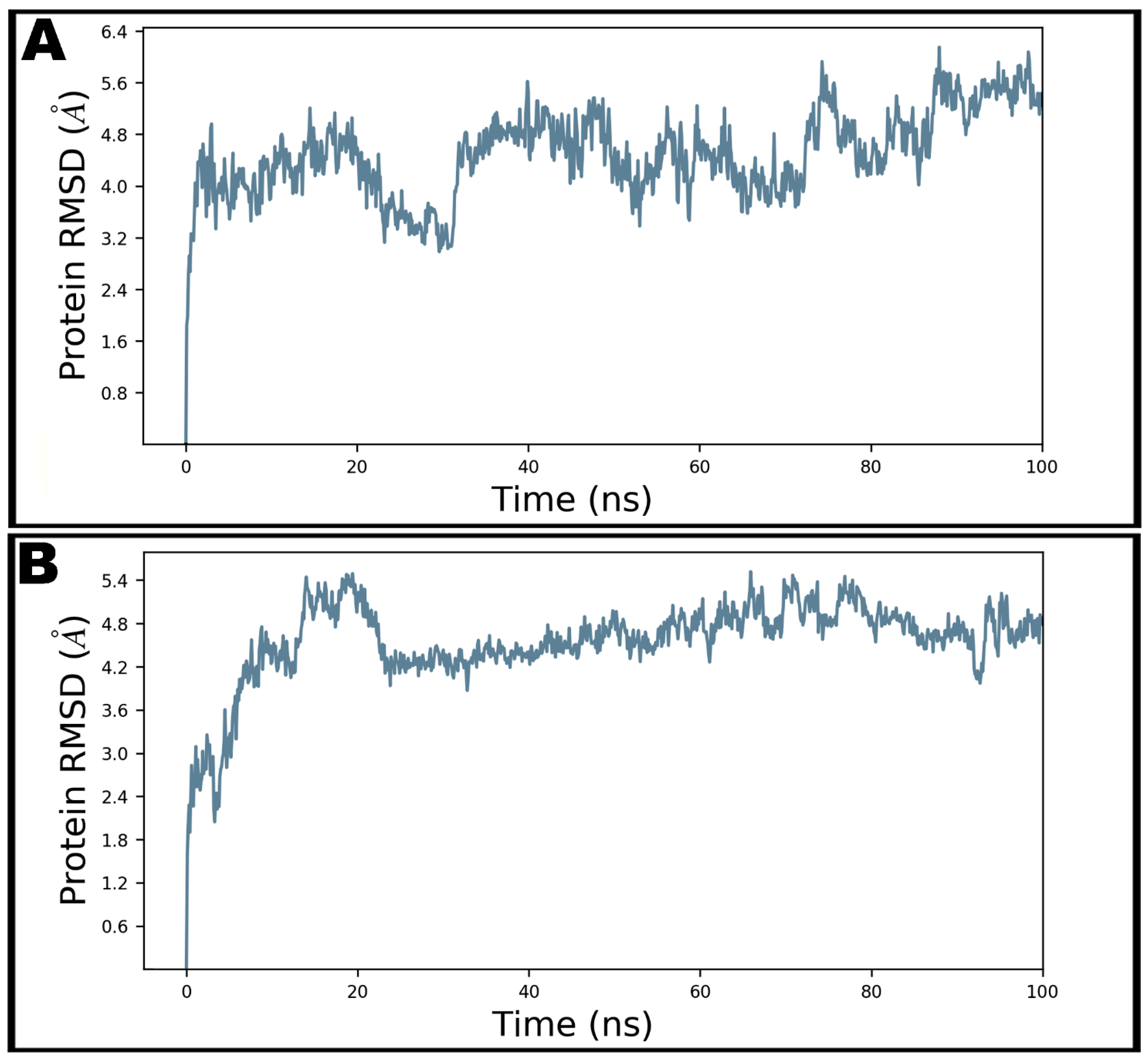
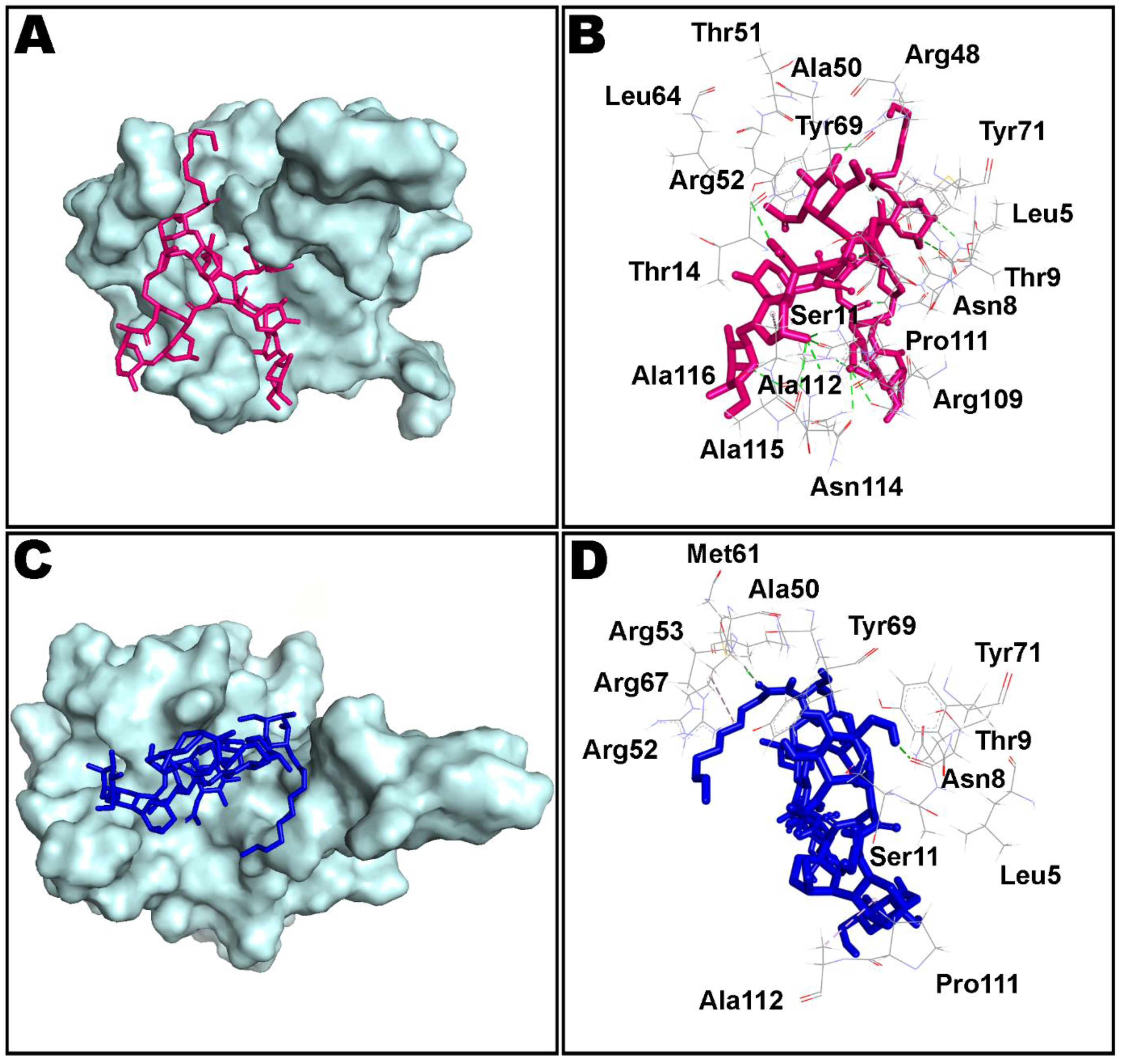
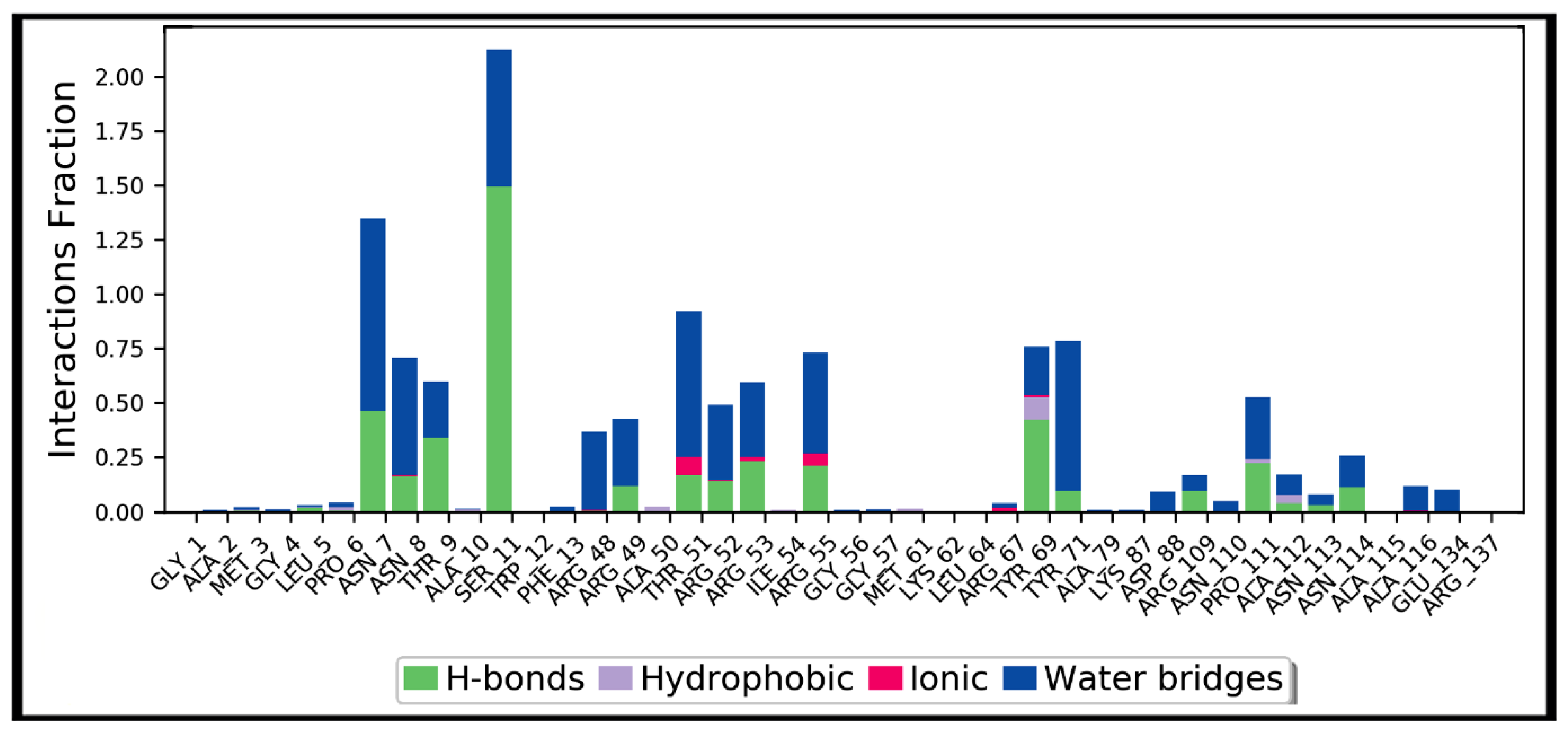
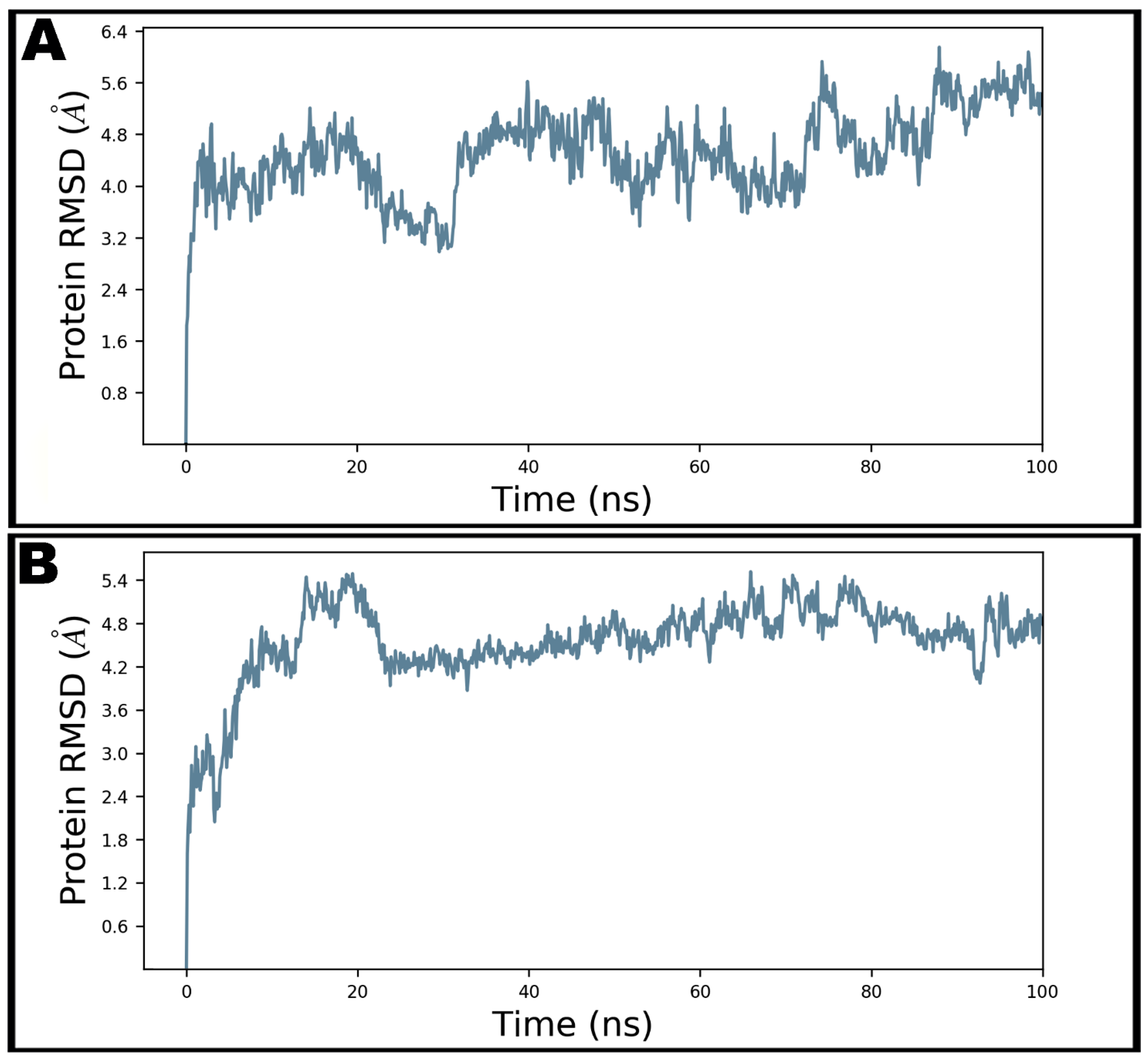
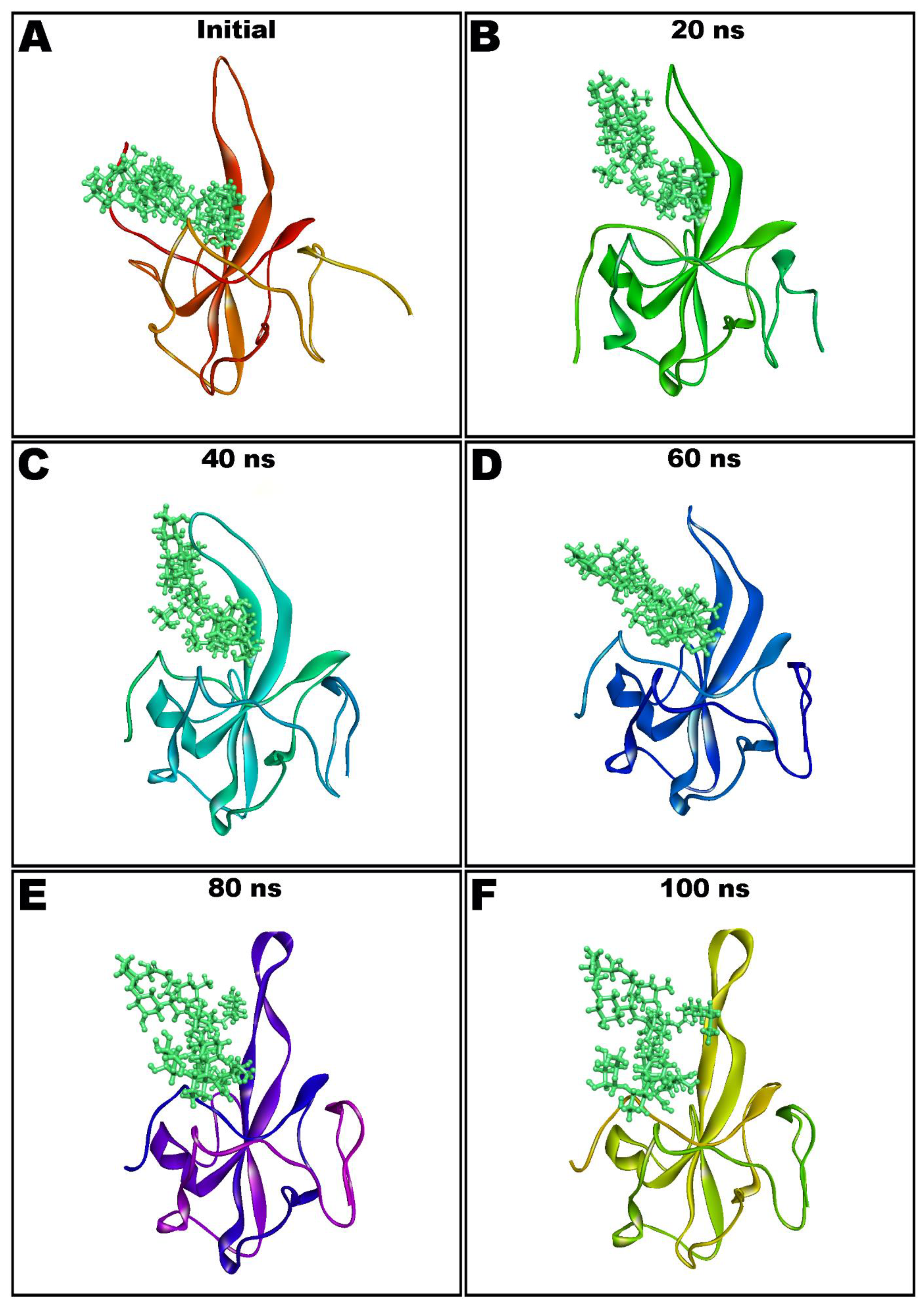
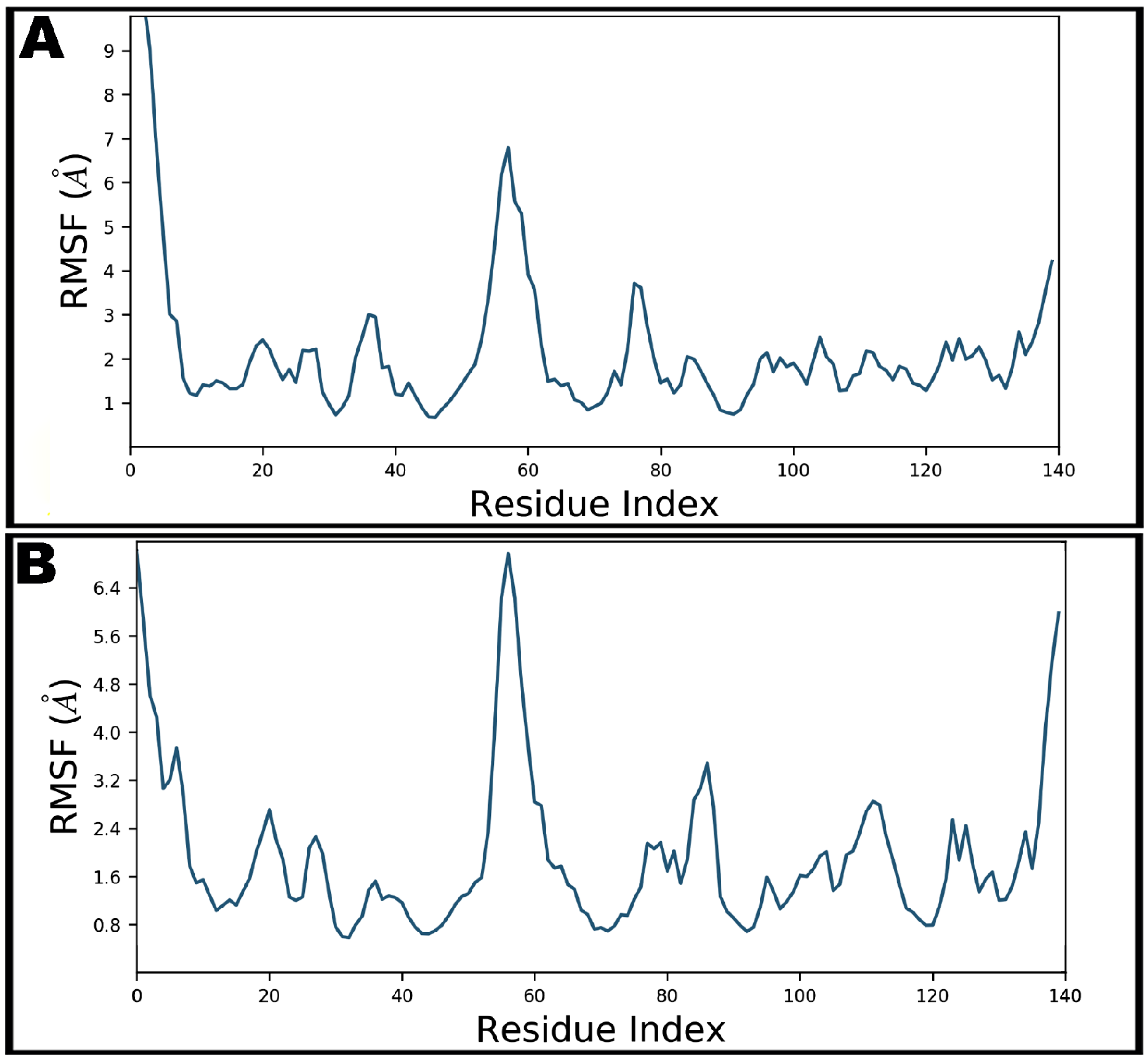
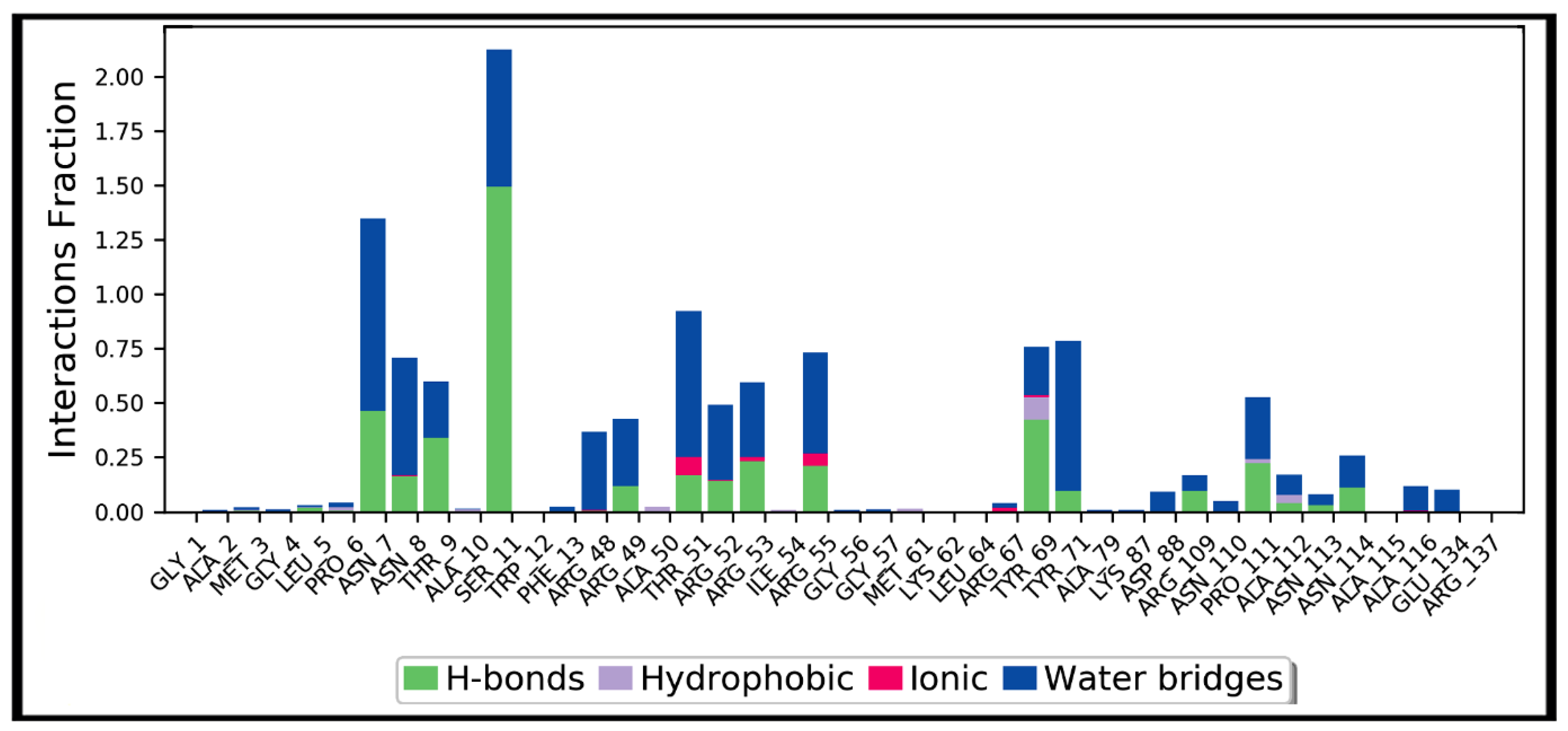
3.4. Molecular Dynamics Simulation Studies
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barkoff, C.M.; Mousa, S.A. Pharmacotherapy in COVID 19: Potential Impact of Targeting the Complement System. Biomedicines 2021, 9, 11. [Google Scholar] [CrossRef]
- Raoult, D.; Hsueh, P.-R.; Stefani, S.; Rolain, J.-M. COVID-19 Therapeutic and Prevention. Int. J. Antimicrob. Agents 2020, 55, 105937. [Google Scholar] [CrossRef]
