
High Risk Clone: A Proposal of Criteria Adapted to the One Health Context with Application to Enterotoxigenic Escherichia coli in the Pig Population



Abstract
1. Introduction
2. Results
2.1. Proposal of Criteria for a High Risk Clone in the One Health Context
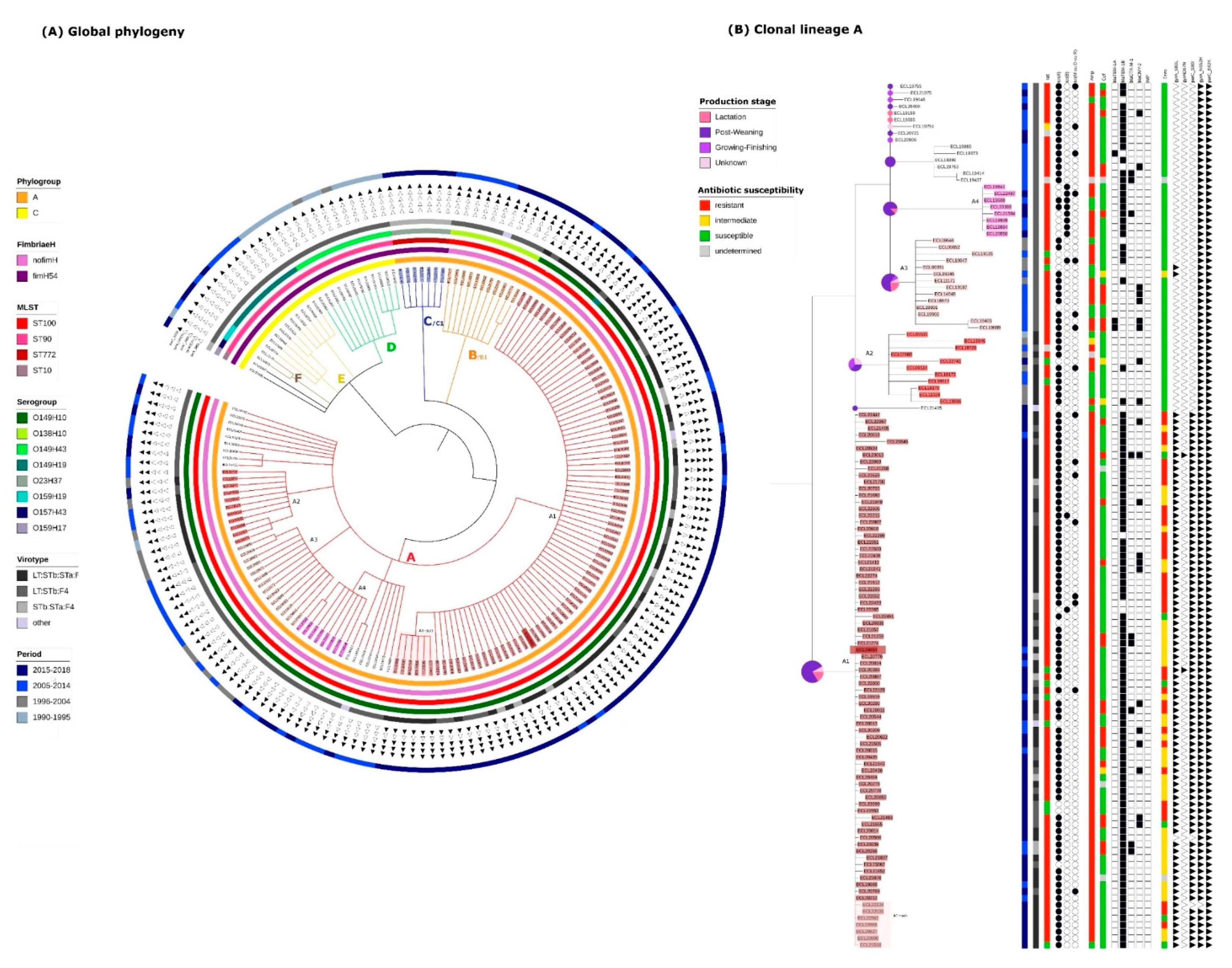
2.2. Identification of Clonal Lineages and Clones
2.3. Virulence Gene Profiles of the Different Clonal Lineages
2.4. Resistance Gene, Multidrug Resistance (MDR) and Replicon Profiles of the Different Clonal Lineages
2.5. Mortality Risk, Production Phase of Affected Pigs, and Persistence on Farm of the Different Clonal Lineages
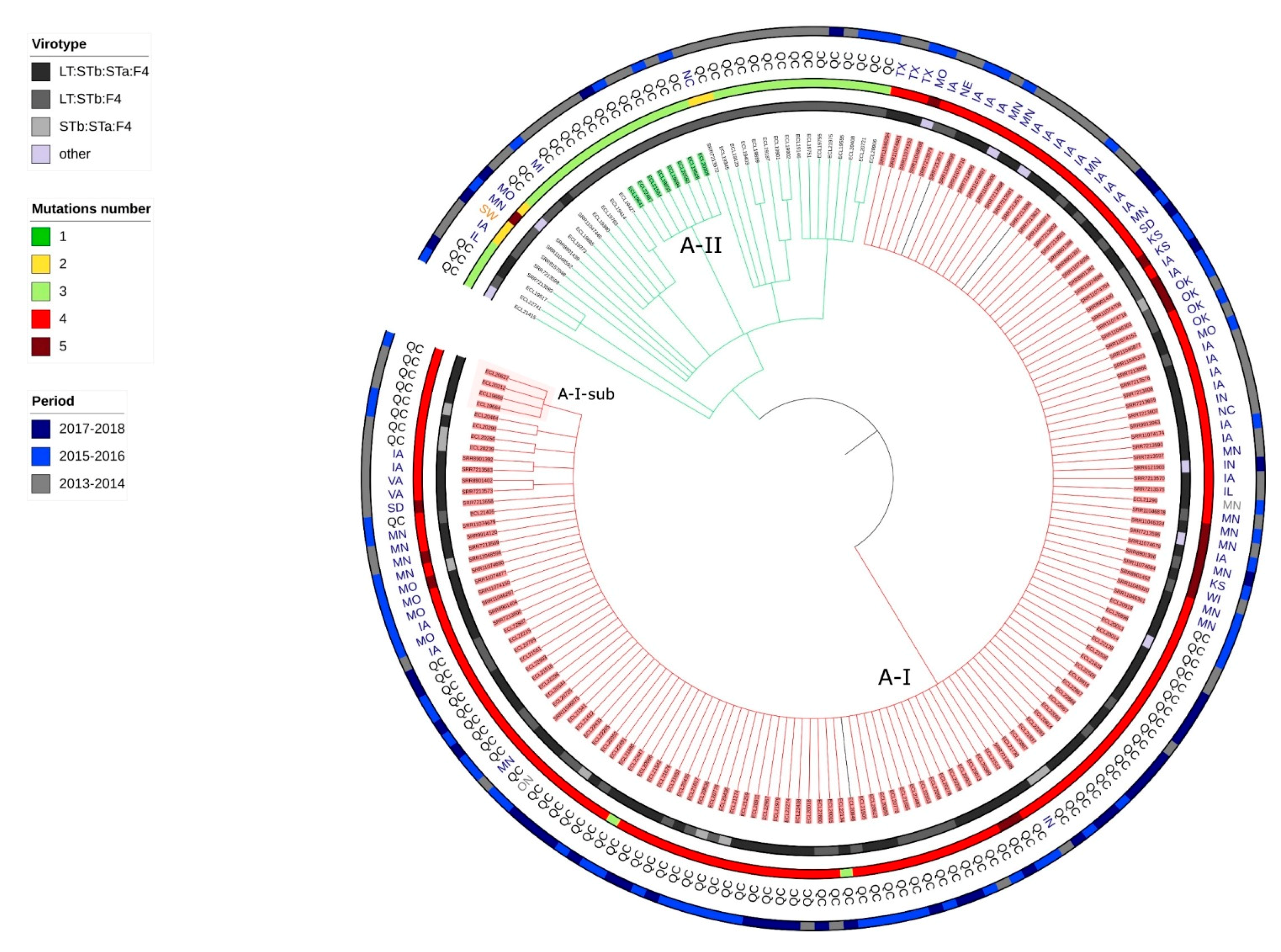
2.6. Presence of Isolates Belonging to the Clonal Lineage A in North America
2.7. Application of the Criteria for “High Risk” Clone to the Clones A-I, A2, A3, A4, C1 and B1
3. Discussion
4. Materials and Methods
4.1. Proposal of Criteria for a High Risk Clone in a One Health Context and Application
4.2. Isolate Selection
4.3. Antimicrobial Susceptibility
4.4. DNA Extraction, Library Preparation and Whole Genome Sequencing
4.5. Quality Assessment and Assembly
4.6. MLST, Serotype, Phylogroup and FimH
4.7. Virulence and Resistance Gene and Replicon Determination
4.8. Phylogenetic Analysis
4.9. Mortality Risk and Stage of Production
4.10. Selection of Other Whole Genome Sequences from GenBank
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Academies, C.o.C. When antibiotics fail. In The Expert Panel on the Potential Socio-Economic Impacts of Antimicrobial Resistance in Canada; Council of Canadian Academies: Ottawa, ON, Canada, 2019. [Google Scholar]
- Hu, Y.; Yang, X.; Li, J.; Lv, N.; Liu, F.; Wu, J.; Lin, I.Y.; Wu, N.; Weimer, B.C.; Gao, G.F.; et al. The Bacterial Mobile Resistome Transfer Network Connecting the Animal and Human Microbiomes. Appl. Environ. Microbiol. 2016, 82, 6672–6681. [Google Scholar] [CrossRef]
- Baquero, F.; Coque, T.M. Multilevel population genetics in antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 705–706. [Google Scholar] [CrossRef]
- Willems, R.J.; Hanage, W.P.; Bessen, D.E.; Feil, E.J. Population biology of Gram-positive pathogens: High-risk clones for dissemination of antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 872–900. [Google Scholar] [CrossRef]
- Woodford, N.; Turton, J.F.; Livermore, D.M. Multiresistant Gram-negative bacteria: The role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 736–755. [Google Scholar] [CrossRef]
- Mathers, A.J.; Peirano, G.; Pitout, J.D. Chapter Four-Escherichia coli ST131: The Quintessential Example of an International Multiresistant High-Risk Clone. Adv. Appl. Microbiol. 2015, 90, 109–154. [Google Scholar] [PubMed]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Mathers, A.J.; Peirano, G.; Pitout, J.D. The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clin. Microbiol. Rev. 2015, 28, 565–591. [Google Scholar] [CrossRef]
