
Carbon-Supported Trimetallic Catalysts (PdAuNi/C) for Borohydride Oxidation Reaction

 , ,
, ,  ,
,  ,
,  ,
, 
Abstract
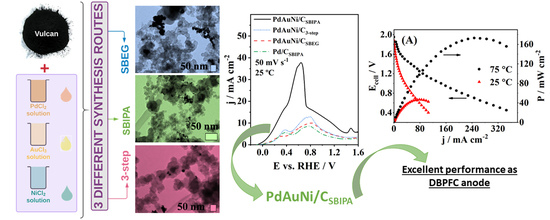
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Catalyst Preparation
2.3. Physical Characterization
2.4. Working Electrode Preparation
2.5. Electrochemical Evaluation
2.6. Fuel Cell Testing
3. Results and Discussion
3.1. Physical Characterization Results
3.2. Borohydride Oxidation Reaction Studies
3.3. Fuel Cell Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Larminie, J.; Dicks, A. Fuel Cell Systems Explained, 3rd ed.; John Wiley & Sons Ltd.: London, UK, 2018. [Google Scholar] [CrossRef]
- Kreuer, K.D. Fuel Cells, Introduction. In Fuel Cells; Kreuer, K.D., Ed.; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Maiyalagan, T.; Saji, V.S. Electrocatalysts for Low Temperature Fuel Cells; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017. [Google Scholar] [CrossRef]
- Akay, R.G.; Yurtcan, A.B. Direct Liquid Fuel Cells: Fundamentals, Advances and Future, 1st ed.; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Demirci, U.B. Direct borohydride fuel cell: Main issues met by the membrane–electrodes-assembly and potential solutions. J. Power Sources 2007, 172, 676–687. [Google Scholar] [CrossRef]
- Indig, M.E.; Snyder, R.N. Sodium Borohydride, An Interesting Anodic Fuel (1). J. Electrochem. Soc. 1962, 109, 1104–1106. [Google Scholar] [CrossRef]
- Vielstich, W.; Lamm, A.; Gasteiger, H. Handbook of Fuel Cells: Fundamentals, Technology, Applications, 4 Volume Set; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Ma, J.; Choudhury, N.A.; Sahai, Y. A comprehensive review of direct borohydride fuel cells. Renew. Sustain. Energy Rev. 2010, 14, 183–199. [Google Scholar] [CrossRef]
- de Leon, C.P.; Walsh, F.; Pletcher, D.; Browning, D.; Lakeman, J. Direct borohydride fuel cells. J. Power Sources 2006, 155, 172–181. [Google Scholar] [CrossRef]
- Ong, B.; Kamarudin, S.; Basri, S. Direct liquid fuel cells: A review. Int. J. Hydrogen Energy 2017, 42, 10142–10157. [Google Scholar] [CrossRef]
- Rostamikia, G.; Janik, M.J. Direct borohydride oxidation: Mechanism determination and design of alloy catalysts guided by density functional theory. Energy Environ. Sci. 2010, 3, 1262–1274. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Cyclic voltammetry investigation of borohydride oxidation at a gold electrode. Electrochim. Acta 2010, 55, 6775–6781. [Google Scholar] [CrossRef]
- Lima, F.H.; Pasqualeti, A.M.; Concha, M.B.M.; Chatenet, M.; Ticianelli, E.A. Borohydride electrooxidation on Au and Pt electrodes. Electrochim. Acta 2012, 84, 202–212. [Google Scholar] [CrossRef]
- Arevalo, R.L.; Escaño, M.C.S.; Wang, A.Y.-S.; Kasai, H. Structure and stability of borohydride on Au(111) and Au3M(111) (M = Cr, Mn, Fe, Co, Ni) surfaces. Dalton Trans. 2013, 42, 770–775. [Google Scholar] [CrossRef]
- He, P.; Wang, Y.; Wang, X.; Pei, F.; Wang, H.; Liu, L.; Yi, L. Investigation of carbon supported Au–Ni bimetallic nanoparticles as electrocatalyst for direct borohydride fuel cell. J. Power Sources 2011, 196, 1042–1047. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, Q.; Wu, C.; Wang, Y.; Wei, W.; Wang, X.; Yi, L. Performance improvement of activated nanoporous carbon supported gold catalyst as an anode for direct borohydride–hydrogen peroxide fuel cells. RSC Adv. 2014, 4, 17129–17135. [Google Scholar] [CrossRef]
- Yi, L.; Liu, L.; Wang, X.; Liu, X.; Yi, W.; Wang, X. Carbon supported Pt–Sn nanoparticles as anode catalyst for direct borohydride–hydrogen peroxide fuel cell: Electrocatalysis and fuel cell performance. J. Power Sources 2013, 224, 6–12. [Google Scholar] [CrossRef]
- Oliveira, R.C.P.; Milikić, J.; Daş, E.; Yurtcan, A.B.; Santos, D.M.F.; Šljukić, B. Platinum/polypyrrole-carbon electrocatalysts for direct borohydride-peroxide fuel cells. Appl. Catal. B Environ. 2018, 238, 454–464. [Google Scholar] [CrossRef]
- Freitas, K.S.; Concha, B.M.; Ticianelli, E.A.; Chatenet, M. Mass transport effects in the borohydride oxidation reaction—Influence of the residence time on the reaction onset and faradaic efficiency. Catal. Today 2011, 170, 110–119. [Google Scholar] [CrossRef]
- Olu, P.-Y.; Barros, C.R.; Job, N.; Chatenet, M. Electrooxidation of NaBH4 in Alkaline Medium on Well-defined Pt Nanoparticles Deposited onto Flat Glassy Carbon Substrate: Evaluation of the Effects of Pt Nanoparticle Size, Inter-Particle Distance, and Loading. Electrocatalysis 2014, 5, 288–300. [Google Scholar] [CrossRef]