- Clinical Management of COVID-19. Available online: https://www.who.int/publications/i/item/clinical-management-of-covid-19 (accessed on 31 December 2020).
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef] [Green Version]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef] [Green Version]
- Baron, S.A.; Devaux, C.; Colson, P.; Raoult, D.; Rolain, J.-M. Teicoplanin: An alternative drug for the treatment of COVID-19? Int. J. Antimicrob. Agents 2020, 55, 105944. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, X.; Yu, F.; Liu, J.; Zou, F.; Pan, T.; Zhang, H. Teicoplanin potently blocks the cell entry of 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.K.; Upadhyay, S.; Singh, M.; Raghavendhar, S.; Bhardwaj, M.; Sharma, P.; Patel, A.K. Screening and evaluation of approved drugs as inhibitors of main protease of SARS-CoV-2. Int. J. Biol. Macromol. 2020, 164, 2622–2631. [Google Scholar] [CrossRef]
- Hussain, M.S.; Azam, F.; Ahamed, K.F.H.N.; Ravichandiran, V.; Alkskas, I. Anti-endotoxin effects of terpenoids fraction from Hygrophila auriculata in lipopolysaccharide-induced septic shock in rats. Pharm. Biol. 2016, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, F.; Alabdullah, N.H.; Ehmedat, H.M.; Abulifa, A.R.; Taban, I.; Upadhyayula, S. NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. J. Biomol. Struct. Dyn. 2018, 36, 2099–2117. [Google Scholar] [CrossRef]
- Azam, F.; Taban, I.M.; Eid, E.E.M.; Iqbal, M.; Alam, O.; Khan, S.; Mahmood, D.; Anwar, M.J.; Khalilullah, H.; Khan, M.U. An in-silico analysis of ivermectin interaction with potential SARS-CoV-2 targets and host nuclear importin α. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Sandomenico, A.; Di Rienzo, L.; Calvanese, L.; Iaccarino, E.; D’Auria, G.; Falcigno, L.; Chambery, A.; Russo, R.; Franzoso, G.; Tornatore, L.; et