Detection of the Inflammation Biomarker C-Reactive Protein in Serum Samples: Towards an Optimal Biosensor Formula



Abstract
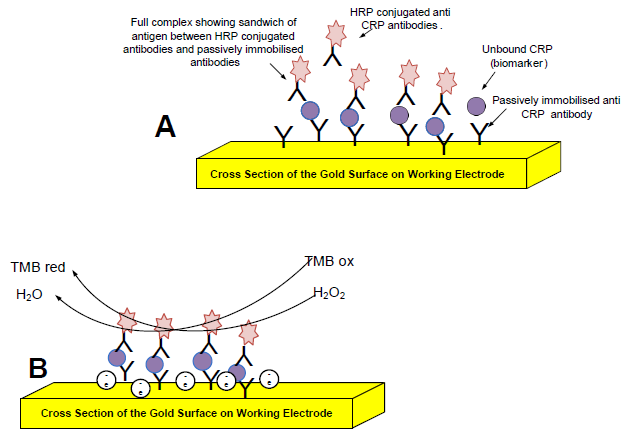
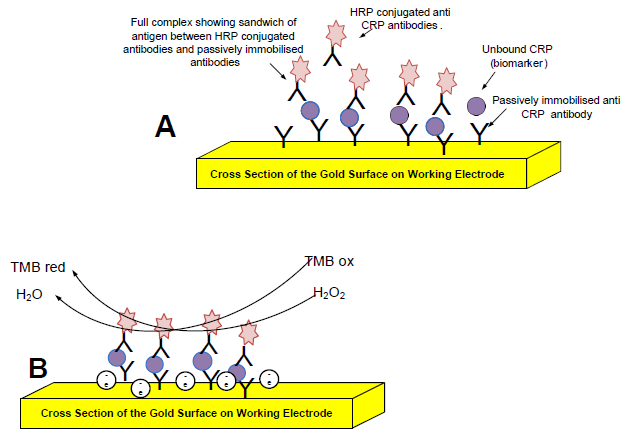
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
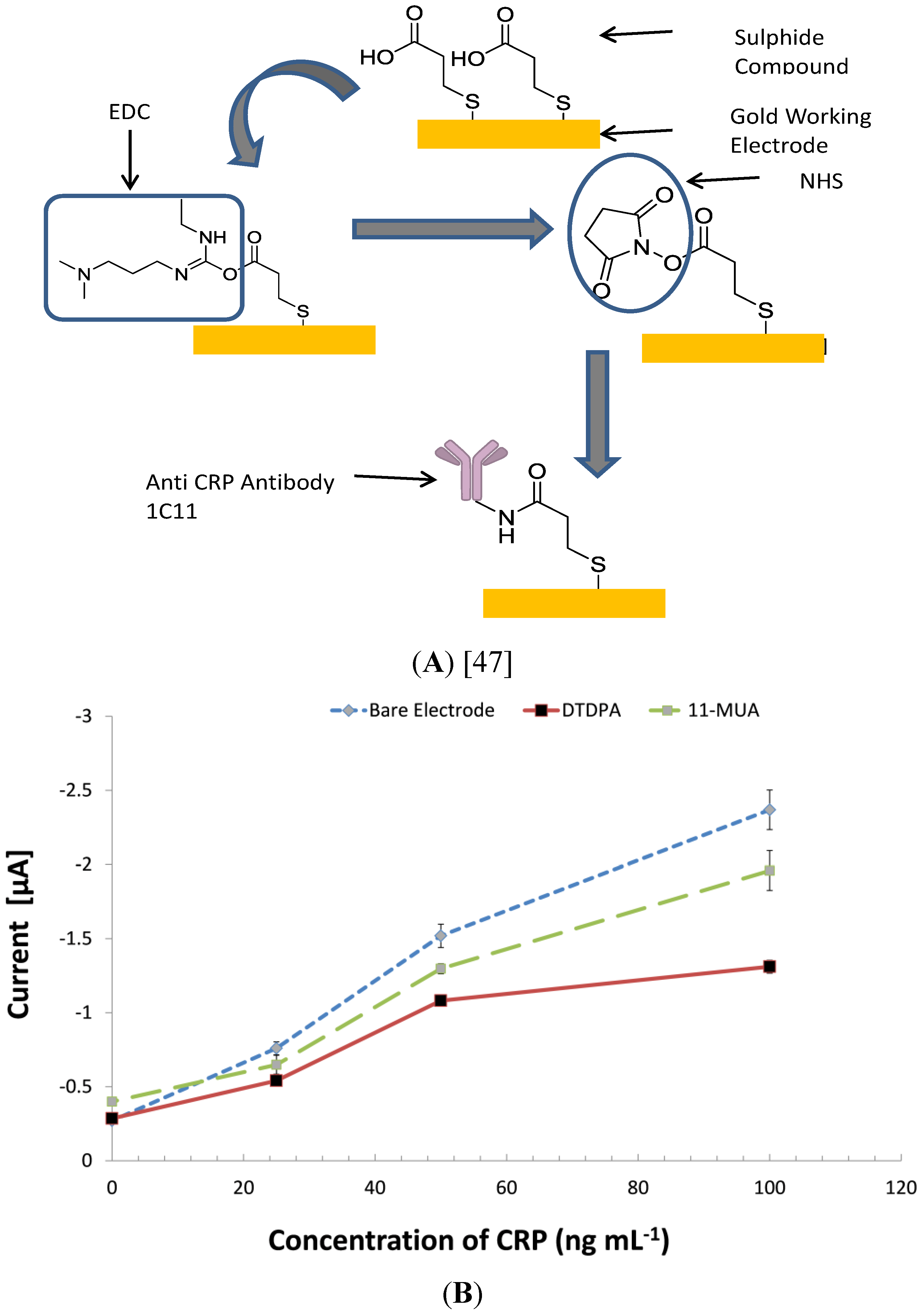{kind=link}
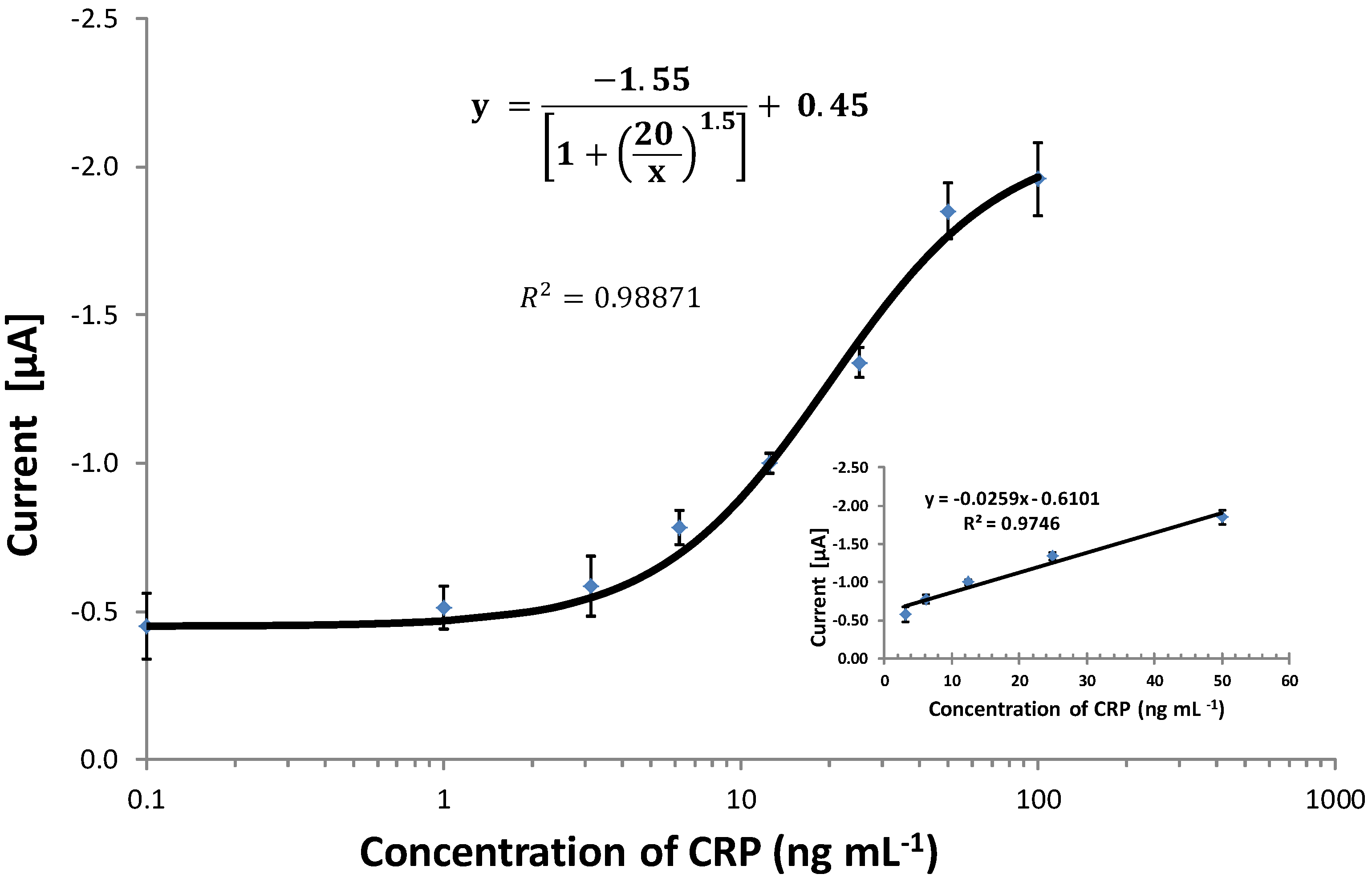{kind=link}
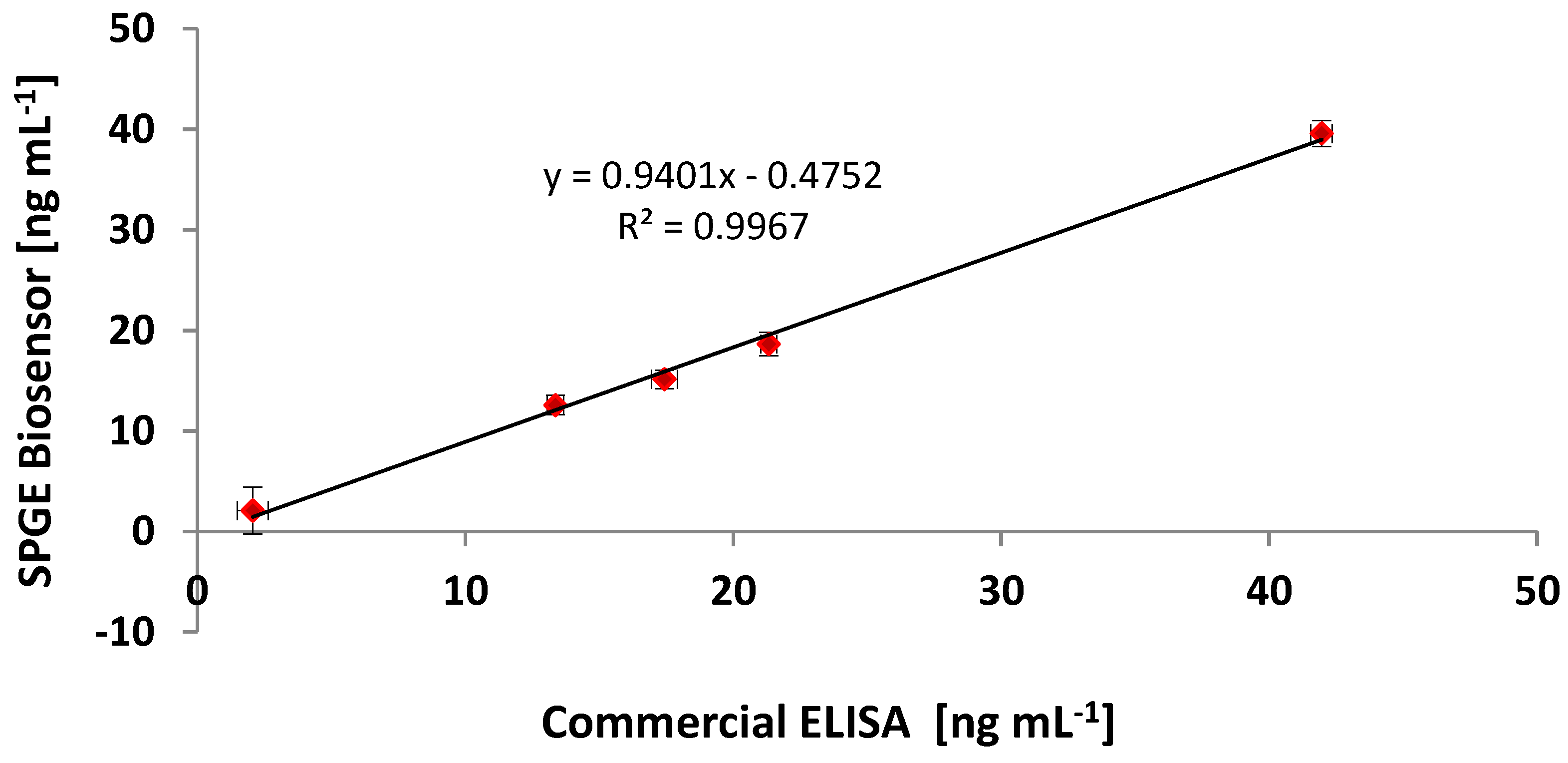{kind=link}
| Biosensor Technology Highlights | CRP Detection Limits | Reference |
|---|---|---|
| Surface plasmon resonance imaging (SPRi) biosensor | 10 pg·mL−1 | [28] |
| RNA aptamers on carbon nanotube’s interdigitated electrodes | 1–8 μmol·L−1 | [29] |
| MOFSET/BJT hybrid transistor-based biosensor | 1 pm·L−1 | [30] |
| Square wave A/stripping voltammetry | 0.5 to 200 μg·mL−1 | [31] |
| Thermally controlled piezoresistive microcantilever | 1 µg·mL−1 | [32] |
| Electrochemical impedance spectroscopy and cyclic voltammetry on carbon nanorods on electrodes | 0.001 µg·mL−1 | [33] |
| Electrochemical Impedance biosensor | 0.04 to 5.84 μg·mL−1 | [34] |
| Amperometric biosensor | 0.3 to 100 μg·mL−1 | [35] |
| Vertical flow immunoassay using flow through films (FTH) and gold nanoparticles | 0.01–10 μg·mL−11 | [36] |
2. Experimental Section
2.1. Chemicals and Materials
2.2. Fabrication of Screen-Printed Gold Electrodes
2.3. Optimizing the CRP Assay on the Gold Electrode
2.4. Cyclic Voltammetry
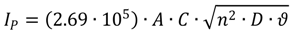
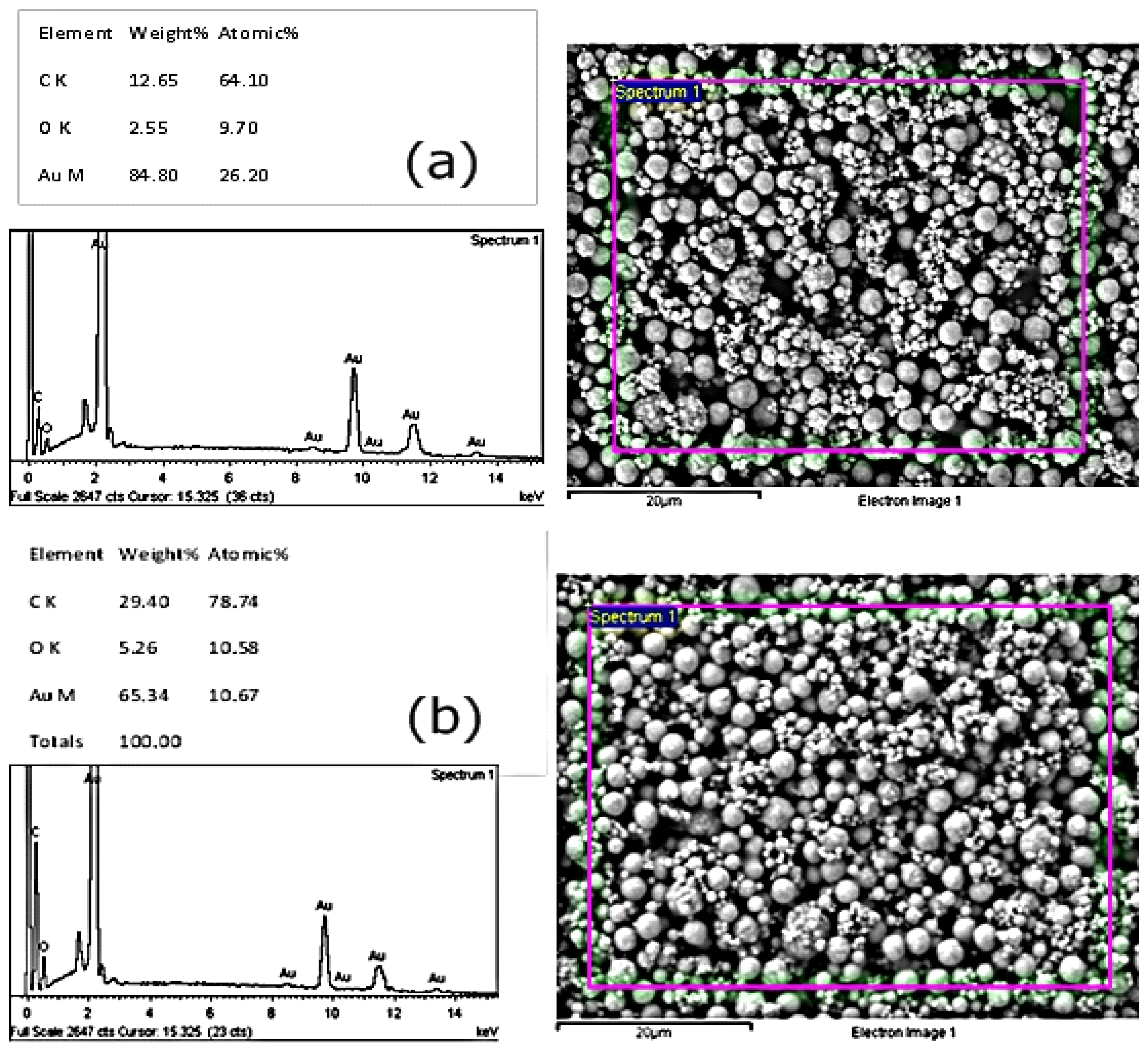
2.5. Investigating Different Ink Compositions and Curing Techniques
2.6. Investigating Different Immobilization Techniques
2.7. Comparing the SPGE with the ELISA
2.8. Calibration and Interpretation of the Data
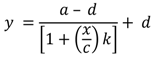
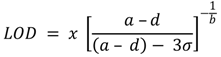
3. Results and Discussion
3.1. Optimization of the ELISA Micro-Plate-Based Assay