- Šljukić, B.; Milikić, J.; Santos, D.M.F.; Sequeira, C.A.C.; Macciò, D.; Saccone, A. Electrocatalytic performance of Pt–Dy alloys for direct borohydride fuel cells. J. Power Sources 2014, 272, 335–343. [Google Scholar] [CrossRef]
- Šljukić, B.; Milikić, J.; Santos, D.M.F.; Sequeira, C.A.C. Carbon-supported Pt0.75M0.25 (M=Ni or Co) electrocatalysts for borohydride oxidation. Electrochim. Acta 2013, 107, 577–583. [Google Scholar] [CrossRef]
- Concha, B.M.; Chatenet, M. Direct oxidation of sodium borohydride on Pt, Ag and alloyed Pt–Ag electrodes in basic media. Electrochim. Acta 2009, 54, 6130–6139. [Google Scholar] [CrossRef]
- Gyenge, E.; Atwan, M.; Northwood, D. Electrocatalysis of Borohydride Oxidation on Colloidal Pt and Pt-Alloys (Pt-Ir, Pt-Ni, and Pt-Au) and Application for Direct Borohydride Fuel Cell Anodes. J. Electrochem. Soc. 2006, 153, A150–A158. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Šljukić, B.; Amaral, L.; Macciò, D.; Saccone, A.; Sequeira, C.A.C. Nickel and Nickel-Cerium Alloy Anodes for Direct Borohydride Fuel Cells. J. Electrochem. Soc. 2014, 161, F594–F599. [Google Scholar] [CrossRef]
- Hosseini, M.G.; Abdolmaleki, M.; Ashrafpoor, S. Electrocatalytic Oxidation of Sodium Borohydride on a Nanoporous Ni/Zn-Ni Electrode. Chin. J. Catal. 2012, 33, 1817–1824. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Zinc Anode for Direct Borohydride Fuel Cells. J. Electrochem. Soc. 2010, 157, B13–B19. [Google Scholar] [CrossRef]
- Vinayan, B.P.; Jafri, R.I.; Nagar, R.; Rajalakshmi, N.; Sethupathi, K.; Ramaprabhu, S. Catalytic activity of platinum–cobalt alloy nanoparticles decorated functionalized multiwalled carbon nanotubes for oxygen reduction reaction in PEMFC. Int. J. Hydrogen Energy 2012, 37, 412–421. [Google Scholar] [CrossRef]
- Rao, C.S.; Singh, D.; Sekhar, R.; Rangarajan, J. Pt–Co electrocatalyst with varying atomic percentage of transition metal. Int. J. Hydrogen Energy 2011, 36, 14805–14814. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Mintsouli, I.; Armyanov, S.; Valova, E.; Kokkinidis, G.; Sotiropoulos, S. Rotating disc electrode studies of borohydride oxidation at Pt and bimetallic Pt–Ni and Pt–Co electrodes. Catal. Today 2011, 170, 126–133. [Google Scholar] [CrossRef]
- Šljukić, B.; Santos, D.M.F. Direct Borohydride Fuel Cells. In Direct Liquid Fuel Cells: Fundamentals. Advances and Future, 1st ed.; Akay, R.G., Yurtcan, A.B., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 203–232. [Google Scholar] [CrossRef]
- Chatenet, M.; Lima, F.H.B.; Ticianelli, E.A. Gold is not a Faradaic-Efficient Borohydride Oxidation Electrocatalyst: An Online Electrochemical Mass Spectrometry Study. J. Electrochem. Soc. 2010, 157, B697–B704. [Google Scholar] [CrossRef]
- Braesch, G.; Bonnefont, A.; Martin, V.; Savinova, E.R.; Chatenet, M. Borohydride oxidation reaction mechanisms and poisoning effects on Au, Pt and Pd bulk electrodes: From model (low) to direct borohydride fuel cell operating (high) concentrations. Electrochim. Acta 2018, 273, 483–494. [Google Scholar] [CrossRef]
- Tamašauskaitė-Tamašiūnaitė, L.; Radomskis, A.; Antanavičiūtė, K.; Jablonskienė, J.; Balciunaite, A.; Žielienė, A.; Naruškevičius, L.; Kondrotas, R.; Norkus, E. Graphene supported platinum–cobalt nanocomposites as electrocatalysts for borohydride oxidation. Int. J. Hydrogen Energy 2014, 39, 4282–4290. [Google Scholar] [CrossRef]
- He, P.; Wang, X.; Liu, Y.; Liu, X.; Yi, L. Comparison of electrocatalytic activity of carbon-supported Au–M (M = Fe, Co, Ni, Cu and Zn) bimetallic nanoparticles for direct borohydride fuel cells. Int. J. Hydrogen Energy 2012, 37, 11984–11993. [Google Scholar] [CrossRef]
- Modibedi, R.M.; Masombuka, T.; Mathe, M. Carbon supported Pd–Sn and Pd–Ru–Sn nanocatalysts for ethanol electro-oxidation in alkaline medium. Int. J. Hydrogen Energy 2011, 36, 4664–4672. [Google Scholar] [CrossRef]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, Y.; Wang, Y.; Yao, J. Synthesis and performance of a novel PdCuPb/C nanocatalyst for ethanol electrooxidation in alkaline medium. Int. J. Hydrogen Energy 2014, 39, 4274–4281. [Google Scholar] [CrossRef]
