Surface Enhanced Raman Spectroscopy of Lactoferrin Adsorbed on Silvered Porous Silicon Covered with Graphene

 , , , , , and
, , , , , and 
Abstract

1. Introduction
2. Materials and Methods
3. Results and Discussion
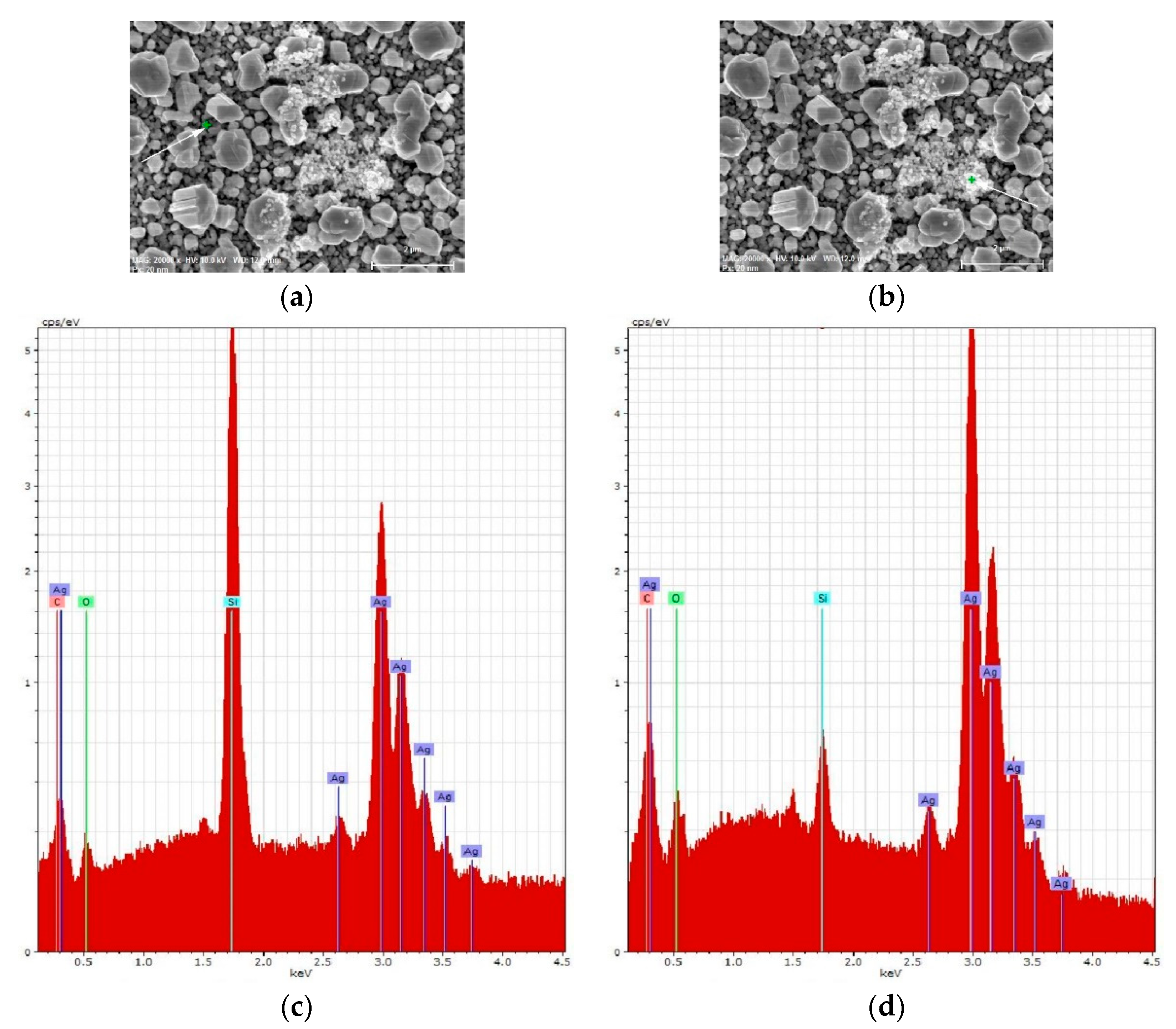
3.1. Characterization of SERS-Active Substrates
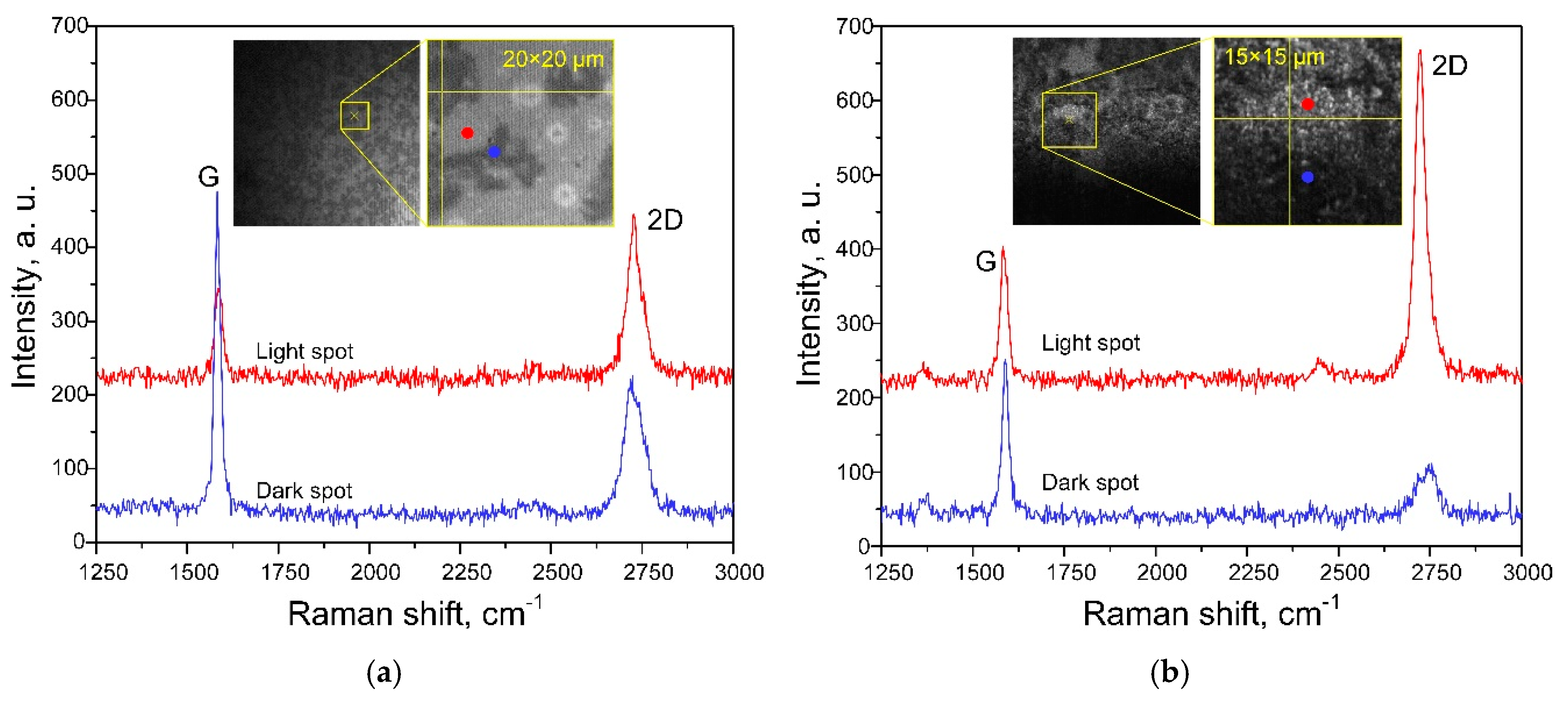
3.2. Characterization of Graphene-Containing Films
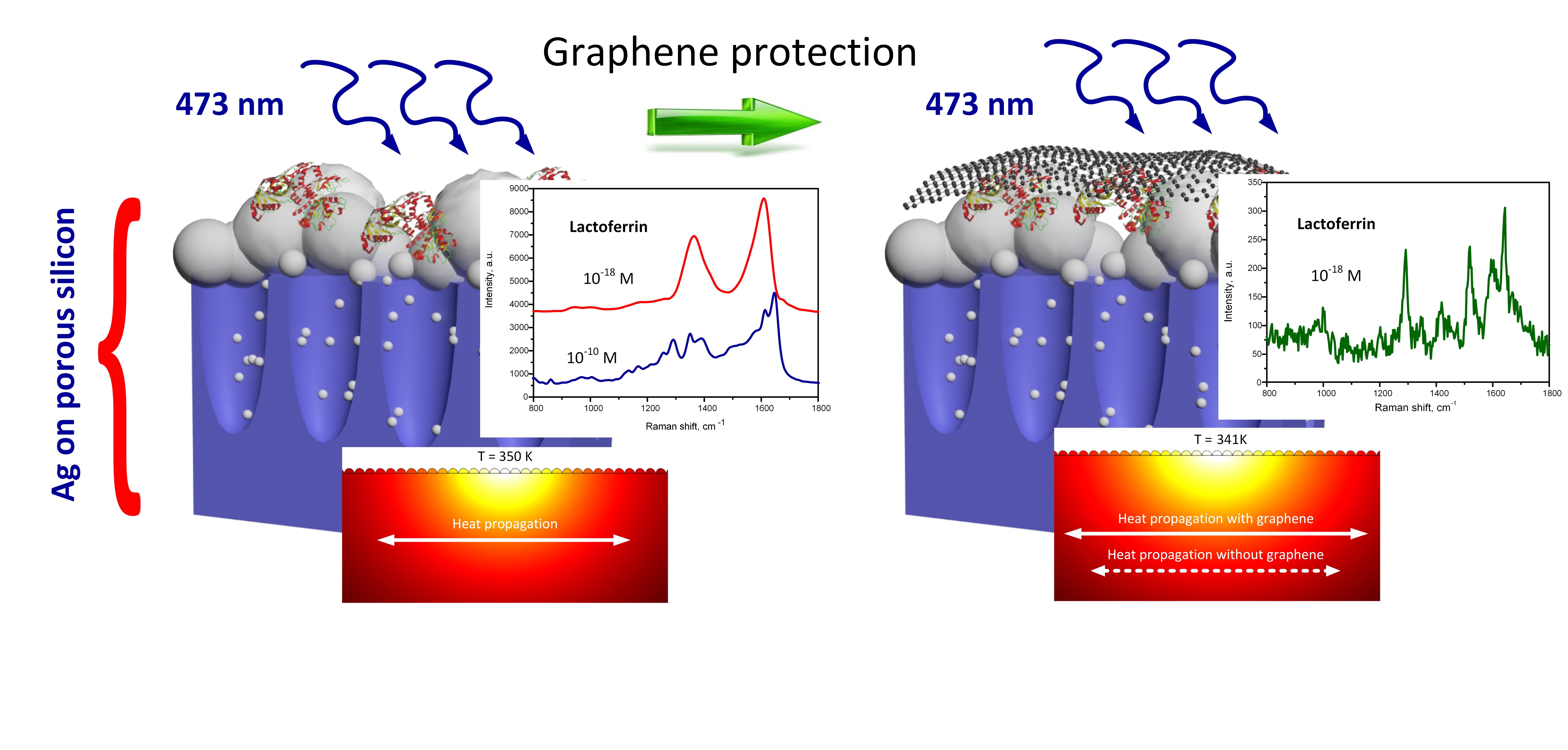
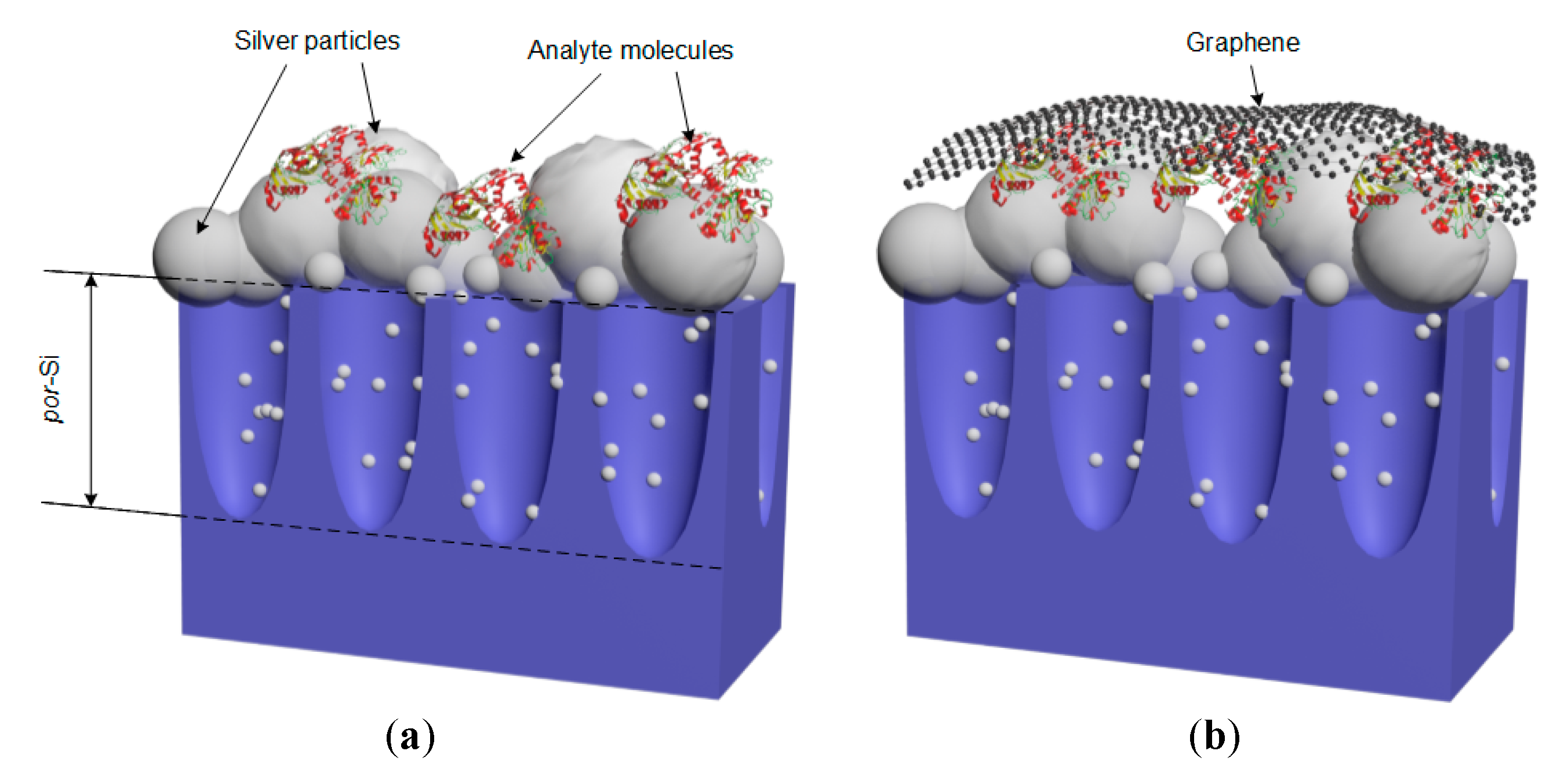
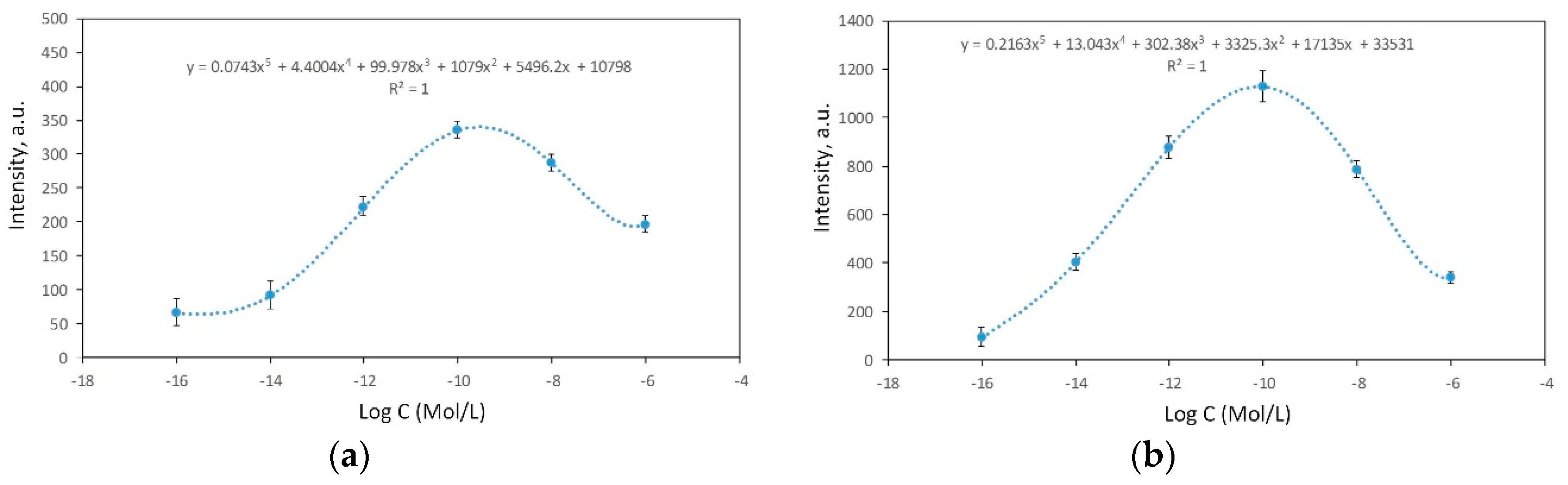
3.3. SERS-Activity of the Silvered por-Si Covered with Graphene
3.4. Simulation of the Silvered por-Si Covered with Graphene
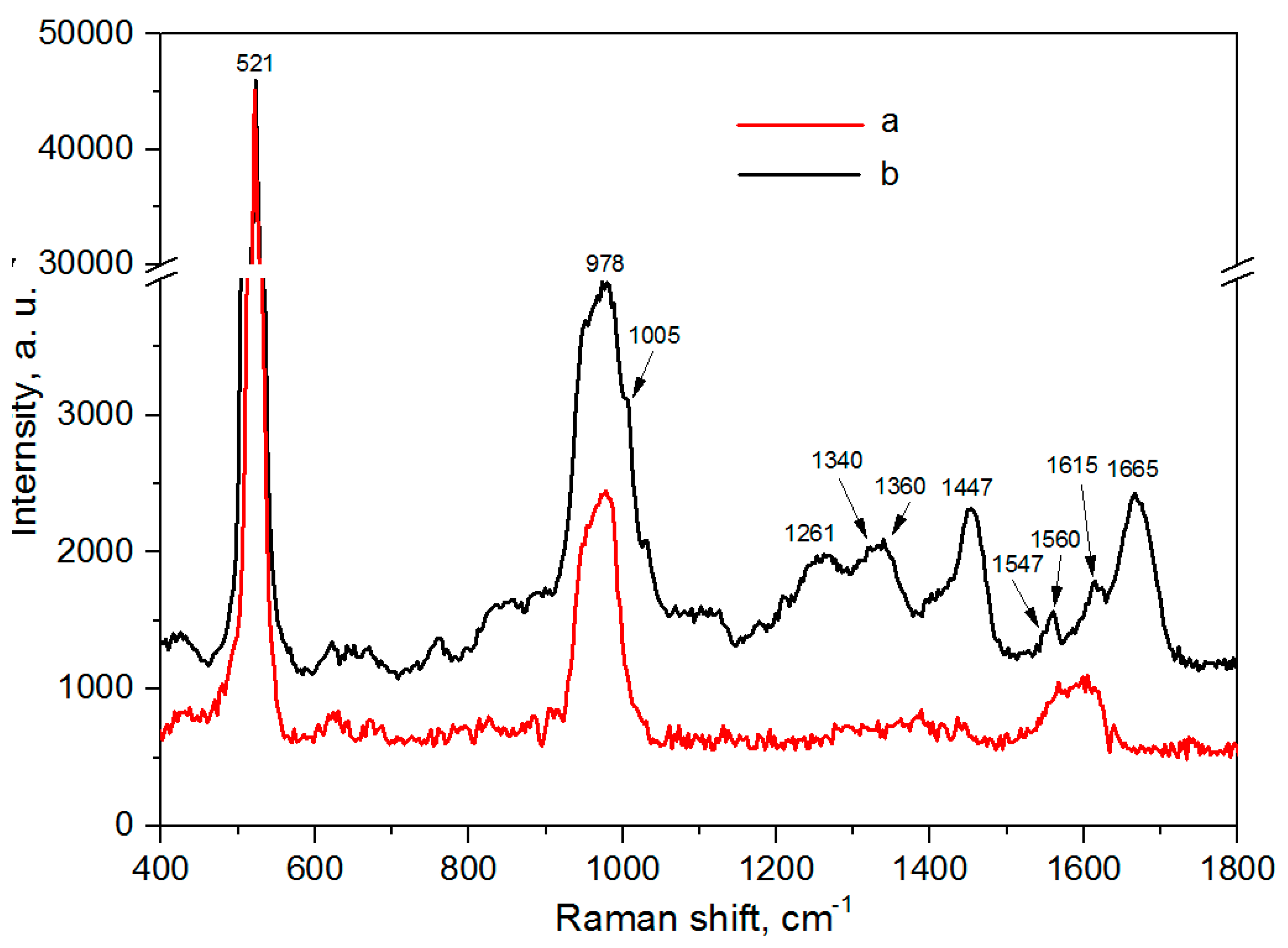
3.5. SERS-Spectroscopy of Lactoferrin Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arduini, F.; Cinti, S.; Scognamiglio, V.; Moscone, D.; Palleschi, G. How cutting-edge technologies impact the design of electrochemical (bio) sensors for environmental analysis. A review. Anal. Chim. Acta 2017, 959, 15–42. [Google Scholar] [CrossRef] [PubMed]
- De Lépinay, L.M.; Pigeau, B.; Besga, B.; Vincent, P.; Poncharal, P.; Arcizet, O. A universal and ultrasensitive vectorial nanomechanical sensor for imaging 2D force fields. Nat. Nanotechnol. 2016, 12, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Turner, N.C. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. Arch. Pathol. Lab. Med. 2018, 36, 1631–1641. [Google Scholar] [CrossRef]
- Nie, S.; Shi, T.; Fillmore, T.L.; Schepmoes, A.A.; Brewer, H.; Gao, Y.; Liu, T. Deep-Dive Targeted Quantification for Ultrasensitive Analysis of Proteins in Nondepleted Human Blood Plasma/Serum and Tissues. Anal. Chem. 2017, 89, 9139–9146. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Yuen, C.; Aniweh, Y.; Preiser, P.; Liu, Q. Towards ultrasensitive malaria diagnosis using surface enhanced Raman spectroscopy. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Diehn, M. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.R.; Moreira, F.T.C.; Fernandes, R.; Sales, M.G.F. Novel and simple electrochemical biosensor monitoring attomolar levels of miRNA-155 in breast cancer. Biosens. Bioelectron. 2016, 80, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W. The two shapes of the Tau protein. Alzheimer’s Dis. 2018, 7, e38516. [Google Scholar] [CrossRef] [PubMed]