al. Insights into the Interaction Mechanism of DTP3 with MKK7 by Using STD-NMR and Computational Approaches. Biomedicines 2021, 9, 20. [Google Scholar] [CrossRef]
- Azam, F.; Abodabos, H.S.; Taban, I.M.; Rfieda, A.R.; Mahmood, D.; Anwar, M.J.; Khan, S.; Sizochenko, N.; Poli, G.; Tuccinardi, T.; et al. Rutin as promising drug for the treatment of Parkinson’s disease: An assessment of MAO-B inhibitory potential by docking, molecular dynamics and DFT studies. Mol. Simul. 2019, 45. [Google Scholar] [CrossRef]
- Shushni, M.A.M.; Azam, F.; Lindequist, U. Oxasetin from Lophiostoma sp. of the Baltic Sea: Identification, in silico binding mode prediction and antibacterial evaluation against fish pathogenic bacteria. Nat. Prod. Commun. 2013, 8, 1223–1226. [Google Scholar] [CrossRef] [Green Version]
- Azam, F.; Vijaya Vara Prasad, M.; Thangavel, N.; Kumar Shrivastava, A.; Mohan, G. Structure-Based Design, Synthesis and Molecular Modeling Studies of Thiazolyl Urea Derivatives as Novel Anti-Parkinsonian Agents. Med. Chem. (Los Angeles) 2012, 8, 1057–1068. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: Jaguar; Schrödinger, LLC: New York, NY, USA, 2020.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, Version 20.1.0.19295; Dassault Systèmes: San Diego, CA, USA, 2020.