3.2. Investigating Different Ink Drying Regime
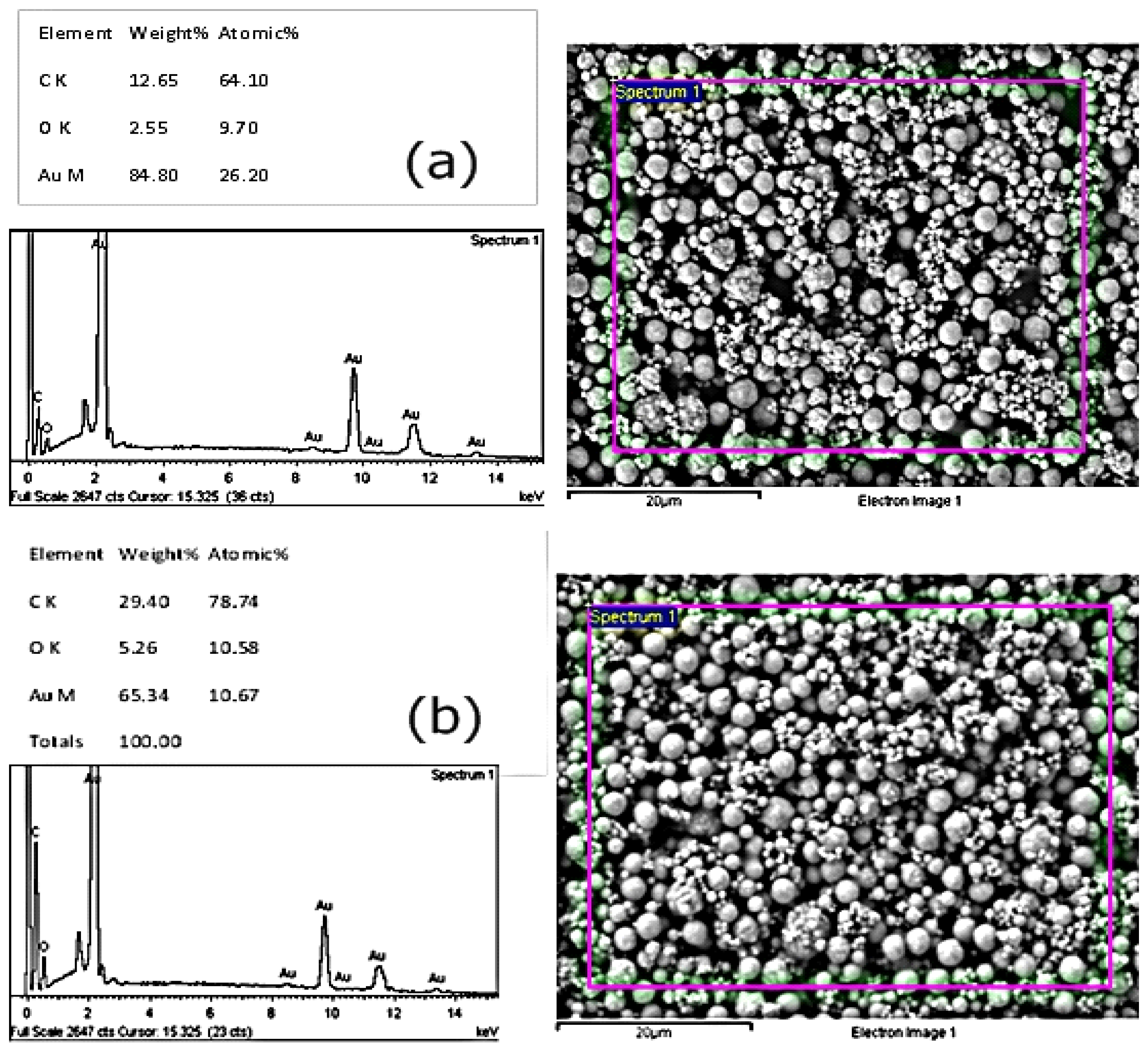
3.3. Investigating Different Ink Compositions

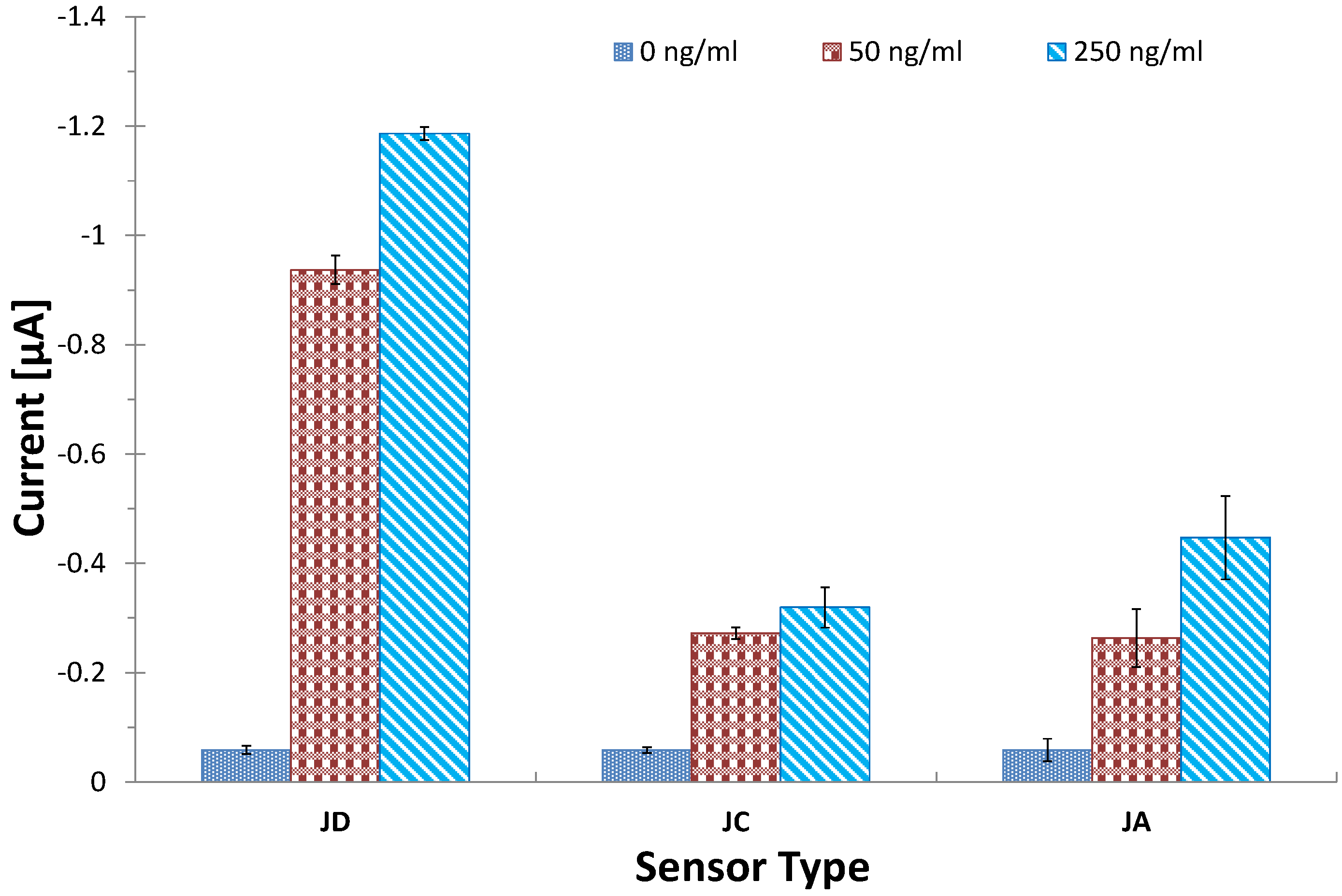
3.4. Optimization of Coating Antibody on SPGEs
3.5. Investigating Other Immobilization Methods
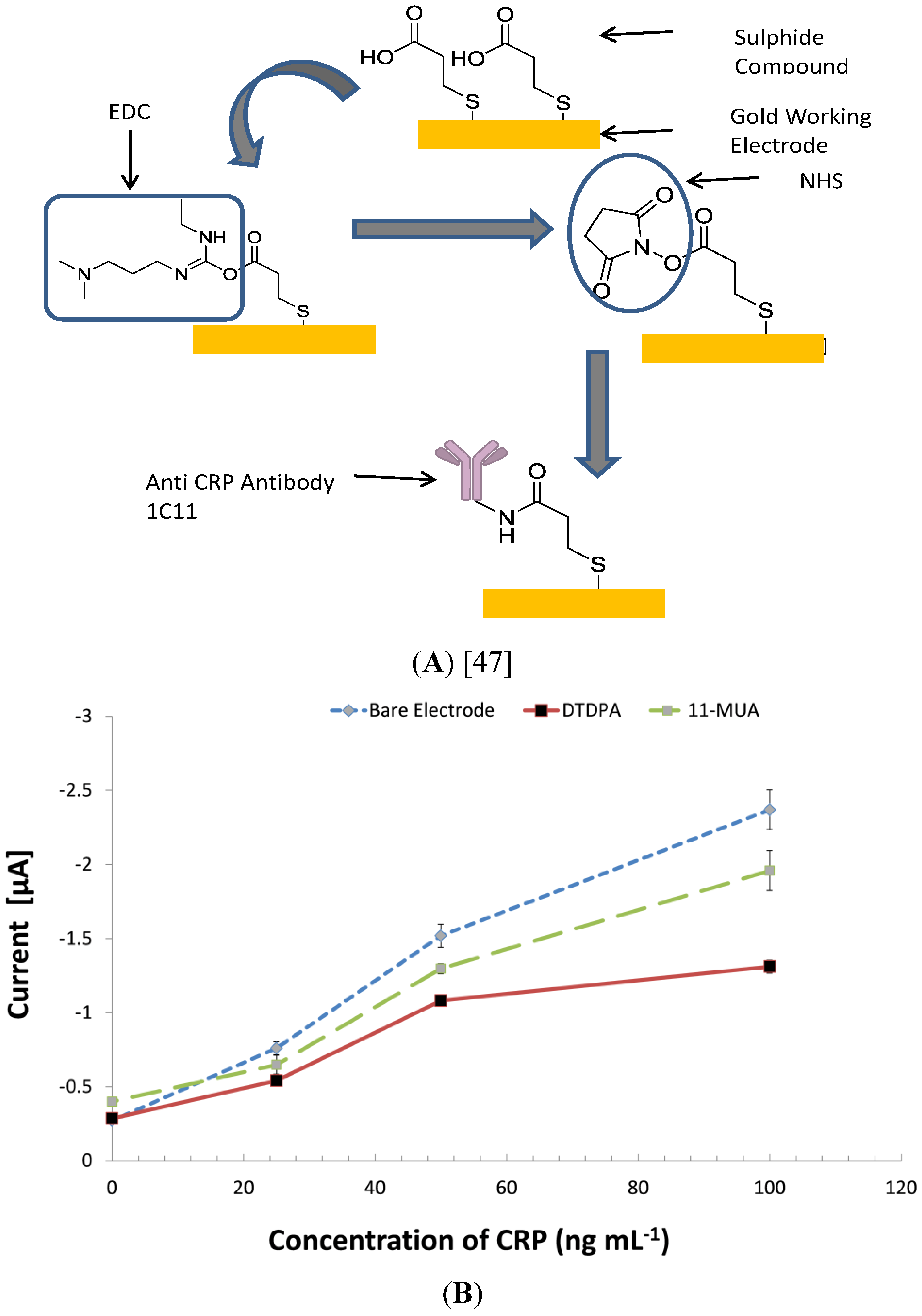
3.6. Comparing the CRP Assay on SPGEs against a Commercial ELISA Kit
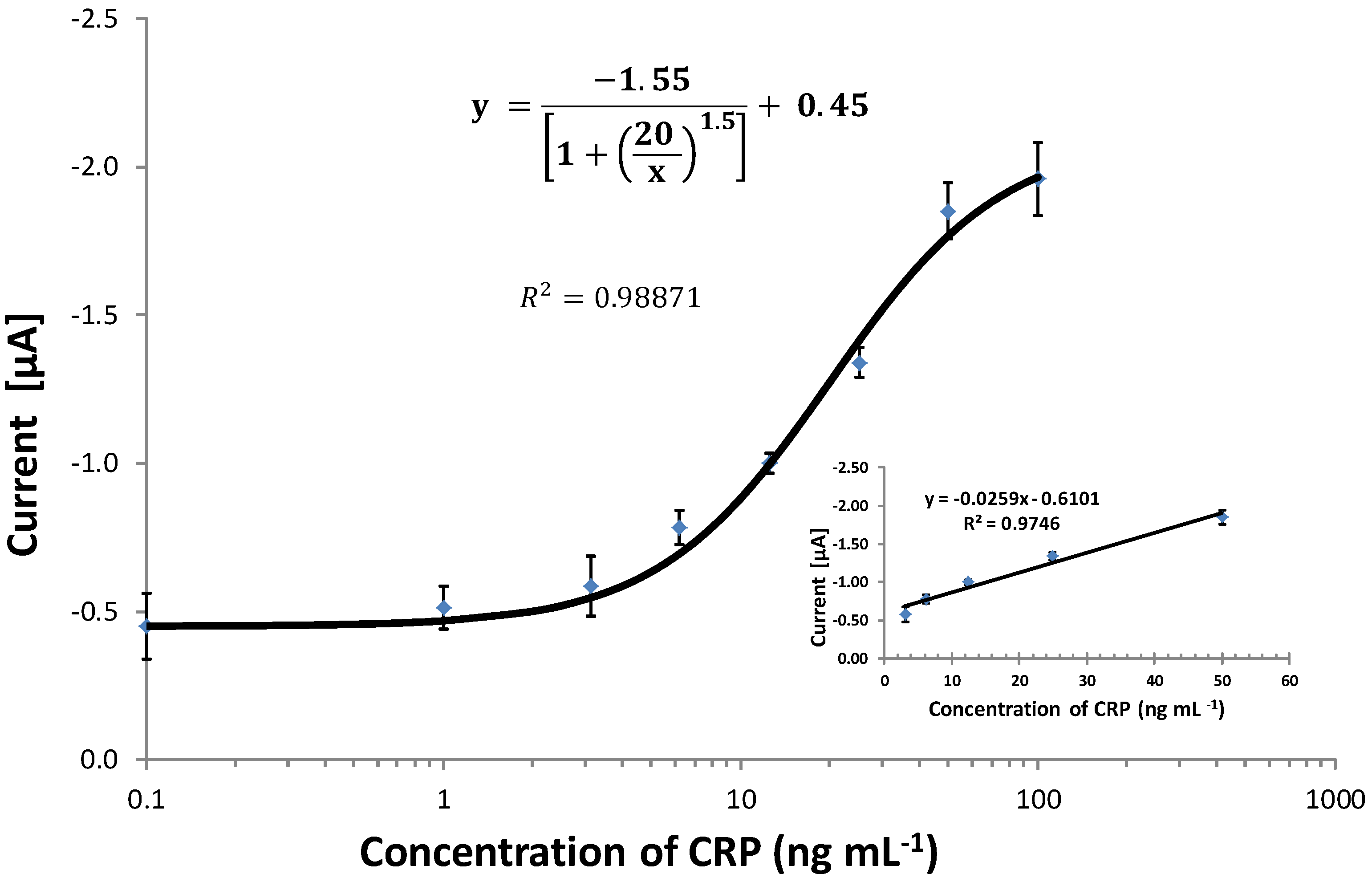
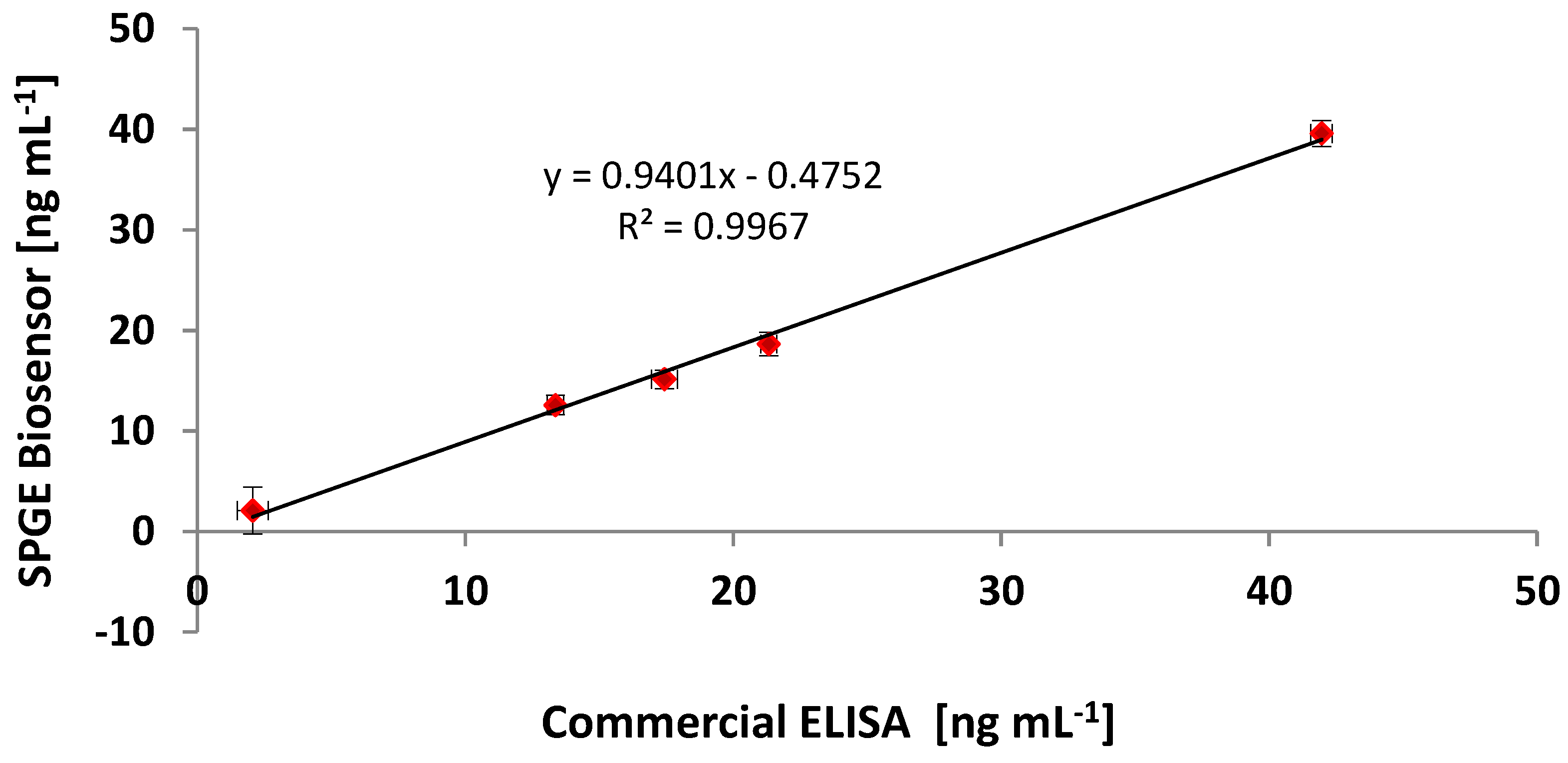
| Sample | Commercial ELISA | Optimised ELISA * | Immunosensor (SPGEs) | |||
|---|---|---|---|---|---|---|
| (ng·mL−1) | CV (%) | (ng·mL−1) | CV (%) | (ng·mL−1) | CV (%) | |
| Negative Control | 1.05 | 8.57 | 1.23 | 13.82 | 0.98 | 11.22 |
| Sample 1 | 21.32 | 5.48 | 20.04 | 6.76 | 18.64 | 1.61 |
| Sample 2 | 17.43 | 5.21 | 16.38 | 6.43 | 15.13 | 3.19 |
| Sample 3 | 13.36 | 7.24 | 12.56 | 8.94 | 12.57 | 2.41 |
| Sample 4 | 2.07 | 9.48 | 1.94 | 11.83 | 2.09 | 9.59 |
| Sample 5 | 41.96 | 3.03 | 39.44 | 3.74 | 39.58 | 1.02 |
| Positive Control | 77.32 | 0.53 | 74.58 | 0.88 | 75.36 | 0.64 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tillett, W.S.; Francis, T., Jr. Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. J. Exp. Med. 1930, 52, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-reactive protein: A critical update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Cliff, W.J.; Schoefl, G.I.; Higgins, G. Coronary C-reactive protein distribution: Its relation to development of atherosclerosis. Atherosclerosis 1999, 145, 375–379. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.D.; Cooper, M.A. A high sensitivity assay for the inflammatory marker C-Reactive protein employing acoustic biosensing. Nanobiotechnology 2008, 6, 1–8. [Google Scholar] [CrossRef]