- Zheng, Y.; Qiao, J.; Yuan, J.; Shen, J.; Wang, A.-J.; Huang, S. Controllable synthesis of PtPd nanocubes on graphene as advanced catalysts for ethanol oxidation. Int. J. Hydrogen Energy 2018, 43, 4902–4911. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, Y.; Pan, H.-B.; Zhu, C.; Fu, S.; Wai, C.M.; Du, D.; Zhu, J.-J.; Lin, Y. Ultrasonic-assisted synthesis of Pd–Pt/carbon nanotubes nanocomposites for enhanced electro-oxidation of ethanol and methanol in alkaline medium. Ultrason. Sonochemistry 2016, 28, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Brouzgou, A.; Podias, A.; Tsiakaras, P. PEMFCs and AEMFCs directly fed with ethanol: A current status comparative review. J. Appl. Electrochem. 2013, 43, 119–136. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Sodium borohydride determination by measurement of open circuit potentials. J. Electroanal. Chem. 2009, 627, 1–8. [Google Scholar] [CrossRef]
- Hong, J.; Fang, B.; Wang, C.; Currie, K. Zn-Air Fuel Cell/Battery Hybrid Power Sources with Addition of Borohydride. ECS Trans. 2006, 3, 89–99. [Google Scholar] [CrossRef]
- Hong, J.; Fang, B.; Wang, C.; Currie, K. Intrinsic borohydride fuel cell/battery hybrid power sources. J. Power Sources 2006, 161, 753–760. [Google Scholar] [CrossRef]
- Wang, X.; Xia, Y. Electrocatalytic performance of PdCo–C catalyst for formic acid oxidation. Electrochem. Commun. 2008, 10, 1644–1646. [Google Scholar] [CrossRef]
- Sheng, G.; Chen, J.; Ye, H.; Hu, Z.; Fu, X.-Z.; Sun, R.; Huang, W.; Wong, C.-P. Hollow PdCo alloy nanospheres with mesoporous shells as high-performance catalysts for methanol oxidation. J. Colloid Interface Sci. 2018, 522, 264–271. [Google Scholar] [CrossRef]
- Martins, M.; Šljukić, B.; Metin, Ö.; Sevim, M.; Sequeira, C.A.C.; Şener, T.; Santos, D.M.F. Bimetallic PdM (M = Fe, Ag, Au) alloy nanoparticles assembled on reduced graphene oxide as catalysts for direct borohydride fuel cells. J. Alloys Compd. 2017, 718, 204–214. [Google Scholar] [CrossRef]
- Martins, M.; Metin, Ö.; Sevim, M.; Šljukić, B.; Sequeira, C.A.C.; Sener, T.; Santos, D.M.F. Monodisperse Pd nanoparticles assembled on reduced graphene oxide-Fe3O4 nanocomposites as electrocatalysts for borohydride fuel cells. Int. J. Hydrogen Energy 2018, 43, 10686–10697. [Google Scholar] [CrossRef]
- Simões, M.; Baranton, S.; Coutanceau, C. Electrooxidation of Sodium Borohydride at Pd, Au, and PdxAu1−x Carbon-Supported Nanocatalysts. J. Phys. Chem. C 2009, 113, 13369–13376. [Google Scholar] [CrossRef]
- Jimenez, I.M.; Janik, M.; De Leon, C.P.; Walsh, F. Pd–Ir alloy as an anode material for borohydride oxidation. J. Power Sources 2014, 269, 498–508. [Google Scholar] [CrossRef]
- Grimmer, C.; Grandi, M.; Zacharias, R.; Cermenek, B.; Weber, H.; Morais, C.; Napporn, T.W.; Weinberger, S.; Schenk, A.; Hacker, V. The electrooxidation of borohydride: A mechanistic study on palladium (Pd/C) applying RRDE, 11B-NMR and FTIR. Appl. Catal. B Environ. 2016, 180, 614–621. [Google Scholar] [CrossRef]
- Šljukić, B.; Martins, M.; Kayhan, E.; Balčiūnaitė, A.; Şener, T.; Sequeira, C.A.C.; Santos, D.M.F. SnO2-C supported PdNi nanoparticles for oxygen reduction and borohydride oxidation. J. Electroanal. Chem. 2017, 797, 23–30. [Google Scholar] [CrossRef]
- Milikić, J.; Ćirić-Marjanović, G.; Mentus, S.; Santos, D.M.F.; Sequeira, C.A.C.; Šljukić, B. Pd/c-PANI electrocatalysts for direct borohydride fuel cells. Electrochim. Acta 2016, 213, 298–305. [Google Scholar] [CrossRef]
- Martins, M.; Šljukić, B.; Sequeira, C.A.C.; Metin, Ö.; Erdem, M.; Sener, T.; Santos, D.M.F. Biobased carbon-supported palladium electrocatalysts for borohydride fuel cells. Int. J. Hydrogen Energy 2016, 41, 10914–10922. [Google Scholar] [CrossRef]
- Uosaki, K. Electrochemical Science for a Sustainable Society: A Tribute to John O’M Bockris, 1st ed.; Springer International Publishing: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Lv, H.; Sun, L.; Zou, L.; Xu, D.; Yao, H.; Liu, B. Size-dependent synthesis and catalytic activities of trimetallic PdAgCu mesoporous nanospheres in ethanol electrooxidation. Chem. Sci. 2019, 10, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Hameed, R.A.; El-Khatib, K.; Youssef, M.E. Electrocatalytic activity of nanostructured Ni and Pd–Ni on Vulcan XC-72R carbon black for methanol oxidation in alkaline medium. Int. J. Hydrogen Energy 2014, 39, 2026–2041. [Google Scholar] [CrossRef]