- Kushibiki, T.; Tu, Y.; Abu-Yousif, A.O.; Hasan, T. Photodynamic activation as a molecular switch to promote osteoblast cell differentiation via AP-1 activation. Sci. Rep. 2015, 5, 13114. [Google Scholar] [CrossRef] [PubMed]
- Tretzel, L.; Görgens, C.; Geyer, H.; Thomas, A.; Dib, J.; Guddat, S.; Thevis, M. Analyses of Meldonium (Mildronate) from Blood, Dried Blood Spots (DBS), and Urine Suggest Drug Incorporation into Erythrocytes. Int. J. Sports Med. 2016, 37, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar] [CrossRef]
- Natan, M.J. Concluding Remarks: Surface enhanced Raman scattering. Faraday Discuss. 2016, 132, 321–328. [Google Scholar] [CrossRef]
- Sharma, H.S.S.; Carmichael, E.; McCall, D. Fabrication of SERS substrate for the detection of rhodamine 6G, glyphosate, melamine and salicylic acid. Vib. Spectrosc. 2016, 83, 159–169. [Google Scholar] [CrossRef]
- Kneipp, K.; Wang, Y.; Kneipp, H.; Perelman, L.T.; Itzkan, I.; Dasari, R.R.; Feld, M.S. Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667–1670. [Google Scholar] [CrossRef]
- Pomastowski, P.; Sprynskyy, M.; Žuvela, P.; Rafińska, K.; Milanowski, M.; Liu, J.J.; Buszewski, B. Silver-Lactoferrin Nanocomplexes as a Potent Antimicrobial Agent. J. Am. Chem. Soc. 2016, 138, 7899–7909. [Google Scholar] [CrossRef] [PubMed]
- Metz-Boutique, M.H.; Jolles, J.; Mazurier, J.; Schoentgen, F.; Legrand, D.; Spik, G.; Montreuil, J.; Jolles, P. Human lactotransferrin: Amino acid sequence and structural comparisons with other transferrins. Eur. J. Biochem. 1984, 145, 659–676. [Google Scholar] [CrossRef]
- Kanwar, J.R.; Roy, K.; Patel, Y.; Zhou, S.F.; Singh, M.R. Multifunctional iron bound lactoferrin and nanomedicinal approaches to enhance its bioactive functions. Molecules 2015, 20, 9703–9731. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B. Isolation of an iron-containing red protein from human milk. Acta Chem. Scand. 1960, 14, 510–512. [Google Scholar] [CrossRef]