- Shomaker, S. OneHealth: A Paradigm for Interdisciplinary Collaboration. Acad. Med. 2015, 90, 997. [Google Scholar] [CrossRef]
- Marcusson, L.L.; Frimodt-Moller, N.; Hughes, D. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog. 2009, 5, e1000541. [Google Scholar] [CrossRef]
- Johnson, J.R.; Porter, S.B.; Thuras, P.; Johnson, T.J.; Price, L.B.; Tchesnokova, V.; Sokurenko, E.V. Greater Ciprofloxacin Tolerance as a Possible Selectable Phenotype Underlying the Pandemic Spread of the H30 Subclone of Escherichia coli Sequence Type 131. Antimicrob. Agents Chemother. 2015, 59, 7132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fuzi, M.; Rodriguez Bano, J.; Toth, A. Global Evolution of Pathogenic Bacteria With Extensive Use of Fluoroquinolone Agents. Front. Microbiol. 2020, 11, 271. [Google Scholar] [CrossRef]
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Sokurenko, E.V.; Tchesnokova, V. Intensity and mechanisms of fluoroquinolone resistance within the H30 and H30Rx subclones of Escherichia coli sequence type 131 compared with other fluoroquinolone-resistant E. coli. Antimicrob. Agents Chemother. 2015, 59, 4471–4480. [Google Scholar] [CrossRef][Green Version]
- Gyles, C.L.; Prescott, J.F.; Songer, J.G.; Thoen, C.O. Pathogenesis of Bacterial Infections in Animals, 4th ed.; Blackwell Publishing, Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Luppi, A. Swine enteric colibacillosis: Diagnosis, therapy and antimicrobial resistance. Porcine Health Manag. 2017, 3, 16. [Google Scholar] [CrossRef]
- de Lagarde, M.; Vanier, G.; Desmarais, G.; Kohan-Ghadr, H.R.; Arsenault, J.; Fairbrother, J.M. A new multidrug-resistant Enterotoxigenic Escherichia coli Pulsed-Field Gel Electrophoresis cluster associated with enrofloxacin non-susceptibility in diseased pigs. J. Appl. Microbiol. 2020. [Google Scholar] [CrossRef]
- Jiang, F.; Wu, Z.; Zheng, Y.; Frana, T.S.; Sahin, O.; Zhang, Q.; Li, G. Genotypes and Antimicrobial Susceptibility Profiles of Hemolytic Escherichia coli from Diarrheic Piglets. Foodborne Pathog. Dis. 2019, 16, 94–103. [Google Scholar] [CrossRef]
- Kusumoto, M.; Hikoda, Y.; Fujii, Y.; Murata, M.; Miyoshi, H.; Ogura, Y.; Gotoh, Y.; Iwata, T.; Hayashi, T.; Akiba, M. Emergence of a Multidrug-Resistant Shiga Toxin-Producing Enterotoxigenic Escherichia coli Lineage in Diseased Swine in Japan. J. Clin. Microbiol. 2016, 54, 1074–1081. [Google Scholar] [CrossRef]