- Nagai, T.; Anzai, T.; Kaneko, H.; Mano, Y.; Anzai, A.; Maekawa, Y.; Takahashi, T.; Meguro, T.; Yoshikawa, T.; Fukuda, K. C-reactive protein overexpression exacerbates pressure overload-induced cardiac remodeling through enhanced inflammatory response. Hypertension 2011, 57, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Cannon, C.P.; Sabatine, M.S.; Jarolim, P.; Sloane, S.; Rifai, N.; Braunwald, E.; Morrow, D.A. Concentrations of C-reactive protein and B-type natriuretic peptide 30 days after acute coronary syndromes independently predict hospitalization for heart failure and cardiovascular death. Clin. Chem. 2009, 55, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.J.; Woodrum, J.E.; Galili, O.; Olson, M.; Samee, S.; Meyer, F.B.; Zhu, X.Y.; Lerman, L.O.; Lerman, A. Concurrent treatment with renin-angiotensin system blockers and acetylsalicylic acid reduces nuclear factor kappaB activation and C-reactive protein expression in human carotid artery plaques. Stroke 2005, 36, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Koike, T.; Ichikawa, T.; Hatakeyama, K.; Shiomi, M.; Zhang, B.; Kitajima, S.; Morimoto, M.; Watanabe, T.; Asada, Y.; Chen, Y.E.; Fan, J. C-reactive protein in atherosclerotic lesions: its origin and pathophysiological significance. Am. J. Pathol. 2005, 167, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.S.; Yusuf, S. C-reactive protein is a bystander of cardiovascular disease. Eur. Heart J. 2010, 31, 2092–2096. [Google Scholar] [CrossRef]
- Schulz, R.; Heusch, G. C-reactive protein: Just a biomarker of inflammation or a pathophysiological player in myocardial function and morphology. Hypertension 2011, 57, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Anand, I.S.; Latini, R.; Florea, V.G.; Kuskowski, M.A.; Rector, T.; Masson, S.; Signorini, S.; Mocarelli, P.; Hester, A.; Glazer, R.; Cohn, J.N. C-reactive protein in heart failure: Prognostic value and the effect of valsartan. Circulation 2005, 112, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Mali, B.; Armbruster, D.; Serediak, E.; Ottenbreit, T. Comparison of immunoturbidimetric and immunonephelometric assays for specific proteins. Clin. Biochem. 