- Birgens, H. Lactoferrin in plasma measured by an ELISA technique: Evidence that plasma lactoferrin is an indicator of neutrophil turnover and bone marrow activity in acute leukaemia. Scand. J. Haematol. 1985, 34, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Jensen, O.L.; Gluud, B.S.; Birgens, H.S. The concentration of lactoferrin in tears of normals and of diabetics. Acta Ophthalmol. 1986, 64, 83–87. [Google Scholar] [CrossRef]
- Ohashi, Y.; Ishida, R.; Kojima, T.; Goto, E.; Matsumoto, Y.; Watanabe, K.; Tsubota, K. Abnormal protein profiles in tears with dry eye syndrome. Am. J. Ophthalmol. 2003, 136, 291–299. [Google Scholar] [CrossRef]
- Buccigrossi, V.; de Marco, G.; Bruzzese, E.; Ombrato, L.; Bracale, I.; Polito, G.; Guarino, A. Lactoferrin Induces Concentration-Dependent Functional Modulation of Intestinal Proliferation and Differentiation. Pediatr. Res. 2007, 61, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Filik, J.; Stone, N. Drop coating deposition Raman spectroscopy of protein mixtures. Analyst 2007, 132, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Zalutski, I.V.; Lukianova, N.Y.; Storchai, D.M.; Burlaka, A.P.; Shvets, Y.V.; Borikun, T.V.; Todor, I.M.; Lukashevich, V.S.; Rudnichenko, Y.A.; Chekhun, V.F. Influence of exogenous lactoferrin on the oxidant/antioxidant balance and molecular profile of hormone receptor-positive and -negative human breast cancer cells in vitro. Exp. Oncol. 2017, 39, 106–111. [Google Scholar] [CrossRef]
- Lukashevich, V.S.; Budzevich, A.I.; Semak, I.V.; Kuznetsova, V.N.; Malyushkova, E.V.; Pyzh, A.E.; Novakovskaya, S.A.; Rudnichenko, J.A.; Popkov, N.A.; Ivashkevich, O.A.; et al. Production of recombinant human lactoferrin from the milk of goat-producers and its physiological effects. Dokl. Natl. Acad. Sci. Belarus 2016, 60, 72–81. [Google Scholar]
- Lai, S.; Wang, S.; Luo, J.; Lee, L.J.; Yang, S.-T.; Madou, M.J. Design of a compact disk-like microfluidic platform for enzyme-linked immunosorbent assay. Anal. Chem. 2004, 76, 1832–1837. [Google Scholar] [CrossRef] [PubMed]
- Satija, J.; Punjabi, N.; Mishra, D.; Mukherji, S. Plasmonic-ELISA: Expanding horizons. RSC Adv. 2016, 6, 85440–85456. [Google Scholar] [CrossRef]