- Obradović, M.; Stančić, Z.; Lačnjevac, U.; Radmilovic, V.; Gavrilović-Wohlmuther, A.; Gojković, S. Electrochemical oxidation of ethanol on palladium-nickel nanocatalyst in alkaline media. Appl. Catal. B Environ. 2016, 189, 110–118. [Google Scholar] [CrossRef]
- Dutta, A.; Datta, J. Energy efficient role of Ni/NiO in PdNi nano catalyst used in alkaline DEFC. J. Mater. Chem. A 2014, 2, 3237–3250. [Google Scholar] [CrossRef]
- Shen, S.; Zhao, T.S.; Xu, J.; Li, Y. High performance of a carbon supported ternary PdIrNi catalyst for ethanol electro-oxidation in anion-exchange membrane direct ethanol fuel cells. Energy Environ. Sci. 2011, 4, 1428–1433. [Google Scholar] [CrossRef]
- Yurderi, M.; Bulut, A.; Zahmakiran, M.; Kaya, M. Carbon supported trimetallic PdNiAg nanoparticles as highly active, selective and reusable catalyst in the formic acid decomposition. Appl. Catal. B Environ. 2014, 160, 514–524. [Google Scholar] [CrossRef]
- Zhu, W.; Ke, J.; Wang, S.-B.; Ren, J.; Wang, H.-H.; Zhou, Z.-Y.; Si, R.; Zhang, Y.-W.; Yan, C.-H. Shaping Single-Crystalline Trimetallic Pt–Pd–Rh Nanocrystals toward High-Efficiency C–C Splitting of Ethanol in Conversion to CO2. ACS Catal. 2015, 5, 1995–2008. [Google Scholar] [CrossRef]
- Hu, S.; Munoz, F.; Noborikawa, J.; Haan, J.; Scudiero, L.; Ha, S. Carbon supported Pd-based bimetallic and trimetallic catalyst for formic acid electrochemical oxidation. Appl. Catal. B Environ. 2016, 180, 758–765. [Google Scholar] [CrossRef]
- Sharma, G.; Kumar, D.; Kumar, A.; Al-Muhtaseb, A.H.; Pathania, D.; Naushad, M.; Mola, G.T. Revolution from monometallic to trimetallic nanoparticle composites, various synthesis methods and their applications: A review. Mater. Sci. Eng. C 2017, 71, 1216–1230. [Google Scholar] [CrossRef]
- Ulas, B.; Caglar, A.; Sahin, O.; Kivrak, H. Composition dependent activity of PdAgNi alloy catalysts for formic acid electrooxidation. J. Colloid Interface Sci. 2018, 532, 47–57. [Google Scholar] [CrossRef]
- Beyhan, S.; Léger, J.-M.; Kadırgan, F. Understanding the influence of Ni, Co, Rh and Pd addition to PtSn/C catalyst for the oxidation of ethanol by in situ Fourier transform infrared spectroscopy. Appl. Catal. B Environ. 2014, 144, 66–74. [Google Scholar] [CrossRef]
- Shang, C.; Hong, W.; Wang, J.; Wang, E. Carbon supported trimetallic nickel–palladium–gold hollow nanoparticles with superior catalytic activity for methanol electrooxidation. J. Power Sources 2015, 285, 12–15. [Google Scholar] [CrossRef]
- Dutta, A.; Datta, J. Outstanding Catalyst Performance of PdAuNi Nanoparticles for the Anodic Reaction in an Alkaline Direct Ethanol (with Anion-Exchange Membrane) Fuel Cell. J. Phys. Chem. C 2012, 116, 25677–25688. [Google Scholar] [CrossRef]
- Su, P.-C.; Chen, H.-S.; Chen, T.-Y.; Liu, C.-W.; Lee, C.-H.; Lee, J.-F.; Chan, T.-S.; Wang, K.-W. Enhancement of electrochemical properties of Pd/C catalysts toward ethanol oxidation reaction in alkaline solution through Ni and Au alloying. Int. J. Hydrogen Energy 2013, 38, 4474–4482. [Google Scholar] [CrossRef]
- Lu, L.; Kang, J. Amperometric nonenzymatic sensing of glucose at very low working potential by using a nanoporous PdAuNi ternary alloy. Microchim. Acta 2018, 185, 111. [Google Scholar] [CrossRef] [PubMed]
- Bulut, A.; Yurderi, M.; Kaya, M.; Aydemir, M.; Baysal, A.; Durap, F.; Zahmakiran, M. Amine-functionalized graphene nanosheet-supported PdAuNi alloy nanoparticles: Efficient nanocatalyst for formic acid dehydrogenation. New J. Chem. 2018, 42, 16103–16114. [Google Scholar] [CrossRef]
- Li, S.; Lai, J.; Luque, R.; Xu, G. Designed multimetallic Pd nanosponges with enhanced electrocatalytic activity for ethylene glycol and glycerol oxidation. Energy Environ. Sci. 2016, 9, 3097–3102. [Google Scholar] [CrossRef]
- Chatenet, M.; Molina-Concha, M.; Diard, J.-P. First insights into the borohydride oxidation reaction mechanism on gold by electrochemical impedance spectroscopy. Electrochim. Acta 2009, 54, 1687–1693. [Google Scholar] [CrossRef]
- Nagle, L.C.; Rohan, J. Nanoporous gold anode catalyst for direct borohydride fuel cell. Int. J. Hydrogen Energy 2011, 36, 10319–10326. [Google Scholar] [CrossRef]
- Tegou, A.; Armyanov, S.; Valova, E.; Steenhaut, O.; Hubin, A.; Kokkinidis, G.; Sotiropoulos, S. Mixed platinum–gold electrocatalysts for borohydride oxidation prepared by the galvanic replacement of nickel deposits. J. Electroanal. Chem. 2009, 634, 104–110. [Google Scholar] [CrossRef]