2009, 42, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.L.; Moulton, L.; Law, T.C.; Farrow, G.; Cooper-Anderson, M.; Savory, J.; Rifai, N. Evaluation of nine automated high-sensitivity c-reactive protein methods: Implications for clinical and epidemiological applications. Part 2. Clin. Chem. 2001, 47, 418–425. [Google Scholar] [PubMed]
- Bjerrum, L.; Gahrn-Hansen, B.; Munck, A.P. C-reactive protein measurement in general practice may lead to lower antibiotic prescribing for sinusitis. Br. J. Gen. Pract. 2004, 54, 659–662. [Google Scholar] [PubMed]
- WHO. The Atlas of Heart Disease and Stroke. Available online: http://www.who.int/cardiovascular_diseases/resources/atlas/en/ (accessed on 16 July 2014).
- Allender, S.; Scarborough, P.; O’flaherty, M.; Capewell, S. Patterns of coronary heart disease mortality over the 20th century in England and Wales: Possible plateaus in the rate of decline. BMC Publ. Health 2008, 8. [Google Scholar] [CrossRef]
- Gibler, W.B.; Cannon, C.P.; Blomkalns, A.L.; Char, D.M.; Drew, B.J.; Hollander, J.E.; Jaffe, A.S.; Jesse, R.L.; Newby, L.K.; Ohman, E.M.; et al. Practical implementation of the guidelines for unstable angina/non-ST-segment elevation myocardial infarction in the emergency department. Ann. Emerg. Med. 2005, 46, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.H. Markers for early detection of cardiac diseases. Scand J. Clin. Lab Invest. Suppl. 2005, 240, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Abbott, R.D.; Yano, K.; Rodriguez, B.L.; Willcox, D.C.; Curb, J.D. C-reactive protein, cardiovascular disease and stroke: New roles for an old biomarker. Expert Rev. Neurother. 2004, 4, 507–518. [Google Scholar] [PubMed]
