Magnetic Tracking of Protein Synthesis in Microfluidic Environments—Challenges and Perspectives

 ,
, 
Abstract
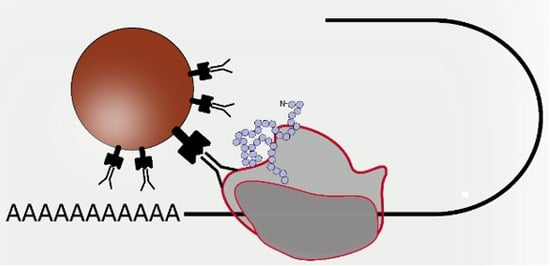
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Process and Significance of Protein Synthesis
2. Recent Progress in Studying Translation and Polysome Function: Need for Novel Analysis Tools
3. Magnetic Tracking of Protein Synthesis
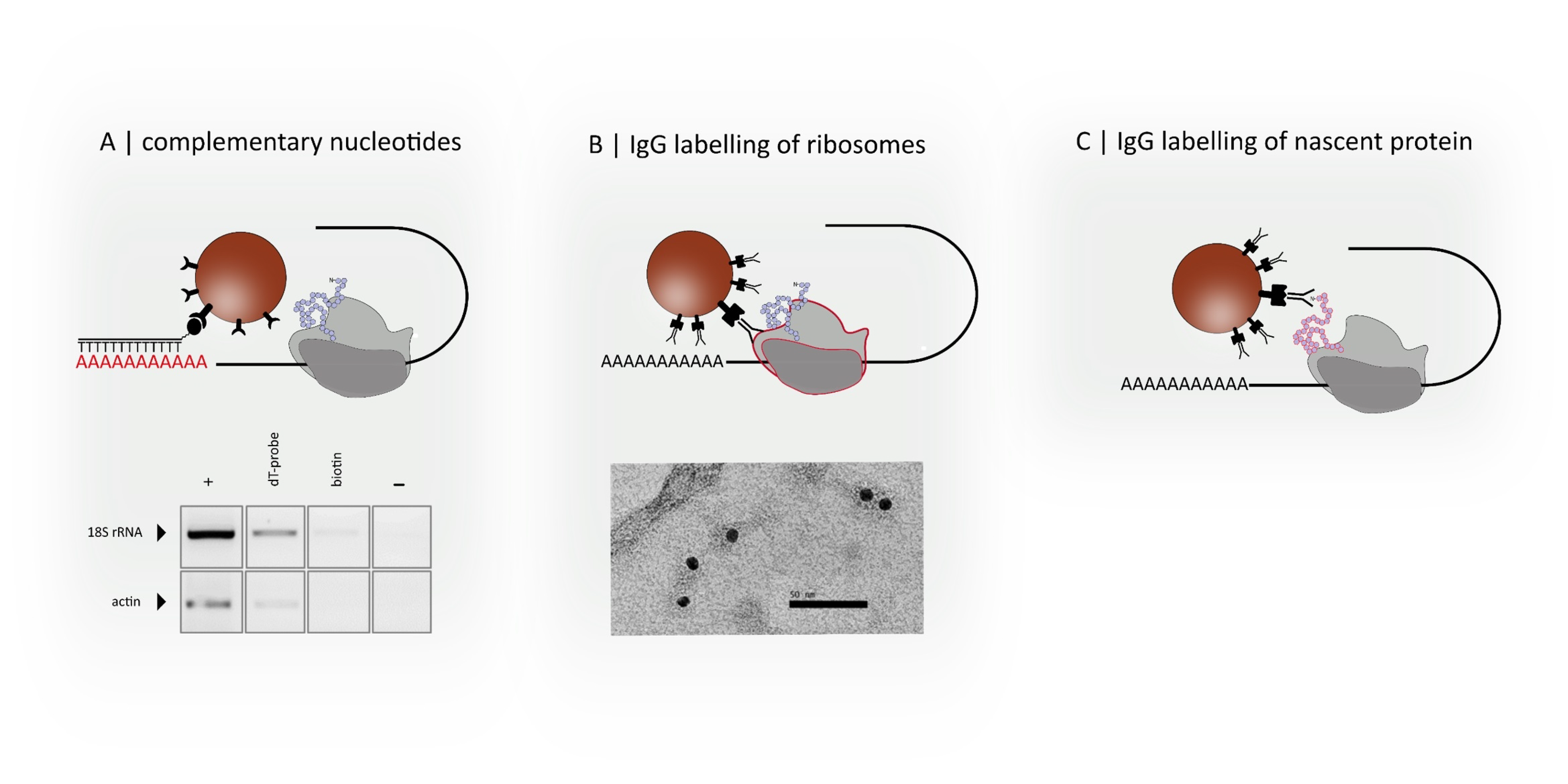
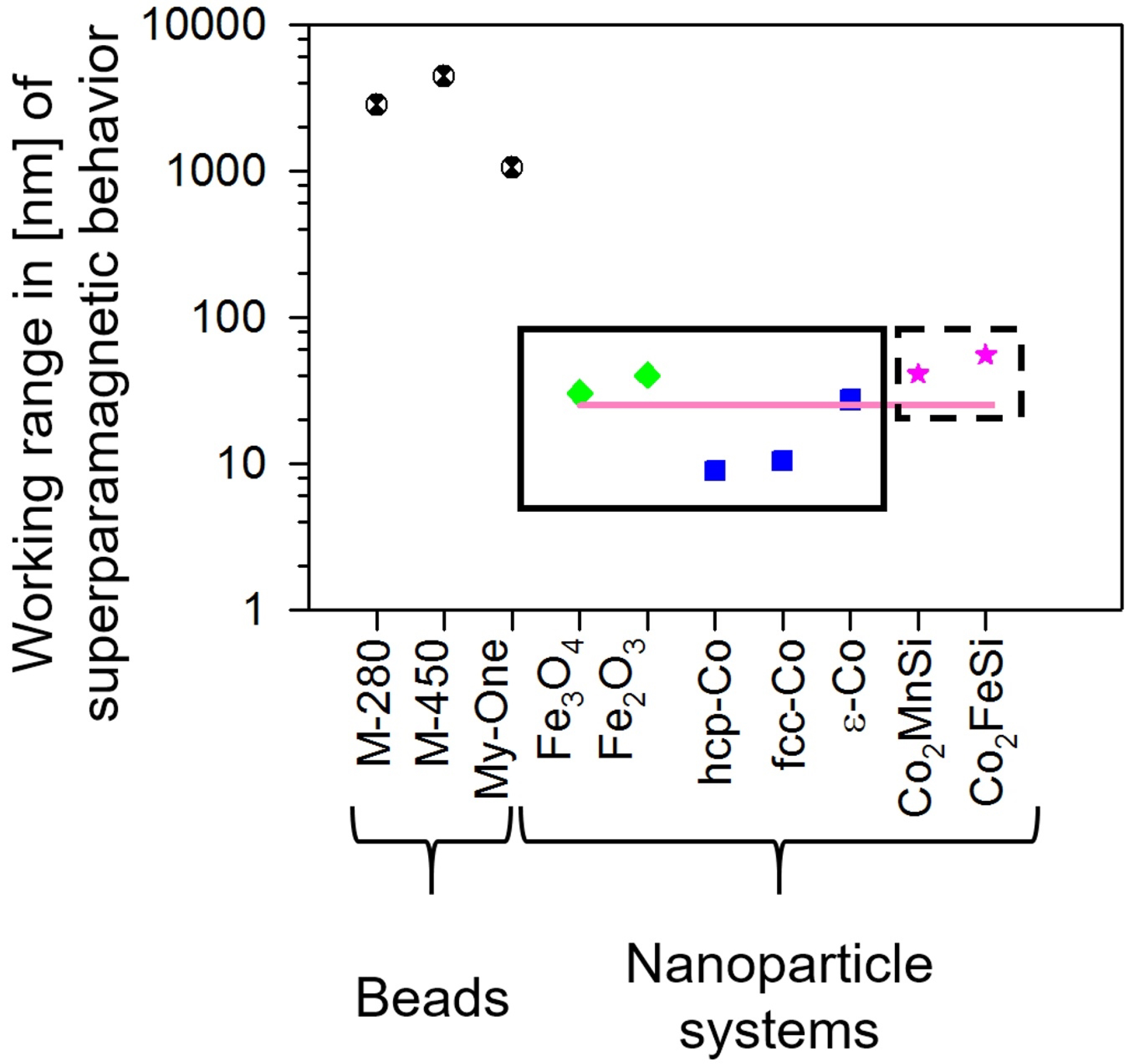
3.1. Functionalization and Specific Labelling
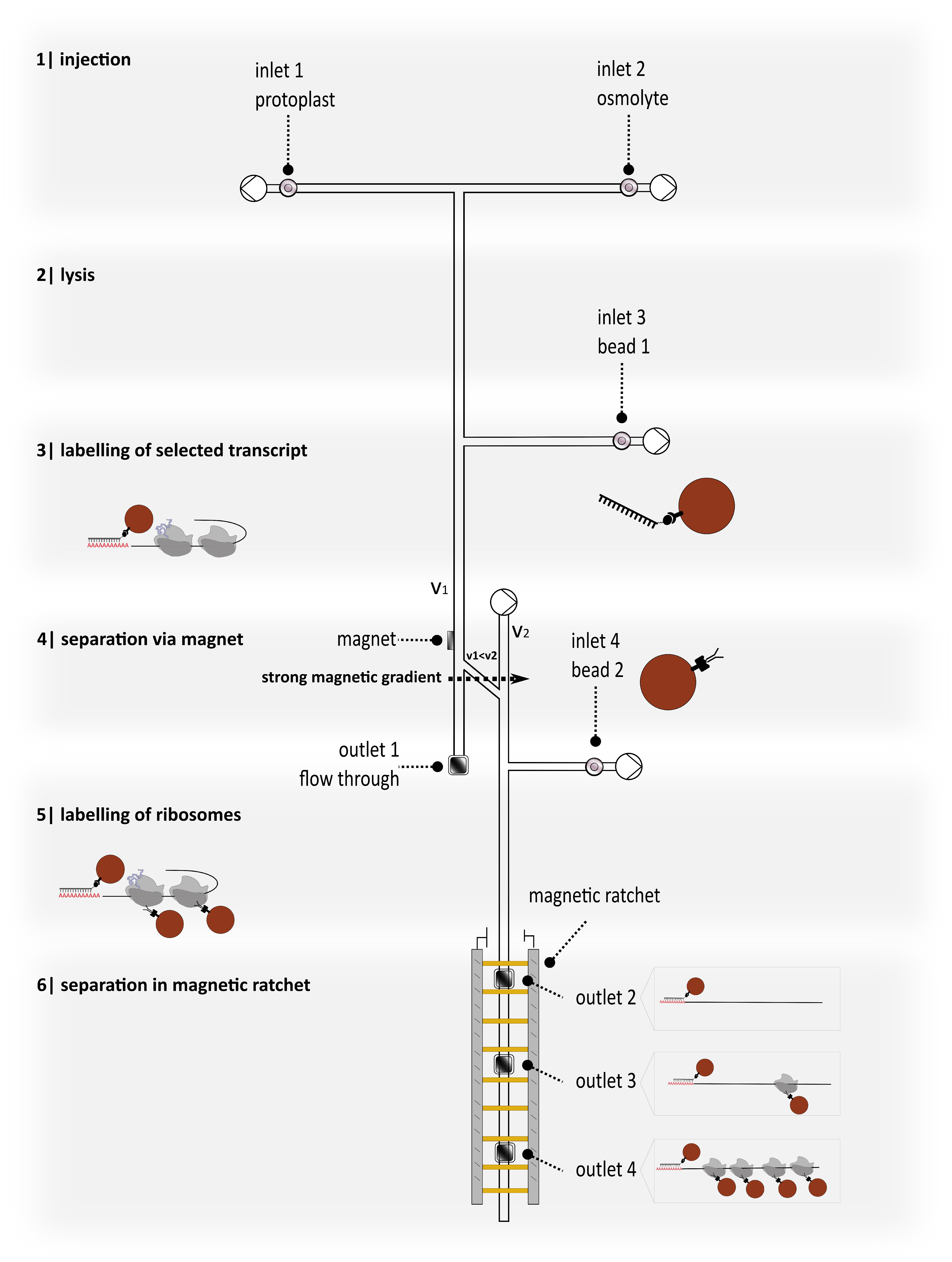
3.2. Proposing a New Device for Separation and Detection Ensuring Further Analysis
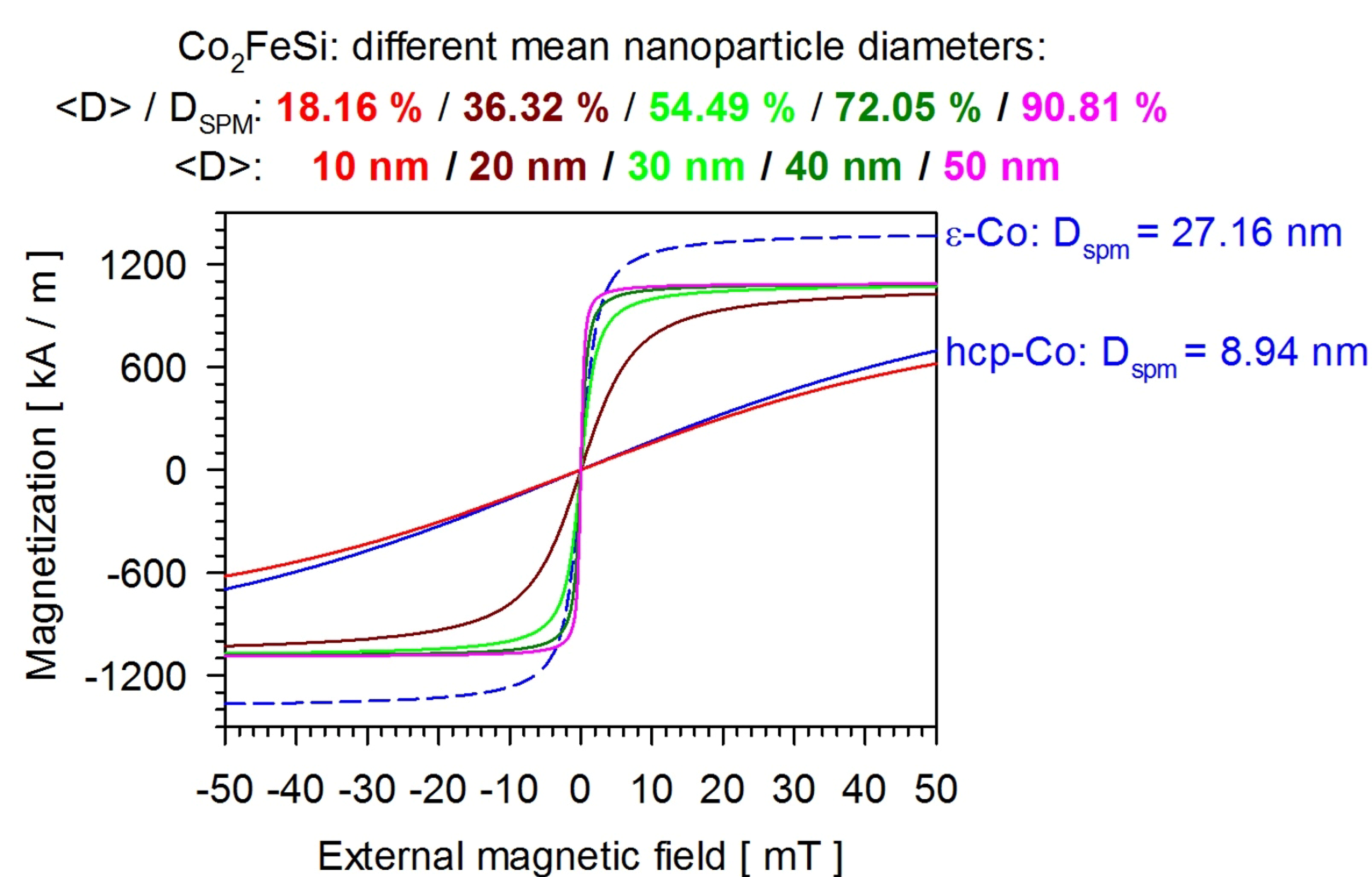
3.3. Microfluidic Design and Physical Parameters
3.4. Challenges and Perspectives
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Sonker, M.; Sahore, V.; Woolley, A.T. Recent Advances in Microfluidic Sample Preparation and Separation Techniques for Molecular Biomarker Analysis: A Critical Review. Anal. Chim. Acta 2017, 986, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Ko, J.; Cha, K.; Ho Jeon, J.; Rhie, G.; Choi, J.; deMello, A.J.; Choo, J. Fast and sensitive detection of an anthrax biomarker using SERS-based solenoid microfluidic sensor. Biosens. Bioelectron. 2015, 72, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, Y.; Zeng, Y.; He, M. A Microfluidic ExoSearch Chip for Multiplexed Exosome Detection Towards Blood-based Ovarian Cancer Diagnosis. Lab. Chip 2016, 16, 489–496. [Google Scholar] [CrossRef]
