
CuZnAl-Oxide Nanopyramidal Mesoporous Materials for the Electrocatalytic CO2 Reduction to Syngas: Tuning of H2/CO Ratio

 ,
,  ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
| Cathode: | E° (V vs. NHE, pH = 0) | |
| 2H+ + 2 e− ⇄ H2 | 0.000 | (1) |
| CO2 + 2H+ + 2 e− ⇄ CO + H2O | −0.105 | (2) |
| CO2 + 2H+ + 2 e− ⇄ HCOOH | −0.169 | (3) |
| CO2 + 6H+ + 6 e− ⇄ CH3OH + 6H2O | −0.017 | (4) |
| CO2 + 8H+ + 8 e− ⇄ CH4 + 2H2O | 0.169 | (5) |
| 2CO2 + 10H+ + 10 e− ⇄ CH3CHO + 3H2O | 0.050 | (6) |
| 2CO2 + 12H+ + 12 e− ⇄ C2H5OH + 3H2O | 0.084 | (7) |
| 3CO2 + 16H+ + 16 e− ⇄ CH3(CO)CH3 + 5H2O | −0.140 | (8) |
| 3CO2 + 16H+ + 16 e− ⇄ CH3CH2CHO + 5H2O | 0.140 | (9) |
| 3CO2 + 18H+ + 18 e− ⇄ C3H7OH + 5H2O | 0.099 | (10) |
2. Materials and Methods
2.1. Materials
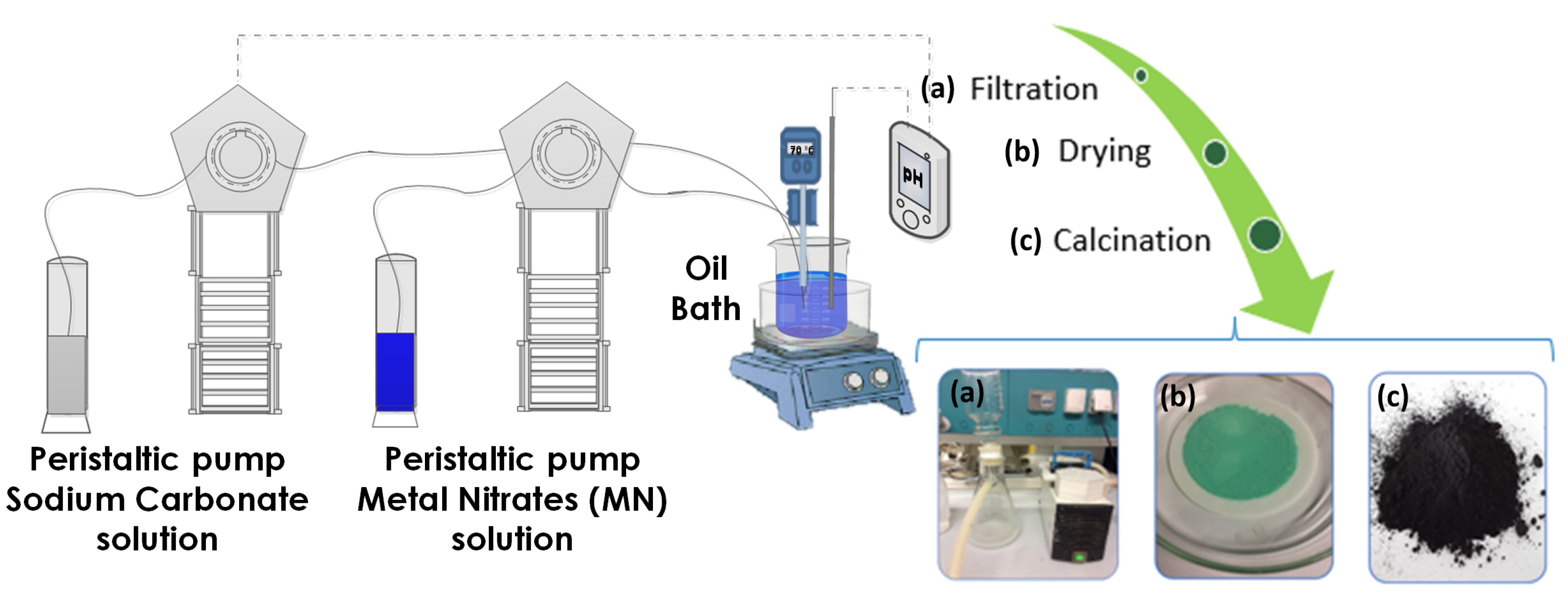
2.2. Synthesis of CuZnAl-Oxide Based Catalysts