- Kaspersen, H.; Urdahl, A.M.; Grontvedt, C.A.; Gulliksen, S.M.; Tesfamichael, B.; Slettemeas, J.S.; Norstrom, M.; Sekse, C. Actinobacillus pleuropneumoniae Eradication with Enrofloxacin May Lead to Dissemination and Long-Term Persistence of Quinolone Resistant Escherichia coli in Pig Herds. Antibiotics 2020, 9, 910. [Google Scholar] [CrossRef]
- Hayer, S.S.; Lim, S.; Hong, S.; Elnekave, E.; Johnson, T.; Rovira, A.; Vannucci, F.; Clayton, J.B.; Perez, A.; Alvarez, J. Genetic Determinants of Resistance to Extended-Spectrum Cephalosporin and Fluoroquinolone in Escherichia coli Isolated from Diseased Pigs in the United States. mSphere 2020, 5. [Google Scholar] [CrossRef]
- Baquero, F.; Tedim, A.P.; Coque, T.M. Antibiotic resistance shaping multi-level population biology of bacteria. Front. Microbiol. 2013, 4, 15. [Google Scholar] [CrossRef]
- Wang, L.; Crameri, G. Emerging zoonotic viral diseases. Rev. Sci. Tech. 2014, 33, 569–581. [Google Scholar] [CrossRef]
- Falkow, S. Is Persistent Bacterial Infection Good for Your Health? Cell 2006, 124, 699–702. [Google Scholar] [CrossRef]
- Blackburn, J.K.; Ganz, H.H.; Ponciano, J.M.; Turner, W.C.; Ryan, S.J.; Kamath, P.; Cizauskas, C.; Kausrud, K.; Holt, R.D.; Stenseth, N.C.; et al. Modeling R(0) for Pathogens with Environmental Transmission: Animal Movements, Pathogen Populations, and Local Infectious Zones. Int. J. Environ. Res. Public Health 2019, 16, 954. [Google Scholar] [CrossRef]
- WHO (World Health Organisation). One Health. Available online: https://www.who.int/features/qa/one-health/en/ (accessed on 1 February 2017).
- McEwen, S.A.; Collignon, P.J. Antimicrobial Resistance: A One Health Perspective. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Vingopoulou, E.I.; Siarkou, V.I.; Batzias, G.; Kaltsogianni, F.; Sianou, E.; Tzavaras, I.; Koutinas, A.; Saridomichelakis, M.N.; Sofianou, D.; Tzelepi, E.; et al. Emergence and maintenance of multidrug-resistant Escherichia coli of canine origin harbouring a blaCMY-2-IncI1/ST65 plasmid and topoisomerase mutations. J. Antimicrob. Chemother. 2014, 69, 2076–2080. [Google Scholar] [CrossRef][Green Version]
- van der Putten, B.C.L.; Remondini, D.; Pasquini, G.; Janes, V.A.; Matamoros, S.; Schultsz, C. Quantifying the contribution of four resistance mechanisms to ciprofloxacin MIC in Escherichia coli: A systematic review. J. Antimicrob. Chemother. 2018, 74, 298–310. [Google Scholar] [CrossRef]
- Carattoli, A. Resistance plasmid families in Enterobacteriaceae. Antimicrob. Agents Chemother. 2009, 53, 2227–2238. [Google Scholar] [CrossRef]
- Carattoli, A. Plasmids in Gram negatives: Molecular typing of resistance plasmids. Int. J. Med. Microbiol. 2011, 301, 654–658. [Google Scholar] [CrossRef]