- Parrour, G.; Chatenet, M.; Diard, J.-P. Electrochemical impedance spectroscopy study of borohydride oxidation reaction on gold—Towards a mechanism with two electrochemical steps. Electrochim. Acta 2010, 55, 9113–9124. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Chronopotentiometric Investigation of Borohydride Oxidation at a Gold Electrode. J. Electrochem. Soc. 2010, 157, F16–F21. [Google Scholar] [CrossRef]
- Zhang, D.; Cheng, K.; Shi, N.; Guo, F.; Wang, G.; Cao, D. Nickel particles supported on multi-walled carbon nanotubes modified sponge for sodium borohydride electrooxidation. Electrochem. Commun. 2013, 35, 128–130. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Šljukić, B.; Amaral, L.; Milikić, J.; Sequeira, C.A.C.; Macciò, D.; Saccone, A. Nickel–rare earth electrodes for sodium borohydride electrooxidation. Electrochim. Acta 2016, 190, 1050–1056. [Google Scholar] [CrossRef]
- Oshchepkov, A.G.; Braesch, G.; Ould-Amara, S.; Rostamikia, G.; Maranzana, G.; Bonnefont, A.; Papaefthimiou, V.; Janik, M.J.; Chatenet, M.; Savinova, E.R. Nickel Metal Nanoparticles as Anode Electrocatalysts for Highly Efficient Direct Borohydride Fuel Cells. ACS Catal. 2019, 9, 8520–8528. [Google Scholar] [CrossRef]
- Chen, A.; Ostrom, C. Palladium-Based Nanomaterials: Synthesis and Electrochemical Applications. Chem. Rev. 2015, 115, 11999–12044. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, Y.; Wang, L.; Wang, S. Scalable synthesis of a Pd nanoparticle loaded hierarchically porous graphene network through multiple synergistic interactions. Chem. Commun. 2015, 51, 8357–8360. [Google Scholar] [CrossRef] [PubMed]
- Corradini, P.G.; Pires, F.I.; Paganin, V.A.; Perez, J.; Antolini, E. Effect of the relationship between particle size, inter-particle distance, and metal loading of carbon supported fuel cell catalysts on their catalytic activity. J. Nanoparticle Res. 2012, 14, 1–9. [Google Scholar] [CrossRef]
- Modibedi, R.M.; Mehlo, T.; Ozoemena, K.I.; Mathe, M. Preparation, characterisation and application of Pd/C nanocatalyst in passive alkaline direct ethanol fuel cells (ADEFC). Int. J. Hydrogen Energy 2015, 40, 15605–15612. [Google Scholar] [CrossRef]
- Kim, P.; Joo, J.B.; Kim, W.; Kim, J.; Song, I.K.; Yi, J. NaBH4-assisted ethylene glycol reduction for preparation of carbon-supported Pt catalyst for methanol electro-oxidation. J. Power Sources 2006, 160, 987–990. [Google Scholar] [CrossRef]
- Henrique, R.S.; Ayoub, J.M.S.; Piasentin, R.M.; Linardi, M.; Santos, M.C. Preparation of Pt/C-In2O3.SnO2 electrocatalysts by borohydride reduction process for ethanol electro-oxidation. Int. J. Electrochem. Sci. 2012, 7, 2036–2046. [Google Scholar] [CrossRef]
- Assumpção, M.; da Silva, S.; De Souza, R.; Buzzo, G.; Spinacé, E.; Santos, M.; Neto, A.; Silva, J. Investigation of PdIr/C electrocatalysts as anode on the performance of direct ammonia fuel cell. J. Power Sources 2014, 268, 129–136. [Google Scholar] [CrossRef]
- Miller, S.D.; İnoğlu, N.; Kitchin, J. Configurational correlations in the coverage dependent adsorption energies of oxygen atoms on late transition metal fcc(111) surfaces. J. Chem. Phys. 2011, 134, 104709. [Google Scholar] [CrossRef]
- Counsell, J.D.P. Surface Science Studies of Adsorption and Reactivity of Pd (111) and Au-Pd (111). Ph.D. Thesis, Cardiff University, Cardiff, Wales, UK, 2010. [Google Scholar]
- Naresh, N.; Wasim, F.G.S.; Ladewig, B.P.; Neergat, M. Removal of surfactant and capping agent from Pd nanocubes (Pd-NCs) using tert-butylamine: Its effect on electrochemical characteristics. J. Mater. Chem. A 2013, 1, 8553–8559. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Zhang, J.-M.; Zhang, P.-Y.; Li, Y.; Xiang, S.; Tang, H.-G.; Fan, Y.-J. Highly active carbon nanotube-supported Ru@Pd core-shell nanostructure as an efficient electrocatalyst toward ethanol and formic acid oxidation. Mol. Catal. 2017, 436, 138–144. [Google Scholar] [CrossRef]
- Qiu, X.; Dai, Y.; Tang, Y.; Lu, T.; Wei, S.; Chen, Y. One-pot synthesis of gold–palladium@palladium core–shell nanoflowers as efficient electrocatalyst for ethanol electrooxidation. J. Power Sources 2015, 278, 430–435. [Google Scholar] [CrossRef]
- Zhou, W.; Lee, J.Y. Highly active core–shell Au@Pd catalyst for formic acid electrooxidation. Electrochem. Commun. 2007, 9, 1725–1729. [Google Scholar] [CrossRef]