- Finka, A.; Goloubinoff, P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 2013, 18, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.; Wieske, M.; Stark, H.; Schatz, M.; Stahl, J.; Zemlin, F.; Lutsch, G.; van Heel, M. The 80S rat liver ribosome at 25 a resolution by electron cryomicroscopy and angular reconstitution. Structure 1998, 6, 389–399. [Google Scholar] [CrossRef]
- Montesano, L.; Glitz, D.G. Wheat germ cytoplasmic ribosomes. Structure of ribosomal subunits and localization of N6,N6-dimethyladenosine by immunoelectron microscopy. J. Biol. Chem. 1988, 263, 4932–4938. [Google Scholar]
- Browning, K.S.; Bailey-Serres, J. Mechanism of Cytoplasmic mRNA Translation. Arab. Book Am. Soc. Plant Biol. 2015, 13. [Google Scholar] [CrossRef]
- Moore, M.; Gossmann, N.; Dietz, K.-J. Redox Regulation of Cytosolic Translation in Plants. Trends Plant Sci. 2016, 21, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, A.; Juntawong, P.; Bailey-Serres, J. Isolation of Plant Polysomal mRNA by Differential Centrifugation and Ribosome Immunopurification Methods. In Plant Systems Biology; Belostotsky, D.A., Ed.; Methods in Molecular BiologyTM; Humana Press: Totowa, NJ, USA, 2009; pp. 109–126. ISBN 978-1-60327-563-7. [Google Scholar]
- Buttgereit, F.; Brand, M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995, 312, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nelson, C.J.; Trösch, J.; Castleden, I.; Huang, S.; Millar, A.H. Protein Degradation Rate in Arabidopsis thaliana Leaf Growth and Development. Plant Cell 2017, 29, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, A.; Castellano, M.M. Regulation of Translation Initiation under Abiotic Stress Conditions in Plants: Is It a Conserved or Not so Conserved Process among Eukaryotes? Comp. Funct. Genom. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.-L.; Muthuramalingam, M.; Vogel, M.; Dietz, K.-J. The link between transcript regulation and de novo protein synthesis in the retrograde high light acclimation response of Arabidopsis thaliana. BMC Genom. 2014, 15, 320. [Google Scholar] [CrossRef]
- Abreu, R.D.S.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2018, a032896. [Google Scholar] [CrossRef]
- Iwasaki, S.; Ingolia, N.T. The Growing Toolbox for Protein Synthesis Studies. Trends Biochem. Sci. 2017, 42, 612–624. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zou, Q.; Liu, Y.; Yang, X. Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun. 2016, 7, 11194. [Google Scholar] [CrossRef]
- Dieterich, D.C.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 2006, 103, 9482–9487. [Google Scholar] [CrossRef]
- Dieterich, D.C.; Hodas, J.J.L.; Gouzer, G.; Shadrin, I.Y.; Ngo, J.T.; Triller, A.; Tirrell, D.A.; Schuman, E.M. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2010, 13, 897–905. [Google Scholar] [CrossRef]
- Howden, A.J.M.; Geoghegan, V.; Katsch, K.; Efstathiou, G.; Bhushan, B.; Boutureira, O.; Thomas, B.; Trudgian, D.C.; Kessler, B.M.; Dieterich, D.C.; et al. QuaNCAT: Quantitating proteome dynamics in primary cells. Nat. Methods 2013, 10, 343–346. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Gossen, M.; Dittmar, G.; Selbach, M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics 2009, 9, 205–209. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Halstead, J.M.; Lionnet, T.; Wilbertz, J.H.; Wippich, F.; Ephrussi, A.; Singer, R.H.; Chao, J.A. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 2015, 347, 1367–1671. [Google Scholar] [CrossRef]
- Katz, Z.B.; English, B.P.; Lionnet, T.; Yoon, Y.J.; Monnier, N.; Ovryn, B.; Bathe, M.; Singer, R.H. Mapping translation “hot-spots” in live cells by tracking single molecules of mRNA and ribosomes. eLife 2016, 5, e10415. [Google Scholar] [CrossRef]
- Wang, C.; Han, B.; Zhou, R.; Zhuang, X. Real-time imaging of translation on single mRNA transcripts in live cells. Cell 2016, 165, 990–1001. [Google Scholar] [CrossRef]
- Yan, X.; Hoek, T.A.; Vale, R.D.; Tanenbaum, M.E. Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 2016, 165, 976–989. [Google Scholar] [CrossRef]
- Morisaki, T.; Lyon, K.; DeLuca, K.F.; DeLuca, J.G.; English, B.P.; Zhang, Z.; Lavis, L.D.; Grimm, J.B.; Viswanathan, S.; Looger, L.L.; et al. Real-time quantification of single RNA translation dynamics in living cells. Science 2016, 352, 1425–1429. [Google Scholar] [CrossRef]
- Wu, B.; Eliscovich, C.; Yoon, Y.J.; Singer, R.H. Translation dynamics of single mRNAs in live cells and neurons. Science 2016, 352, 1430–1435. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity of Mammalian Proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef]