- Cantón, R.; González-Alba, J.; Galán, J. CTX-M enzymes: Origin and diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- Price, L.B.; Johnson, J.R.; Aziz, M.; Clabots, C.; Johnston, B.; Tchesnokova, V.; Nordstrom, L.; Billig, M.; Chattopadhyay, S.; Stegger, M.; et al. The epidemic of extended-spectrum-beta-lactamase-producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. MBio 2013, 4. [Google Scholar] [CrossRef]
- Garnett, J.A.; Martínez-Santos, V.I.; Saldaña, Z.; Pape, T.; Hawthorne, W.; Chan, J.; Simpson, P.J.; Cota, E.; Puente, J.L.; Girón, J.A.; et al. Structural insights into the biogenesis and biofilm formation by the Escherichia coli common pilus. Proc. Natl. Acad. Sci. USA 2012, 109, 3950–3955. [Google Scholar] [CrossRef] [PubMed]
- Xicohtencatl-Cortes, J.; Monteiro-Neto, V.; Saldaña, Z.; Ledesma, M.A.; Puente, J.L.; Girón, J.A. The Type 4 Pili of Enterohemorrhagic Escherichia coli O157:H7 Are Multipurpose Structures with Pathogenic Attributes. J. Bacteriol. 2009, 191, 411–421. [Google Scholar] [CrossRef]
- Journet, L.; Cascales, E. The type VI secretion system in Escherichia coli and related species. EcoSal Plus 2016, 7, 1–34. [Google Scholar] [CrossRef]
- Krishnan, S.; Prasadarao, N.V. Outer membrane protein A and OprF: Versatile roles in Gram-negative bacterial infections. FEBS J. 2012, 279, 919–931. [Google Scholar] [CrossRef]
- Sniegowski, P.D.; Gerrish, P.J.; Lenski, R.E. Evolution of high mutation rates in experimental populations of E. coli. Nature 1997, 387, 703–705. [Google Scholar] [CrossRef]
- Grad, Y.H.; Lipsitch, M.; Feldgarden, M.; Arachchi, H.M.; Cerqueira, G.C.; FitzGerald, M.; Godfrey, P.; Haas, B.J.; Murphy, C.I.; Russ, C. Genomic epidemiology of the Escherichia coli O104: H4 outbreaks in Europe, 2011. Proc. Natl. Acad. Sci. USA 2012, 109, 3065–3070. [Google Scholar] [CrossRef]
- Ribot, E.M.; Fair, M.A.; Gautom, R.; Cameron, D.N.; Hunter, S.B.; Swaminathan, B.; Barrett, T.J. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog. Dis. 2006, 3, 59–67. [Google Scholar] [CrossRef]
- Jahanbakhsh, S.; Kabore, K.P.; Fravalo, P.; Letellier, A.; Fairbrother, J.M. Impact of medicated feed along with clay mineral supplementation on Escherichia coli resistance to antimicrobial agents in pigs after weaning in field conditions. Res. Vet. Sci. 2015, 102, 72–79. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.J.; Kim, S.S.; Mollenkopf, D.F.; Mathys, D.A.; Schuenemann, G.M.; Daniels, J.B.; Wittum, T.E. Antimicrobial-resistant Enterobacteriaceae recovered from companion animal and livestock environments. Zoonoses Public Health 2018, 16, 519–527. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Joensen, K.G.; Tetzschner, A.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 2015, 53, 2410–2426. [Google Scholar] [CrossRef]