- Pan, Y.; Sonn, G.A.; Sin, M.L.Y.; Mach, K.E.; Shih, M.-C.; Gau, V.; Wong, P.K.; Liao, J.C. Electrochemical immunosensor detection of urinary lactoferrin in clinical samples for urinary tract infection diagnosis. Biosens. Bioelectron. 2010, 26, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Krizkova, S.; Skalickova, S.; Dospivova, D.; Adam, V.; Kizek, R. Microfluidic tool coupled with electrochemical assay for detection of lactoferrin isolated by antibody-modified paramagnetic beads. Electrophoresis 2013, 34, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Fei, L.; Wei, W.; Xin, D.; Jia, R.; Qiuguang, S. Determination of Bovine Lactoferrin in Food by HPLC with a Heparin Affinity Column for Sample Preparation. J. AOAC Int. 2017, 100, 133–138. [Google Scholar] [CrossRef]
- Król, J.; Litwińczuk, Z.; Brodziak, A.; Barłowska, J. Lactoferrin, lysozyme and immunoglobulin G content in milk of four breeds of cows managed under intensive production system. Pol. J. Vet. Sci. 2010, 13, 357–361. [Google Scholar] [PubMed]
- Lee, S.; Kumar, P.; Hu, Y.; Cheng, G.J.; Irudayaraj, J. Graphene laminated gold bipyramids as sensitive detection platforms for antibiotic molecules. Chem. Commun. 2015, 51, 15494–15497. [Google Scholar] [CrossRef] [PubMed]
- Koglin, E.; Séquaris, J.M.; Fritz, J.C.; Valenta, P. Surface enhanced Raman scattering (SERS) of nucleic acid bases adsorbed on silver colloids. J. Mol. Struct. 1984, 114, 219–223. [Google Scholar] [CrossRef]
- Bantz, K.C.; Meyer, A.F.; Wittenberg, N.J.; Im, H.; Kurtuluş, Ö.; Lee, S.H.; Haynes, C.L. Recent progress in SERS biosensing. Phys. Chem. Chem. Phys. 2011, 13, 11551. [Google Scholar] [CrossRef] [PubMed]
- Cialla, D.; März, A.; Böhme, R.; Theil, F.; Weber, K.; Schmitt, M.; Popp, J. Surface-enhanced Raman spectroscopy (SERS): Progress and trends. Anal. Bioanal. Chem. 2011, 403, 27–54. [Google Scholar] [CrossRef] [PubMed]
- Miclăuş, T.; Beer, C.; Chevallier, J.; Scavenius, C.; Bochenkov, V.E.; Enghild, J.J.; Sutherland, D.S. Dynamic protein coronas revealed as a modulator of silver nanoparticle sulphidation in vitro. Nat. Commun. 2016, 7, 11770. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kwon, S.; Koo, T.W.; Lee, L.P.; Berlin, A.A. Surface-Enhanced Raman Scattering of Small Molecules from Silver-Coated Silicon Nanopores. Adv. Mater. 2003, 15, 1595–1598. [Google Scholar] [CrossRef]