- Osaka, T.; Matsunaga, T.; Nakanishi, T.; Arakaki, A.; Niwa, D.; Iida, H. Synthesis of magnetic nanoparticles and their application to bioassays. Anal. Bioanal. Chem. 2006, 384, 593–600. [Google Scholar] [CrossRef]
- Schmitz-Linneweber, C.; Williams-Carrier, R.; Barkan, A. RNA Immunoprecipitation and Microarray Analysis Show a Chloroplast Pentatricopeptide Repeat Protein to Be Associated with the 5′ Region of mRNAs Whose Translation It Activates. Plant Cell 2005, 17, 2791–2804. [Google Scholar] [CrossRef]
- Orlando, V.; Paro, R. Mapping polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell 1993, 75, 1187–1198. [Google Scholar] [CrossRef]
- Smith, J.E.; Baker, K.E. Purification of Transcript-Specific mRNP Complexes Formed In Vivo from Saccharomyces cerevisiae. Methods Mol. Biol. 2017, 1648, 201–220. [Google Scholar] [CrossRef]
- Wagner, G.J. Content and Vacuole/Extravacuole Distribution of Neutral Sugars, Free Amino Acids, and Anthocyanin in Protoplasts 1. Plant Physiol. 1979, 64, 88–93. [Google Scholar] [CrossRef]
- Le-Quoc, K.; Le-Quoc, D.; Pugin, A. An Efficient Method for Plant Vacuole Isolation using Digitonin for Plasmalemma Lysis. J. Plant Physiol. 1987, 126, 329–335. [Google Scholar] [CrossRef]
- Weddemann, A.; Wittbracht, F.; Auge, A.; Hütten, A. A hydrodynamic switch: Microfluidic separation system for magnetic beads. Appl. Phys. Lett. 2009, 94, 173501. [Google Scholar] [CrossRef]
- Auge, A.; Weddemann, A.; Wittbracht, F.; Hütten, A. Magnetic ratchet for biotechnological applications. Appl. Phys. Lett. 2009, 94, 183507. [Google Scholar] [CrossRef]