- Roer, L.; Tchesnokova, V.; Allesøe, R.; Muradova, M.; Chattopadhyay, S.; Ahrenfeldt, J.; Thomsen, M.C.F.; Lund, O.; Hansen, F.; Hammerum, A.M.; et al. Development of a Web Tool for Escherichia coli Subtyping Based on fimH Alleles. J. Clin. Microbiol. 2017, 55, 2538–2543. [Google Scholar] [CrossRef]
- Beghain, J.; Bridier-Nahmias, A.; Le Nagard, H.; Denamur, E.; Clermont, O. ClermonTyping: An easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a New Bioinformatic Tool To Discover Antibiotic Resistance Genes in Bacterial Genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. PlasmidFinder and pMLST: In silico detection and typing of plasmids. Antimicrob. Agents Chemother. 2014, 02412–02414. [Google Scholar]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. sRNAbench and sRNAtoolbox 2019: Intuitive fast small RNA profiling and differential expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef]
- Croucher, N.J.; Harris, S.R.; Grad, Y.H.; Hanage, W.P. Bacterial genomes in epidemiology--present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120202. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2014, 43, e15. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.F.; Mohamed, K.; Fan, Y.; The Agama Study Group; Achtman, M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Criterion | Definition | Application to ETEC:F4 Clone A-I Detected in Pigs in Quebec |
|---|---|---|
| 1/Emergence | The clone is newly recognized, newly evolved or has occurred previously but shows an increase in incidence or expansion in geographical, host or vector range. | The clone A-I emerged in North America in 2013. |
| 2/Carriage of multiple antimicrobial characteristics | The clone carries multiple resistance genes associated with phenotypic multidrug resistance. The resistance genes can be carried by mobile genetic elements or by the chromosome (and then results from mutations) or both. | The clone A-I carries at least the genes tet(A) and the blaTEM-1 which are associated with phenotypic resistance to tetracyclines and penicillins, respectively. Several replicons such as the IncFII have been identified in the clone A-I. Moreover, it also carries parC and gyrA mutations responsible for non-susceptibility to fluoroquinolones. |
| 3/High capacity of dissemination through one or a combination of the following characteristics: | The clone is likely to disseminate due to: | The clone A-I has been detected in at least one province of Canada as well as in many states of the USA. |
| 3a/ High infectivity |
| The clone A-I has been observed in different batches of pigs on the same farm for 6 months. |
| 3b/ Long-term persistence |