- Lin, H.; Mock, J.; Smith, D.; Gao, T.; Sailor, M.J. Surface-Enhanced Raman Scattering from Silver-Plated Porous Silicon. J. Phys. Chem. B 2004, 108, 11654–11659. [Google Scholar] [CrossRef]
- Giorgis, F.; Destrovi, E.; Chiodoni, A.; Froner, E.; Scarpa, M.; Venturello, A.; Geobaldo, F. Porous silicon as efficient surface enhanced Raman scattering (SERS) substrate. Appl. Surf. Sci. 2008, 254, 7494–7497. [Google Scholar] [CrossRef]
- Terekhov, S.N.; Panarin, A.Y.; Kholostov, K.I.; Bondarenko, V.P.; Turpin, P.Y. Nanostructured silver films on meso- and macroporous silicon layers as a substrate for surface-enhanced Raman scattering. Phys. Chem. Appl. Nanostruct. 2009, 507–510. [Google Scholar] [CrossRef]
- Kosović, M.; Balarin, M.; Ivanda, M.; Đerek, V.; Marciuš, M.; Ristić, M.; Gamulin, O. Porous Silicon Covered with Silver Nanoparticles as Surface-Enhanced Raman Scattering (SERS) Substrate for Ultra-Low Concentration Detection. Appl. Spectrosc. 2015, 69, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Hakonen, A.; Rindzevicius, T.; Schmidt, M.S.; Andersson, P.O.; Juhlin, L.; Svedendahl, M.; Käll, M. Detection of nerve gases using surface-enhanced Raman scattering substrates with high droplet adhesion. Nanoscale 2016, 8, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Arzumanyan, G.; Doroshkevich, N.; Mamatkulov, K.; Shashkov, S.; Girel, K.; Bandarenka, H.; Borisenko, V. Phospholipid detection by surface-enhanced Raman scattering using silvered porous silicon substrates. Phys. Status Solidi A 2017, 214, 1600915. [Google Scholar] [CrossRef]
- Yakimchuk, D.; Kaniukov, E.; Bundyukova, V.; Osminkina, L.; Teichert, S.; Demyanov, S.; Sivakov, V. Silver nanostructures evolution in porous SiO2/p-Si matrices for wide wavelength surface-enhanced Raman scattering applications. MRS Commun. 2018, 8, 95–99. [Google Scholar] [CrossRef]