- Benyettou, F.; Milosevic, I.; Olsen, J.; Motte, J.; Trabolsi, A. Ultra-Small Superparamagnetic Iron Oxide Nanoparticles Made to Order. J. Bioanal. Biomed. 2012, 5, 2. [Google Scholar] [CrossRef]
- Wang, C.; Meyer, J.; Teichert, N.; Auge, A.; Rausch, E.; Balke, B.; Hütten, A.; Fecher, G.H.; Felser, C. Heusler nanoparticles for spintronics and ferromagnetic shape memory alloys. J. Vac. Sci. Technol. B 2014, 32, 020802. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wegener, M.; Ennen, I.; Walhorn, V.; Anselmetti, D.; Hütten, A.; Dietz, K.-J. Magnetic Tracking of Protein Synthesis in Microfluidic Environments—Challenges and Perspectives. Nanomaterials 2019, 9, 585. https://doi.org/10.3390/nano9040585
Wegener M, Ennen I, Walhorn V, Anselmetti D, Hütten A, Dietz K-J. Magnetic Tracking of Protein Synthesis in Microfluidic Environments—Challenges and Perspectives. Nanomaterials. 2019; 9(4):585. https://doi.org/10.3390/nano9040585
Chicago/Turabian StyleWegener, Melanie, Inga Ennen, Volker Walhorn, Dario Anselmetti, Andreas Hütten, and Karl-Josef Dietz. 2019. "Magnetic Tracking of Protein Synthesis in Microfluidic Environments—Challenges and Perspectives" Nanomaterials 9, no. 4: 585. https://doi.org/10.3390/nano9040585
APA StyleWegener, M., Ennen, I., Walhorn, V., Anselmetti, D., Hütten, A., & Dietz, K.-J. (2019). Magnetic Tracking of Protein Synthesis in Microfluidic Environments—Challenges and Perspectives. Nanomaterials, 9(4), 585. https://doi.org/10.3390/nano9040585