- Apple, F.S. Cardiac troponin monitoring for detection of myocardial infarction: Newer generation assays are here to stay. Clin. Chim. Acta 2007, 380, 1–3, discussion 245–246. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Chambers, J.C.; Zhang, W.; Clarke, R.; Hopewell, J.C.; Peden, J.F.; Erdmann, J.; Braund, P.; Engert, J.C.; Bennett, D.; et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA 2009, 302, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Haverkate, F.; Thompson, S.G.; Pyke, S.D.; Gallimore, J.R.; Pepys, M.B. Production of C-reactive protein and risk of coronary events in stable and unstable angina. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. Lancet 1997, 349, 462–466. [Google Scholar] [PubMed]
- Morrow, D.A.; Cannon, C.P.; Jesse, R.L.; Newby, L.K.; Ravkilde, J.; Storrow, A.B.; Wu, A.H.B.; Christenson, R.H. National Academy of Clinical Biochemistry Laboratory medicine practice guidelines: Clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Clin. Chem. 2007, 53, 552–574. [Google Scholar] [CrossRef] [PubMed]
- Salam, F.; Tothill, I.E. Detection of Salmonella typhimurium using an electrochemical immunosensor. Biosens. Bioelectron. 2009, 24, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Abdul Kadir, M.K.; Tothill, I.E. Development of an electrochemical immunosensor for fumonisins detection in foods. Toxins 2010, 2, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Tothill, I.E. Biosensors for cancer markers diagnosis. Semin. Cell Dev. Biol. 2009, 20, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Baskeyfield, D.E.H.; Davis, F.; Magan, N.; Tothill, I.E. A membrane-based immunosensor for the analysis of the herbicide Isoproturon. Anal. Chim. Acta 2011, 699, 223–231. [Google Scholar] [PubMed]
- Chammem, H.; Hafaid, I.; Meilhac, O.; Menaa, F.; Mora, L.; Abdelghani, A. Surface Plasmon Resonance for C-reactive protein detection in human plasma. J. Biomater. Nanobiotech. 2014, 5. [Google Scholar] [CrossRef]