- Akhairi, M.; Kamarudin, S. Catalysts in direct ethanol fuel cell (DEFC): An overview. Int. J. Hydrogen Energy 2016, 41, 4214–4228. [Google Scholar] [CrossRef]
- Feng, Y.-Y.; Liu, Z.-H.; Xu, Y.; Wang, P.; Wang, W.-H.; Kong, D.-S. Highly active PdAu alloy catalysts for ethanol electro-oxidation. J. Power Sources 2013, 232, 99–105. [Google Scholar] [CrossRef]
- Feng, Y.; Bin, D.; Yan, B.; Du, Y.; Majima, T.; Zhou, W. Porous bimetallic PdNi catalyst with high electrocatalytic activity for ethanol electrooxidation. J. Colloid Interface Sci. 2017, 493, 190–197. [Google Scholar] [CrossRef]
- Akhtar, K.; Khan, S.A.; Khan, S.B.; Asiri, A.M. Scanning Electron Microscopy: Principle and Applications in Nanomaterials Characterization. In Handbook of Materials Characterization; Akhtar, K., Khan, S.A., Khan, S.B., Asiri, A.M., Eds.; Springer International Publishing: Bassel, Switzerland, 2018; pp. 113–145. [Google Scholar] [CrossRef]
- Pasqualeti, A.M.; Olu, P.-Y.; Chatenet, M.; Lima, F.H.B. Borohydride Electrooxidation on Carbon-Supported Noble Metal Nanoparticles: Insights into Hydrogen and Hydroxyborane Formation. ACS Catal. 2015, 5, 2778–2787. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Saturnino, P.; Lobo, R.F.M.; Sequeira, C.A.C. Direct borohydride/peroxide fuel cells using Prussian Blue cathodes. J. Power Sources 2012, 208, 131–137. [Google Scholar] [CrossRef]
- Santos, D.M.F.; Sequeira, C.A.C. Effect of Membrane Separators on the Performance of Direct Borohydride Fuel Cells. J. Electrochem. Soc. 2011, 159, B126–B132. [Google Scholar] [CrossRef]
- Lam, B.T.X.; Chiku, M.; Higuchi, E.; Inoue, H. Preparation of PdAg and PdAu nanoparticle-loaded carbon black catalysts and their electrocatalytic activity for the glycerol oxidation reaction in alkaline medium. J. Power Sources 2015, 297, 149–157. [Google Scholar] [CrossRef]
- Zhang, S.; Qing, M.; Zhang, H.; Tian, Y. Electrocatalytic oxidation of formic acid on functional MWCNTs supported nanostructured Pd–Au catalyst. Electrochem. Commun. 2009, 11, 2249–2252. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.-H.; Sun, Q.-J.; Chang, J.-B.; Gao, X.; Liu, Y.; Lee, S.-T.; Kang, Z.-H.; Wang, S.-D. Controlled synthesis and synergistic effects of graphene-supported PdAu bimetallic nanoparticles with tunable catalytic properties. Nanoscale 2015, 7, 6356–6362. [Google Scholar] [CrossRef]
- Geraldes, A.N.; da Silva, D.F.; Pino, E.S.; da Silva, J.C.M.; de Souza, R.F.B.; Hammer, P.; Spinacé, E.V.; Neto, A.O.; Linardi, M.; dos Santos, M.C. Ethanol electro-oxidation in an alkaline medium using Pd/C, Au/C and PdAu/C electrocatalysts prepared by electron beam irradiation. Electrochim. Acta 2013, 111, 455–465. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Y.; Wang, X.; Chen, Y.; Zhou, Y.; Tang, Y.; Lu, L.; Bao, J.; Lu, T. Preparation of Pd–Au/C catalysts with different alloying degree and their electrocatalytic performance for formic acid oxidation. Appl. Catal. B Environ. 2011, 102, 614–619. [Google Scholar] [CrossRef]
- Dutta, A.; Mondal, A.; Broekmann, P.; Datta, J. Optimal level of Au nanoparticles on Pd nanostructures providing remarkable electro-catalysis in direct ethanol fuel cell. J. Power Sources 2017, 361, 276–284. [Google Scholar] [CrossRef]
- Qin, Y.-H.; Jiang, Y.; Niu, D.-F.; Zhang, X.-S.; Zhou, X.-G.; Niu, L.; Yuan, W.-K. Carbon nanofiber supported bimetallic PdAu nanoparticles for formic acid electrooxidation. J. Power Sources 2012, 215, 130–134. [Google Scholar] [CrossRef]
- Yin, Z.; Chi, M.; Zhu, Q.; Ma, D.; Sun, J.; Bao, X. Supported bimetallic PdAu nanoparticles with superior electrocatalytic activity towards methanol oxidation. J. Mater. Chem. A 2013, 1, 9157–9163. [Google Scholar] [CrossRef]
- Zhang, Z.; Xin, L.; Sun, K.; Li, W. Pd–Ni electrocatalysts for efficient ethanol oxidation reaction in alkaline electrolyte. Int. J. Hydrogen Energy 2011, 36, 12686–12697. [Google Scholar] [CrossRef]
- Holade, Y.; Sahin, N.E.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Recent Advances in Carbon Supported Metal Nanoparticles Preparation for Oxygen Reduction Reaction in Low Temperature Fuel Cells. Catalysts 2015, 5, 310–348. [Google Scholar] [CrossRef]