- Bandarenka, H.; Artsemyeva, K.; Redko, S.; Panarin, A.; Terekhov, S.; Bondarenko, V. Effect of swirl-like resistivity striations in n+-type Sb doped Si wafers on the properties of Ag/porous silicon SERS substrates. Phys. Status Solidi C 2013, 10, 624–627. [Google Scholar] [CrossRef]
- Wu, K.; Rindzevicius, T.; Schmidt, M.S.; Mogensen, K.B.; Hakonen, A.; Boisen, A. Wafer-scale leaning silver nanopillars for molecular detection at ultra-low concentrations. J. Phys. Chem. C 2015, 119, 2053–2062. [Google Scholar] [CrossRef]
- Bandarenka, H.; Girel, K.; Zavatski, S.; Panarin, A.; Terekhov, S. Progress in the Development of SERS-Active Substrates Based on Metal-Coated Porous Silicon. Materials 2018, 11, 852. [Google Scholar] [CrossRef] [PubMed]
- Cullis, A.G.; Canham, L.T.; Calcott, P.D.J. The structural and luminescence properties of porous silicon. J. Appl. Phys. 1997, 82, 909–965. [Google Scholar] [CrossRef]
- Ling, X.; Xie, L.; Fang, Y.; Xu, H.; Zhang, H.; Kong, J.; Liu, Z. Can Graphene be used as a Substrate for Raman Enhancement? Nano Lett. 2010, 10, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shim, S.; Kim, B.; Shin, H.S. Surface-Enhanced Raman Scattering of Single- and Few-Layer Graphene by the Deposition of Gold Nanoparticles. Chemistry 2011, 17, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Mao, N.; Zhang, J. Graphene: A Platform for Surface-Enhanced Raman Spectroscopy. Small 2013, 9, 1206–1224. [Google Scholar] [CrossRef] [PubMed]
- Dolgiy, A.; Redko, S.; Bandarenka, H.; Prischepa, S.; Yanushkevich, K.; Nenzi, P.; Bondarenko, V. Electrochemical Deposition and Characterization of Ni in Mesoporous Silicon. J. Electrochem. Soc. 2012, 159, D623–D627. [Google Scholar] [CrossRef]
- Bandarenka, H.V.; Girel, K.V.; Bondarenko, V.P.; Khodasevich, I.A.; Panarin, A.Y.; Terekhov, S.N. Formation Regularities of Plasmonic Silver Nanostructures on Porous Silicon for Effective Surface-Enhanced Raman Scattering. Nanoscale Res. Lett. 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Fransson, G.B.; Lonnerdal, B. Iron in human milk. J. Pediatr. 1980, 96, 380–384. [Google Scholar] [CrossRef]
- Abe, H.; Saito, H.; Miyakawa, H.; Tayura, Y.; Shimamura, S.; Nagao, E.; Tomita, M. Heat Stability of Bovine Lactoferrin at Acidic pH. J. Dairy Sci. 1991, 74, 65–71. [Google Scholar] [CrossRef]