- Qureshi, A.; Roci, I.; Gurbuz, Y.; Niazi, J.H. An aptamer based competition assay for protein detection using CNT activated gold-interdigitated capacitor arrays. Biosens. Bioelectron. 2012, 34, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Kwon, H.C.; Yeom, S.H.; Kwon, D.H.; Kang, S.W. MOSFET-BJT hybrid mode of the gated lateral bipolar junctiontransistor for C-reactive protein detection. Biosens. Bioelectron. 2011, 28, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Lu, M.; Wang, W.; Bian, Z.P.; Zhang, J.R.; Zhu, J.J. electrochemical immunosensor for simultaneous detection of dual cardiac markers based on a poly(dimethylsiloxane)-gold nanoparticles composite microfluidic chip: A proof of principle. Clin. Chem. 2010, 56, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.-K.; Lai, Y.-C.; Hong, W.-T.; Pheanpanitporn, Y.; Chen, C.-S.; Huang, L.-S. Electrical detection of C-reactive protein using a single free-standing, thermally controlled piezoresistive microcantilever for highly reproducible and accurate measurements. Sensors 2013, 13, 9653–9668. [Google Scholar] [PubMed]
- Gupta, R.K.; Periyakaruppan, A.; Meyyappan, M.; Koehne, J.E. Label-free detection of C-reactive protein using a carbon nanofiber based biosensor. Biosens. Bioelectron. 2014, 59, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, S.V.; Tanwar, V.K.; Biradar, A.M. Electrochemical impedance immunosensor for the detection of C-reactive protein inaqueous solution. Sens. Lett. 2010, 8, 362–369. [Google Scholar]
- Shaolin, Z.; Hankun, Z.; Ning, G.; Yuting, C. A renewable C reactive protein amperometric immunosensor based on magnetic multiwalled carbon nanotubes probes modified electrode. Appl. Mech. Mater. 2011, 80–81, 452–456. [Google Scholar]
- Oh, Y.K.; Joung, H.A.; Kim, S.; Kim, M.G. Vertical flow immunoassay (VFA) biosensor for a rapid one-step immunoassay. Lab Chip 2013, 13, 768–772. [Google Scholar] [PubMed]