- Zhu, C.; Wen, D.; Oschatz, M.; Holzschuh, M.; Liu, W.; Herrmann, A.-K.; Simon, F.; Kaskel, S.; Eychmüller, A. Kinetically Controlled Synthesis of PdNi Bimetallic Porous Nanostructures with Enhanced Electrocatalytic Activity. Small 2015, 11, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.; Wei, X. Catalytic performances of PdNi/MWCNT for electrooxidations of methanol and ethanol in alkaline media. Int. J. Hydrogen Energy 2015, 40, 1154–1162. [Google Scholar] [CrossRef]
- Ramulifho, T.; Ozoemena, K.I.; Modibedi, R.M.; Jafta, C.J.; Mathe, M. Electrocatalytic oxidation of ethylene glycol at palladium-bimetallic nanocatalysts (PdSn and PdNi) supported on sulfonate-functionalised multi-walled carbon nanotubes. J. Electroanal. Chem. 2013, 692, 26–30. [Google Scholar] [CrossRef]
- Chen, L.Y.; Chen, N.; Hou, Y.; Wang, Z.C.; Lv, S.H.; Fujita, T.; Jiang, J.H.; Hirata, A.; Chen, M. Geometrically Controlled Nanoporous PdAu Bimetallic Catalysts with Tunable Pd/Au Ratio for Direct Ethanol Fuel Cells. ACS Catal. 2013, 3, 1220–1230. [Google Scholar] [CrossRef]
- Behmenyar, G.; Akın, A.N. Investigation of carbon supported Pd–Cu nanoparticles as anode catalysts for direct borohydride fuel cell. J. Power Sources 2014, 249, 239–246. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, D.-W.; Park, I.S.; Hong, Y.-K.; Park, Y.-M.; Lee, K.-Y. The effective bimetallic component of Pd–Au/C for electrochemical oxidation of borohydrides. Curr. Appl. Phys. 2010, 10, S40–S43. [Google Scholar] [CrossRef]
- He, P.; Wang, X.; Liu, Y.; Yi, L.; Liu, X. Reverse micelle synthesis of AuNi alloy as electrocatalyst of borohydride oxidation. Int. J. Hydrogen Energy 2012, 37, 1254–1262. [Google Scholar] [CrossRef]
- Yang, J.Q.; Liu, B.H.; Wu, S. Carbon-supported Pd catalysts: Influences of nanostructure on their catalytic performances for borohydride electrochemical oxidation. J. Power Sources 2009, 194, 824–829. [Google Scholar] [CrossRef]
- Duan, D.; Yin, X.; Wang, Q.; Liu, S.; Wang, Y. Performance evaluation of borohydride electrooxidation reaction with ternary alloy Au–Ni–Cu/C catalysts. J. Appl. Electrochem. 2018, 48, 835–847. [Google Scholar] [CrossRef]
- Antolini, E. Structural parameters of supported fuel cell catalysts: The effect of particle size, inter-particle distance and metal loading on catalytic activity and fuel cell performance. Appl. Catal. B Environ. 2016, 181, 298–313. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Wei, J.; Wang, X.; Wang, Y.; Chen, Q.; Pei, F.; Wang, Y. Investigation of carbon-supported Au hollow nanospheres as electrocatalyst for electrooxidation of sodium borohydride. Int. J. Hydrogen Energy 2009, 34, 3360–3366. [Google Scholar] [CrossRef]
- Wang, K.; Lu, J.; Zhuang, L. A Current−Decomposition Study of the Borohydride Oxidation Reaction at Ni Electrodes. J. Phys. Chem. C 2007, 111, 7456–7462. [Google Scholar] [CrossRef]
- Hosseini, M.G.; Abdolmaleki, M. Synthesis and characterization of porous nanostructured Ni/PdNi electrode towards electrooxidation of borohydride. Int. J. Hydrogen Energy 2013, 38, 5449–5456. [Google Scholar] [CrossRef]
- Oliveira, R.C.P.; Vasić, M.; Santos, D.M.F.; Babić, B.; Hercigonja, R.; Sequeira, C.A.C.; Šljukić, B. Performance assessment of a direct borohydride-peroxide fuel cell with Pd-impregnated faujasite X zeolite as anode electrocatalyst. Electrochim. Acta 2018, 269, 517–525. [Google Scholar] [CrossRef]
- Yi, L.; Song, Y.; Wang, X.; Yi, L.; Hu, J.; Su, G.; Yi, W.; Yan, H. Carbon supported palladium hollow nanospheres as anode catalysts for direct borohydride-hydrogen peroxide fuel cells. J. Power Sources 2012, 205, 63–70. [Google Scholar] [CrossRef]
- Song, C.; Wang, G.; Li, B.; Miao, C.; Ma, K.; Zhu, K.; Cheng, K.; Ye, K.; Yan, J.; Cao, D.; et al. A novel electrode of ternary CuNiPd nanoneedles decorated Ni foam and its catalytic activity toward NaBH4 electrooxidation. Electrochim. Acta 2019, 299, 395–404. [Google Scholar] [CrossRef]
- Oh, T.H.; Jang, B.; Kwon, S. Performance evaluation of direct borohydride–hydrogen peroxide fuel cells with electrocatalysts supported on multiwalled carbon nanotubes. Energy 2014, 76, 911–919. [Google Scholar] [CrossRef]
- Pei, F.; Wang, Y.; Wang, X.; He, P.; Chen, Q.; Wang, X.; Wang, H.; Yi, L.; Guo, J. Performance of supported Au–Co alloy as the anode catalyst of direct borohydride-hydrogen peroxide fuel cell. Int. J. Hydrogen Energy 2010, 35, 8136–8142. [Google Scholar] [CrossRef]