- Bandarenka, H.; Shapel, A.; Balucani, M. Cu-Si Nanocomposites Based on Porous Silicon Matrix. Solid State Phenom. 2009, 151, 222–226. [Google Scholar] [CrossRef]
- Girel, K.; Yantcevich, E.; Arzumanyan, G.; Doroshkevich, N.; Bandarenka, H. Detection of DNA molecules by SERS spectroscopy with silvered porous silicon as an active substrate. Phys. Status Solidi A 2016, 213, 2911–2915. [Google Scholar] [CrossRef]
- Xiu, Z.; Zhang, Q.; Puppala, H.L.; Colvin, V.L.; Alvarez, P.J.J. Negligible Particle-Specific Antibacterial Activity of Silver Nanoparticles. Nano Lett. 2012, 12, 4271–4275. [Google Scholar] [CrossRef] [PubMed]
- Calizo, I.; Balandin, A.A.; Bao, W.; Miao, F.; Lau, C.N. Temperature Dependence of the Raman Spectra of Graphene and Graphene Multilayers. Nano Lett. 2007, 7, 2645–2649. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Chakraboty, B.; Sood, A. Raman spectroscopy of graphene on different substrates and influence of defects. Bull. Mater. Sci. 2008, 31, 579–584. [Google Scholar] [CrossRef]
- Saito, R.; Hofmann, M.; Dresselhaus, G.; Jorio, A.; Dresselhaus, M.S. Raman spectroscopy of graphene and carbon nanotubes. Adv. Phys. 2011, 60, 413–550. [Google Scholar] [CrossRef]
- Iafisco, M.; Foltran, I.; Di Foggia, M.; Bonora, S.; Roveri, N. Calorimetric and Raman investigation of cow’s milk lactoferrin. J. Therm. Anal. Calorim. 2010, 103, 41–47. [Google Scholar] [CrossRef]
- Khrustalev, V.V.; Khrustaleva, T.A.; Kahanouskaya, E.Y.; Rudnichenko, Y.A.; Bandarenka, H.V.; Arutyunyan, A.M.; Kordyukova, L.V. The alpha helix 1 from the first conserved region of HIV1 gp120 is reconstructed in the short NQ21 peptide. Arch. Biochem. Biophys. 2018, 638, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Anderson, B.F.; Norris, G.E.; Baker, E.N.; Lesk, A.M.; Chothia, C. Domain Closure in Lactoferrin. J. Mol. Biol. 1993, 234, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Mantel, C.; Miyazawa, K.; Broxmeyer, H.E. Physical characteristics and polymerization during iron saturation of lactoferrin, a myelopoietic regulatory molecule with suppressor activity. Adv. Exp. Med. Biol. 1994, 357, 121–132. [Google Scholar] [PubMed]