- Heurich, M.; Abdul Kadir, M.K.; Tothill, I.E. An electrochemical sensor based on carboxymethylated dextran modified gold surface for ochratoxin A analysis. Sens. Actuator B Chem. 2011, 156, 162–168. [Google Scholar] [CrossRef]
- Tothill, I.E.; Turner, A.P.F. Biosensors. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Finglas, P., Trugo, L., Eds.; Elsevier B.V.: Amsterdam, The Netherlands, 2003; p. 250. [Google Scholar]
- Parker, C.O.; Lanyon, Y.H.; Manning, M.; Arrigan, D.W.; Tothill, I.E. Electrochemical immunochip sensor for aflatoxin M1 detection. Anal. Chem. 2009, 81, 5291–5298. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.F.; Tothill, I.E. Development and characterisation of disposable gold electrodes, and their use for lead (II) analysis. Anal. Bioanal. Chem. 2006, 386, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Gosser, D.K.J. Cyclic Voltammetry: Simulation and Analysis of Reaction Mechanisms; Wiley-VCH: Berlin, Germany, 1993. [Google Scholar]
- Bard, J.F.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; John Wiley and Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Warwick, M.J. Standerdisation of immunoassays. In Immunoassay, A Practical Guide; Brian, L., Ed.; Tylor and Francis Ltd.: London, UK, 1996. [Google Scholar]
- Grennan, K.; Killard, A.J.; Smyth, M.R. Physical characterizations of a screen-printed electrode for use in an amperometric biosensor system. Electroanalysis 2001, 13, 745–750. [Google Scholar] [CrossRef]
- Hobby, A. Gwent Group Advanced Material Systems. Technical Infomation Sheet. Available online: http://www.gwent.org/gem_thick_film.html (accessed on 16 July 2014).
- Leach, R.H.; Pierce, R.J. Radiation Curing Processes. In The Printing Ink Manual, 5th ed.; Springer: London, UK, 2007. [Google Scholar]