- The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2020.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmy, N.M.; Al-Sayed, E.; Moghannem, S.; Azam, F.; El-Shazly, M.; Singab, A.N. Breaking Down the Barriers to a Natural Antiviral Agent: Antiviral Activity and Molecular Docking of Erythrina speciosa Extract, Fractions, and the Major Compound. Chem. Biodivers. 2020, 17. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3; Schrödinger, LLC: New York, NY, USA, 2020; pp. 19–27.
- Vijayakumar, B.; Parasuraman, S.; Raveendran, R.; Velmurugan, D. Identification of natural inhibitors against angiotensin I converting enzyme for cardiac safety using induced fit docking and MM-GBSA studies. Pharmacogn. Mag. 2014, 10, S639. [Google Scholar]
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2020. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Method for simulations in the canonical ensemble. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A multiple-time-step molecular dynamics algorithm for macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Dill, K.A. Additivity Principles in Biochemistry. J. Biol. Chem. 1997, 272, 701–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azam, F.; Amer, A.M.; Rabulifa, A.; Elzwawi, M.M. Ginger components as new leads for the design and development of novel multi-targeted anti-Alzheimer’s drugs: A computational investigation. Drug Des. Dev. Ther. 2014, 8, 2045–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Kong, R.; Yang, G.; Xue, R.; Liu, M.; Wang, F.; Hu, J.; Guo, X.; Chang, S. COVID-19 Docking Server: A meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sgobba, M.; Caporuscio, F.; Anighoro, A.; Portioli, C.; Rastelli, G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440. [Google Scholar] [CrossRef]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural basis of RNA recognition by the SARS-CoV-2 nucleocapsid phosphoprotein. PLoS Pathog. 2020, 16, 1–16. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [PubMed] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Number | Targets | PDB ID | Grid Center for AutoDock Vina Program | Docking ΔGBind (kcal/mol) | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| 1. | Main protease | 6LU7 | −9.732 | 11.403 | 68.925 | −5.4 |
| 2. | Papain-like protease | 6WUU | 22.225 | 68.703 | 4.704 | −5.4 |
| 3. | RdRp (RTP site) | 7BV2 | 91.776 | 91.560 | 104.863 | −9.8 |
| 4. | RdRp (RNA site) | 7BV2 | 71.227 | 92.269 | 112.852 | −7.7 |
| 5. | Spike protein (RBD) | 6M0J | −36.193 | 37.260 | −5.752 | −6.1 |
| 6. | Spike monomer | 6VXX | 219.061 | 220.947 | 261.311 | −5.4 |
| 7. | Spike trimer | 6VYB | 251.872 | 195.411 | 243.040 | −6.9 |
| 8. | S2 protein (post fusion state) | 6LXT | −0.641 | 11.084 | 28.359 | −5.3 |
| 9. | N-protein (C-domain) | 6YUN | −10.288 | 12.683 | 7.740 | −5.6 |
| 10. | N-protein (N-domain) | 6YI3 | 16.299 | 11.628 | 6.638 | −7.4 |
| 11. | Nsp3 (AMP site) | 6W6Y | 9.124 | −8.677 | 16.220 | −7.3 |
| 12. | Nsp3 (MES site) | 6W6Y | 23.830 | 9.255 | 54.812 | −5.9 |
| 13. | Nsp7 | 7BV2 | 104.786 | 80.343 | 127.861 | −4.4 |
| 14. | Nsp8 | 7BV2 | 108.168 | 116.454 | 120.901 | −5.9 |
| 15. | Nsp9 | 6WXD | 53.119 | −10.095 | 22.482 | −4.5 |
| 16. | Nsp10 | 6WVN | 64.644 | 15.650 | 9.522 | −2.2 |
| 17. | Nsp12 | 7BV2 | 97.382 | 97.966 | 93.920 | −7.8 |
| 18. | Nsp13 (helicase ADP site) | 6JYT | 405.020 | 47.480 | 62.350 | −6.5 |
| 19. | Nsp13 (helicase NCB site) | 6JYT | 423.816 | 33.797 | 56.132 | −5.4 |