- Bagby, G.C.; Bennet, R.M. Feedback regulation of granulopoiesis: Polymerization of lactoferrin abrogates its ability to inhibit CSA production. Blood 1982, 60, 108–112. [Google Scholar] [PubMed]
- Babina, S.E.; Tuzikov, F.V.; Tuzikova, N.A.; Buneva, V.N.; Nevinskii, G.A. Effect of nucleotides on the oligomeric state of human lactoferrin. Mol. Biol. 2006, 40, 121–131. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Series | At. % | |||
|---|---|---|---|---|---|
| Fresh Ag/c-Si | Aged Ag/c-Si | Fresh Ag/por-Si | Aged Ag/por-Si | ||
| C | K | 18.32 | 20.71 | 16.74 | 20.70 |
| O | K | 2.03 | 3.92 | 4.25 | 4.30 |
| Si | K | 57.46 | 48.08 | 42.46 | 43.20 |
| Ag | L | 22.13 | 18.48 | 36.55 | 31.71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavatski, S.; Khinevich, N.; Girel, K.; Redko, S.; Kovalchuk, N.; Komissarov, I.; Lukashevich, V.; Semak, I.; Mamatkulov, K.; Vorobyeva, M.; et al. Surface Enhanced Raman Spectroscopy of Lactoferrin Adsorbed on Silvered Porous Silicon Covered with Graphene. Biosensors 2019, 9, 34. https://doi.org/10.3390/bios9010034
Zavatski S, Khinevich N, Girel K, Redko S, Kovalchuk N, Komissarov I, Lukashevich V, Semak I, Mamatkulov K, Vorobyeva M, et al. Surface Enhanced Raman Spectroscopy of Lactoferrin Adsorbed on Silvered Porous Silicon Covered with Graphene. Biosensors. 2019; 9(1):34. https://doi.org/10.3390/bios9010034
Chicago/Turabian StyleZavatski, Sergey, Nadia Khinevich, Kseniya Girel, Sergey Redko, Nikolai Kovalchuk, Ivan Komissarov, Vladimir Lukashevich, Igor Semak, Kahramon Mamatkulov, Maria Vorobyeva, and et al. 2019. "Surface Enhanced Raman Spectroscopy of Lactoferrin Adsorbed on Silvered Porous Silicon Covered with Graphene" Biosensors 9, no. 1: 34. https://doi.org/10.3390/bios9010034
APA StyleZavatski, S., Khinevich, N., Girel, K., Redko, S., Kovalchuk, N., Komissarov, I., Lukashevich, V., Semak, I., Mamatkulov, K., Vorobyeva, M., Arzumanyan, G., & Bandarenka, H. (2019). Surface Enhanced Raman Spectroscopy of Lactoferrin Adsorbed on Silvered Porous Silicon Covered with Graphene. Biosensors, 9(1), 34. https://doi.org/10.3390/bios9010034