- Park, I.; Kima, D.K.; Adanyi, N.; Varadi, M.; Kim, N. Development of a direct-binding chloramphenicol sensor based on thiol or sulfide mediated self-assembled antibody monolayers. Biosens. Bioelectron. 2004, 19, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.J.; Eapen, S.S.; Carlisle, S.J. Handbook of Biosensors and Biochips; John Wiley: Chichester, UK, 2007. [Google Scholar]
- O’dwyer, C.; Gay, B.; Viaris De Lesegno, B.; Weiner, J. The nature of alkanethiol self-assembled monolayer adsorption on sputtered gold substrates. Langmuir 2004, 20, 8172–8182. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Haramura, M.; Tanaka, A. Reduction of nonspecific binding proteins to self-assembled monolayer on gold surface. Surf. Bioorganic Med. Chem. 2006, 14, 537–543. [Google Scholar] [CrossRef]
- Choi, S.; Chae, J. Methods of reducing non-specific adsorption in microfluidic biosensors. J. Micromech. Microeng. 2010, 20, 1–9. [Google Scholar] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakanya, W.M.; Tothill, I.E. Detection of the Inflammation Biomarker C-Reactive Protein in Serum Samples: Towards an Optimal Biosensor Formula. Biosensors 2014, 4, 340-357. https://doi.org/10.3390/bios4040340
Fakanya WM, Tothill IE. Detection of the Inflammation Biomarker C-Reactive Protein in Serum Samples: Towards an Optimal Biosensor Formula. Biosensors. 2014; 4(4):340-357. https://doi.org/10.3390/bios4040340
Chicago/Turabian StyleFakanya, Wellington M., and Ibtisam E. Tothill. 2014. "Detection of the Inflammation Biomarker C-Reactive Protein in Serum Samples: Towards an Optimal Biosensor Formula" Biosensors 4, no. 4: 340-357. https://doi.org/10.3390/bios4040340
APA StyleFakanya, W. M., & Tothill, I. E. (2014). Detection of the Inflammation Biomarker C-Reactive Protein in Serum Samples: Towards an Optimal Biosensor Formula. Biosensors, 4(4), 340-357. https://doi.org/10.3390/bios4040340