- Mahmoodi, R.; Hosseini, M.G.; Rasouli, H. Enhancement of output power density and performance of direct borohydride-hydrogen peroxide fuel cell using Ni-Pd core-shell nanoparticles on polymeric composite supports (rGO-PANI) as novel electrocatalysts. Appl. Catal. B Environ. 2019, 251, 37–48. [Google Scholar] [CrossRef]
- Hosseini, M.; Mahmoodi, R.; Amjadi, M.S. Carbon supported Ni1Pt1 nanocatalyst as superior electrocatalyst with increased power density in direct borohydride-hydrogen peroxide and investigation of cell impedance at different temperatures and discharging currents. Energy 2017, 131, 137–148. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | 2θ° | FWHM° | d-Space (Å) | a (Å) | τXRD (nm) |
|---|---|---|---|---|---|
| Pd/CSBIPA | 39.8 | 2.19 | 2.26 | 3.92 | 4.3 |
| PdAuNi/CSBIPA | 39.4 | 2.47 | 2.26 | 3.92 | 3.8 |
| PdAuNi/CSBEG | 39.6 | 2.32 | 2.26 | 3.92 | 4.9 |
| PdAuNi/C3-step | 38.5 | 1.11 | 2.33 | 4.03 | 8.9 |
| Catalyst | Pd/wt.% | Au/wt.% | Ni/wt.% |
|---|---|---|---|
| Pd/CSBIPA | 12 | - | - |
| PdAuNi/CSBIPA | 5.1 | 2.6 | 0.7 |
| PdAuNi/CSBEG | 4.8 | 0.8 | 9.9 |
| PdAuNi/C3-step | 7.5 | 5.5 | 9.3 |
| PdAuNi/CSBEG | PdAuNi/C3-step | PdAuNi/CSBIPA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 kV | 20 kV | 10 kV | 20 kV | 10 kV | 20 kV | |||||||
| wt.% | at.%. | wt.% | at.% | wt.% | at.%. | wt.% | at.% | wt.% | at.%. | wt.% | at.% | |
| C | 63.9 | 82.7 | 62.3 | 80.6 | 62.6 | 84.5 | 61.8 | 82.8 | 60.1 | 83.6 | 59.3 | 81.5 |
| O | 6.67 | 6.48 | 8.48 | 8.25 | 4.44 | 4.50 | 5.42 | 5.46 | 4.63 | 4.84 | 6.56 | 6.77 |
| Ni | 13.4 | 3.54 | 12.3 | 3.25 | 8.51 | 2.35 | 6.74 | 1.85 | 10.2 | 2.90 | 8.97 | 2.52 |
| Pd | 6.03 | 0.88 | 6.23 | 0.91 | 10.4 | 1.58 | 10.7 | 1.62 | 11.5 | 1.80 | 11.3 | 1.75 |
| Au | 3.82 | 0.30 | 3.93 | 0.31 | 7.96 | 0.66 | 8.17 | 0.67 | 7.92 | 0.67 | 7.56 | 0.63 |
| Catalyst | α | n | β | Ea/kJ mol−1 |
|---|---|---|---|---|
| PdAuNi/CSBEG | 0.60 | 2.8 | 0.91 | 34.0 |
| PdAuNi/CSBIPA | 0.75 | 7.1 | 1.00 | 16.7 |
| PdAuNi/C3-step | 0.73 | 7.4 | 1.12 | 29.9 |
| Pd/CSBIPA | 0.70 | 1.2 | 0.94 | 28.9 |
| Anode | Cathode | Fuel | Oxidant | T/°C | OCV/V | P/mW cm−2 | Ecell, peak/V | Ref. |
|---|---|---|---|---|---|---|---|---|
| PdAuNi/CSBIPA | Pt | 1 M NaBH4 + 4 M NaOH | 5 M H2O2 + 1.5 M HCl | 25 | 1.90 | 47.5 | 0.60 | This work |
| 75 | 1.90 | 175 | 0.70 | |||||
| Au/C | Pt | 0.5 M NaBH4 + 2 M NaOH | 4.5 M H2O2 + 2 M HCl | 20 | 1.41 | 8.72 | 0.42 | [119] |
| AuCu/C | Pt | 0.5 M NaBH4 + 2 M NaOH | 4.5 M H2O2 + 2 M HCl | 20 | 1.50 | 31.3 | 0.49 | [119] |
| AuNi/C | Pt | 0.5 M NaBH4 + 2 M NaOH | 4.5 M H2O2 + 2 M HCl | 20 | 1.59 | 40.6 | 0.57 | [119] |
| Au1.5NiCu/C | Pt | 0.5 M NaBH4 + 2 M NaOH | 4.5 M H2O2 + 2 M HCl | 20 | 1.62 | 47.4 | 0.70 | [119] |
| Au2NiCu/C | Pt | 0.5 M NaBH4 + 2 M NaOH | 4.5 M H2O2 + 2 M HCl | 20 | 1.78 | 60.5 | 0.88 | [119] |
| Au/C | Au/C | 1 M NaBH4 + 3 M NaOH | 2 M H2O2 + 0.5 M H2SO4 | 25 | 1.87 | 28.2 | 0.59 | [129] |
| Au45Co55/C | Au/C | 1 M NaBH4 + 3 M NaOH | 2 M H2O2 + 0.5 M H2SO4 | 25 | 1.92 | 66.5 | 0.77 | [129] |
| Ni@Pd/PANI 1 | Pt/C | 1 M NaBH4 + 2 M NaOH | 2 M H2O2 + 1.5 M HCl | 60 | 1.76 | 120 | 0.61 | [130] |
| PtNi/C | Pt/C | 1 M NaBH4 + 2 M NaOH | 2 M H2O2 + 0.5 M H2SO4 | 60 | 1.77 | 107 | 0.82 | [131] |
| Pd/MWCNT | Pt/MWCNTs 2 | 5 wt.% NaBH4 + 10 wt.% NaOH | 5 wt.% H2O2 + 5 wt.% H2PO4 | 25 | 1.80 | 119 | 0.58 | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
ElSheikh, A.M.A.; Backović, G.; Oliveira, R.C.P.; Sequeira, C.A.C.; McGregor, J.; Šljukić, B.; Santos, D.M.F. Carbon-Supported Trimetallic Catalysts (PdAuNi/C) for Borohydride Oxidation Reaction. Nanomaterials 2021, 11, 1441. https://doi.org/10.3390/nano11061441
ElSheikh AMA, Backović G, Oliveira RCP, Sequeira CAC, McGregor J, Šljukić B, Santos DMF. Carbon-Supported Trimetallic Catalysts (PdAuNi/C) for Borohydride Oxidation Reaction. Nanomaterials. 2021; 11(6):1441. https://doi.org/10.3390/nano11061441
Chicago/Turabian StyleElSheikh, Ahmed M. A., Gordana Backović, Raisa C. P. Oliveira, César A. C. Sequeira, James McGregor, Biljana Šljukić, and Diogo M. F. Santos. 2021. "Carbon-Supported Trimetallic Catalysts (PdAuNi/C) for Borohydride Oxidation Reaction" Nanomaterials 11, no. 6: 1441. https://doi.org/10.3390/nano11061441
APA StyleElSheikh, A. M. A., Backović, G., Oliveira, R. C. P., Sequeira, C. A. C., McGregor, J., Šljukić, B., & Santos, D. M. F. (2021). Carbon-Supported Trimetallic Catalysts (PdAuNi/C) for Borohydride Oxidation Reaction. Nanomaterials, 11(6), 1441. https://doi.org/10.3390/nano11061441