| 20. | Nsp14 (ExoN) | 5C8S | −39.712 | −50.654 | 15.5594 | −7.1 |
| 21. | Nsp14 (N7mtase) | 5C8S | −10.273 | −42.259 | −7.644 | −3.0 |
| 22. | Nsp15 (Exoribonuclease) | 6WLC | 94.134 | −19.803 | −25.857 | −6.5 |
| 23. | Nsp16 (GTA site) | 6WVN | 84.158 | 24.757 | 37.836 | −6.2 |
| 24. | Nsp16 (MGP site) | 6WVN | 100.029 | 38.995 | 18.481 | −7.4 |
| 25. | Nsp16 (SAM site) | 6WVN | 84.156 | 15.450 | 26.991 | −6.5 |
| Target Number | Target | ΔGBind a | ΔGCoul b | ΔGHBond c | ΔGLipo d | SolvGB e | ΔGvdw f | Lig SE g |
|---|---|---|---|---|---|---|---|---|
| 1. | Main protease | −97.55 | −19.11 | −0.68 | −59.81 | 44.25 | −68.46 | 3.87 |
| 2. | Papain-like protease | −78.75 | −39.06 | −3.33 | −58.73 | 54.34 | −42.74 | 39.85 |
| 3. | RdRp (RTP site) | 75.76 | −124.58 | −4.94 | −91.90 | 209.45 | −17.65 | 184.45 |
| 4. | RdRp (RNA site) | −86.33 | −68.97 | −6.24 | −13.97 | 78.93 | −83.45 | 10.54 |
| 5. | Spike protein (RBD) | −95.76 | −32.67 | −2.13 | −54.25 | 48.60 | −66.71 | 18.19 |
| 6. | Spike monomer | −68.38 | −61.51 | −4.39 | −13.35 | 55.98 | −50.51 | 12.74 |
| 7. | Spike trimer | −66.87 | −27.37 | −4.11 | −21.53 | 53.95 | −65.20 | 11.90 |
| 8. | S2 protein (post fusion state) | −36.93 | −23.22 | −2.74 | −14.11 | 55.19 | −61.27 | 24.45 |
| 9. | N-protein (C-domain) | −42.31 | −30.82 | −5.06 | −16.05 | 64.88 | −62.43 | 26.93 |
| 10. | N-protein (N-domain) | −102.13 | −40.95 | −6.98 | −24.95 | 61.07 | −78.69 | −0.90 |
| 11. | Nsp3 (AMP site) | −58.33 | −42.82 | −4.38 | −25.86 | 64.42 | −71.09 | 32.93 |
| 12. | Nsp3 (MES site) | −57.60 | −12.61 | −2.42 | −30.13 | 34.62 | −75.40 | 37.34 |
| 13. | Nsp7 | −66.61 | −54.39 | −4.15 | −19.17 | 55.49 | −60.78 | 8.43 |
| 14. | Nsp8 | −67.93 | −10.55 | −2.28 | −25.51 | 28.75 | −68.85 | 16.42 |
| 15. | Nsp9 | −66.83 | −43.87 | −3.59 | −19.55 | 46.75 | −57.02 | 10.15 |
| 16. | Nsp10 | −47.56 | −29.62 | −2.47 | −23.97 | 57.87 | −62.53 | 40.47 |
| 17. | Nsp12 | −87.84 | −96.33 | −8.48 | −19.26 | 135.75 | −108.20 | 8.52 |
| 18. | Nsp13 (helicase ADP site) | −41.57 | −63.94 | −7.00 | −19.36 | 88.70 | −53.79 | 16.23 |
| 19. | Nsp13 (helicase NCB site) | −53.99 | −42.21 | −4.05 | −26.40 | 87.73 | −78.57 | 7.39 |
| 20. | Nsp14 (ExoN) | −49.41 | −32.04 | −4.56 | −20.91 | 64.52 | −80.34 | 28.66 |
| 21. | Nsp14 (N7mtase) | −38.83 | −21.49 | −4.69 | −15.49 | 47.57 | −44.45 | 1.51 |
| 22. | Nsp15 (Exoribonuclease) | −64.40 | −48.67 | −6.39 | −23.20 | 47.29 | −55.77 | 43.43 |
| 23. | Nsp16 (GTA site) | −63.57 | −55.69 | −5.17 | −12.18 | 65.46 | −62.63 | 8.44 |
| 24. | Nsp16 (MGP site) | −58.04 | −31.42 | −3.89 | −22.67 | 59.61 | −79.15 | 30.67 |
| 25. | Nsp16 (SAM site) | −54.88 | −34.65 | −4.69 | −21.69 | 74.67 | −74.43 | 15.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, F. Elucidation of Teicoplanin Interactions with Drug Targets Related to COVID-19. Antibiotics 2021, 10, 856. https://doi.org/10.3390/antibiotics10070856
Azam F. Elucidation of Teicoplanin Interactions with Drug Targets Related to COVID-19. Antibiotics. 2021; 10(7):856. https://doi.org/10.3390/antibiotics10070856
Chicago/Turabian StyleAzam, Faizul. 2021. "Elucidation of Teicoplanin Interactions with Drug Targets Related to COVID-19" Antibiotics 10, no. 7: 856. https://doi.org/10.3390/antibiotics10070856
APA StyleAzam, F. (2021). Elucidation of Teicoplanin Interactions with Drug Targets Related to COVID-19. Antibiotics, 10(7), 856. https://doi.org/10.3390/antibiotics10070856