| |
| 3c/ Multiplicity of sources |
| |
| 4/High Pathogenicity | The clone can cause severe disease in animals or/and in humans. | The clone A-I is associated with a higher risk of mortality that observed for other clonal lineages. |
| Genes Clonal Lineage | Enterohemolysin (ehxA) | Fimbrial Major Protein (lfpA) | Adhesin (Iha) | Siderophore Yersiniabactin * | Type VI Secretion System * | E. coli Common Pilus (ECP) * | Hemorrhagic E. coli Pilus (HEP) * | ompA Outer Membrane Protein A | rpoS Sigma S (Sigma 38) Factor | tar/cheM Methyl-Accepting Chemotaxis Protein II [Peritrichous Flagella] |
|---|---|---|---|---|---|---|---|---|---|---|
| A (ST100/O149) | Absent | Absent | Present | Absent | Present | Present | Present | Present | Present | Present |
| B ST772/O23 | Present | Absent | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| C ST100/O138 | Absent | Absent | Present | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| D ST90/O149H19 | Absent | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| E ST90/O157 | Absent | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| F ST90/O149H43 | Absent | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| Criteria/Clone | A-I | A2 | A3 | A4 | B1 | C1 |
|---|---|---|---|---|---|---|
| Emergence | yes | no | no | no | no | yes |
| Multidrug resistance | yes | no | no | no | no | yes |
| Potent dissemination | yes | no | no | no | no | no |
| Pathogenicity | yes | yes | yes | yes | yes | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Lagarde, M.; Vanier, G.; Arsenault, J.; Fairbrother, J.M. High Risk Clone: A Proposal of Criteria Adapted to the One Health Context with Application to Enterotoxigenic Escherichia coli in the Pig Population. Antibiotics 2021, 10, 244. https://doi.org/10.3390/antibiotics10030244
de Lagarde M, Vanier G, Arsenault J, Fairbrother JM. High Risk Clone: A Proposal of Criteria Adapted to the One Health Context with Application to Enterotoxigenic Escherichia coli in the Pig Population. Antibiotics. 2021; 10(3):244. https://doi.org/10.3390/antibiotics10030244
Chicago/Turabian Stylede Lagarde, Maud, Ghyslaine Vanier, Julie Arsenault, and John Morris Fairbrother. 2021. "High Risk Clone: A Proposal of Criteria Adapted to the One Health Context with Application to Enterotoxigenic Escherichia coli in the Pig Population" Antibiotics 10, no. 3: 244. https://doi.org/10.3390/antibiotics10030244
APA Stylede Lagarde, M., Vanier, G., Arsenault, J., & Fairbrother, J. M. (2021). High Risk Clone: A Proposal of Criteria Adapted to the One Health Context with Application to Enterotoxigenic Escherichia coli in the Pig Population. Antibiotics, 10(3), 244. https://doi.org/10.3390/antibiotics10030244