2.3. Characterization of Powder Catalysts
2.4. Electrochemical Cell and Experimental Conditions
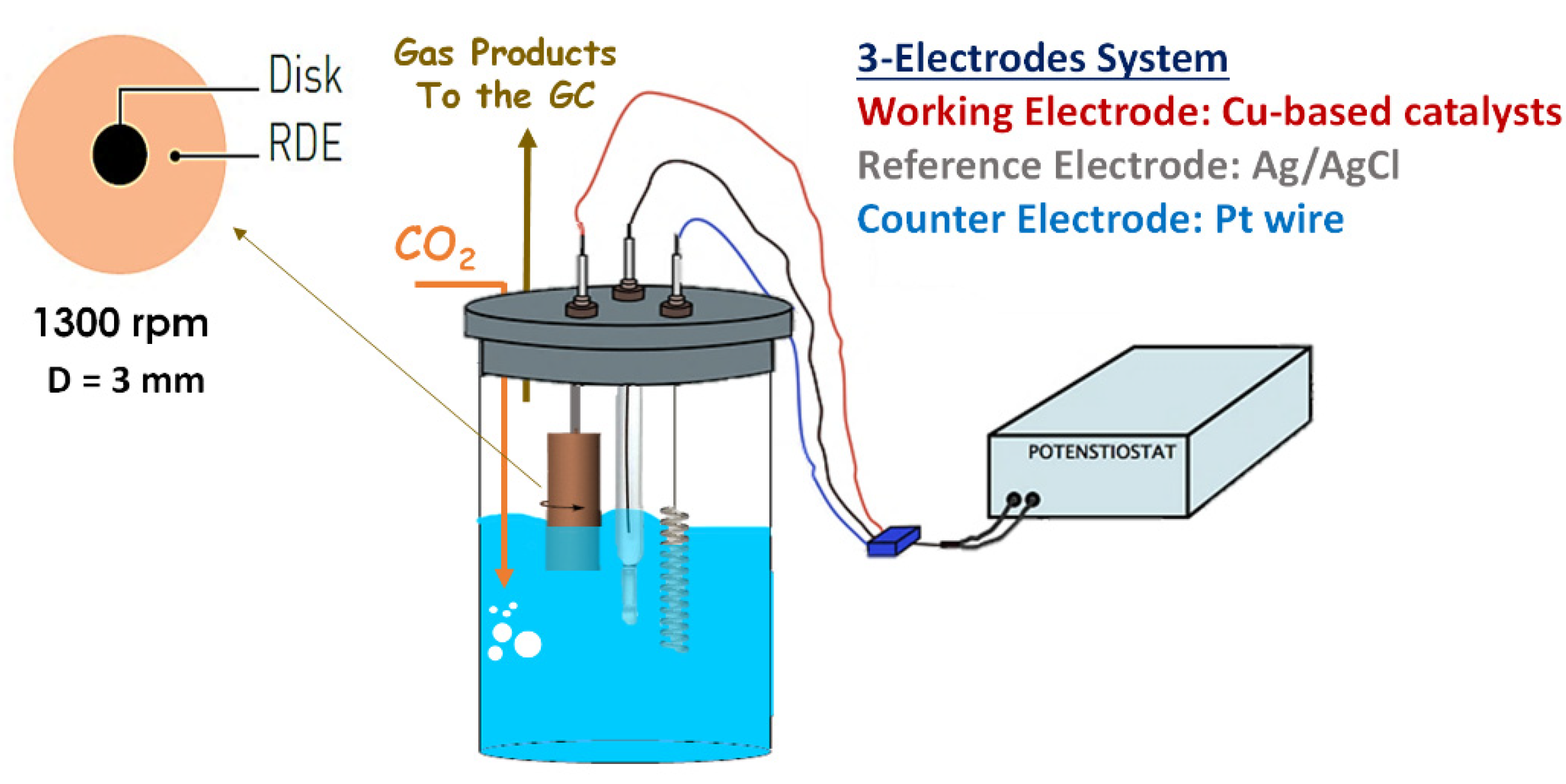
2.4.1. RDE System
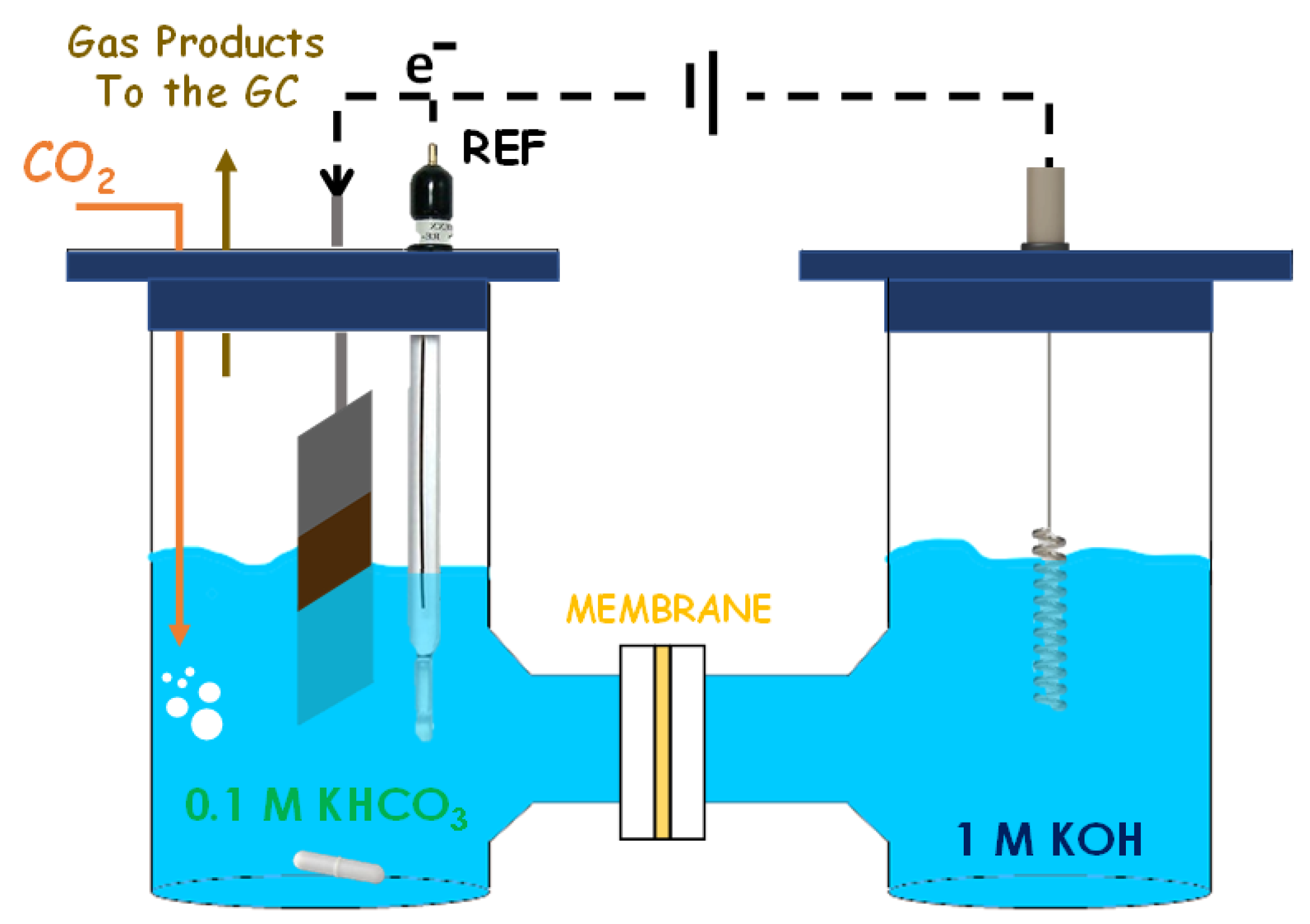
2.4.2. H-Cell System
3. Results and Discussion
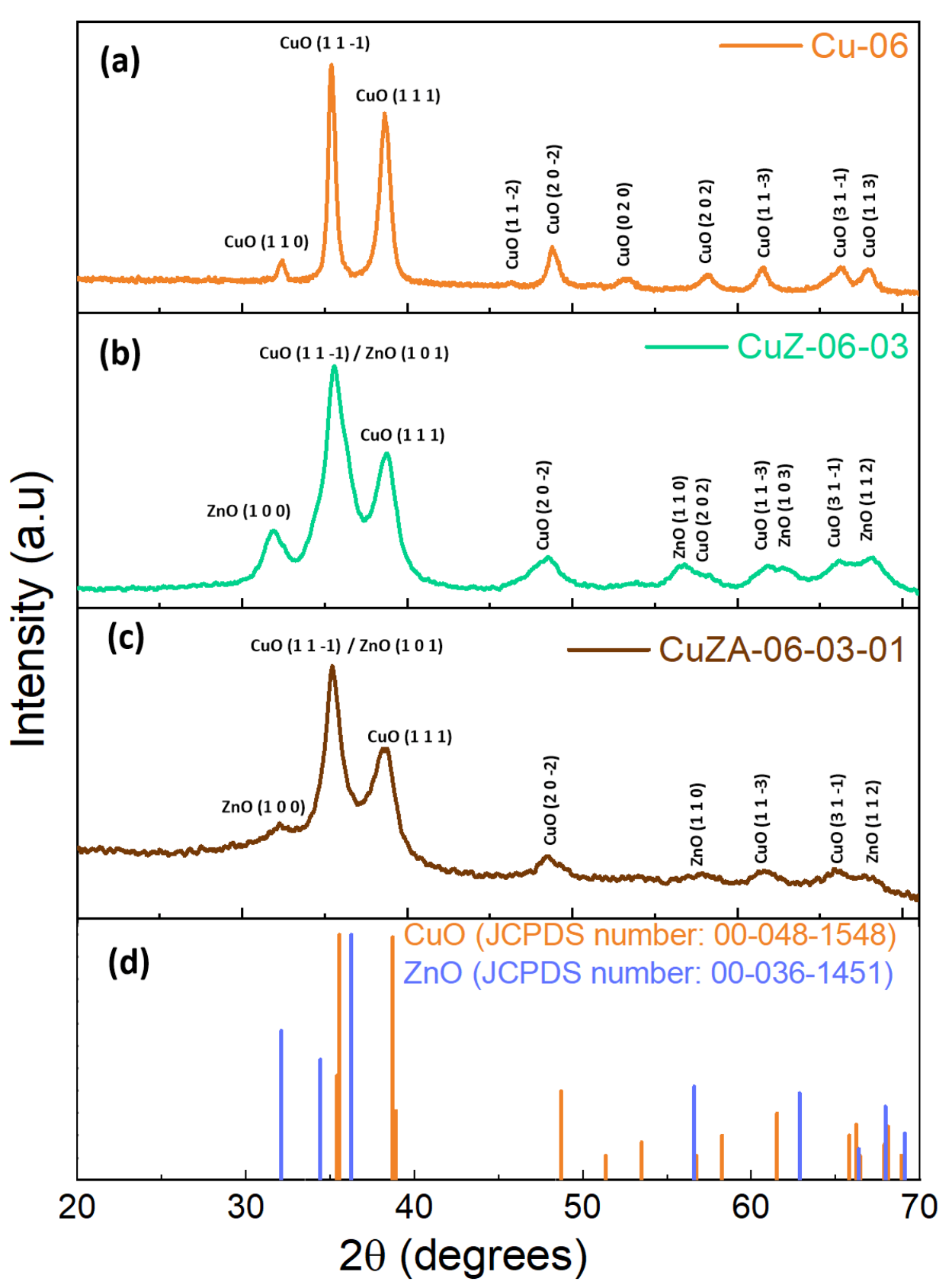
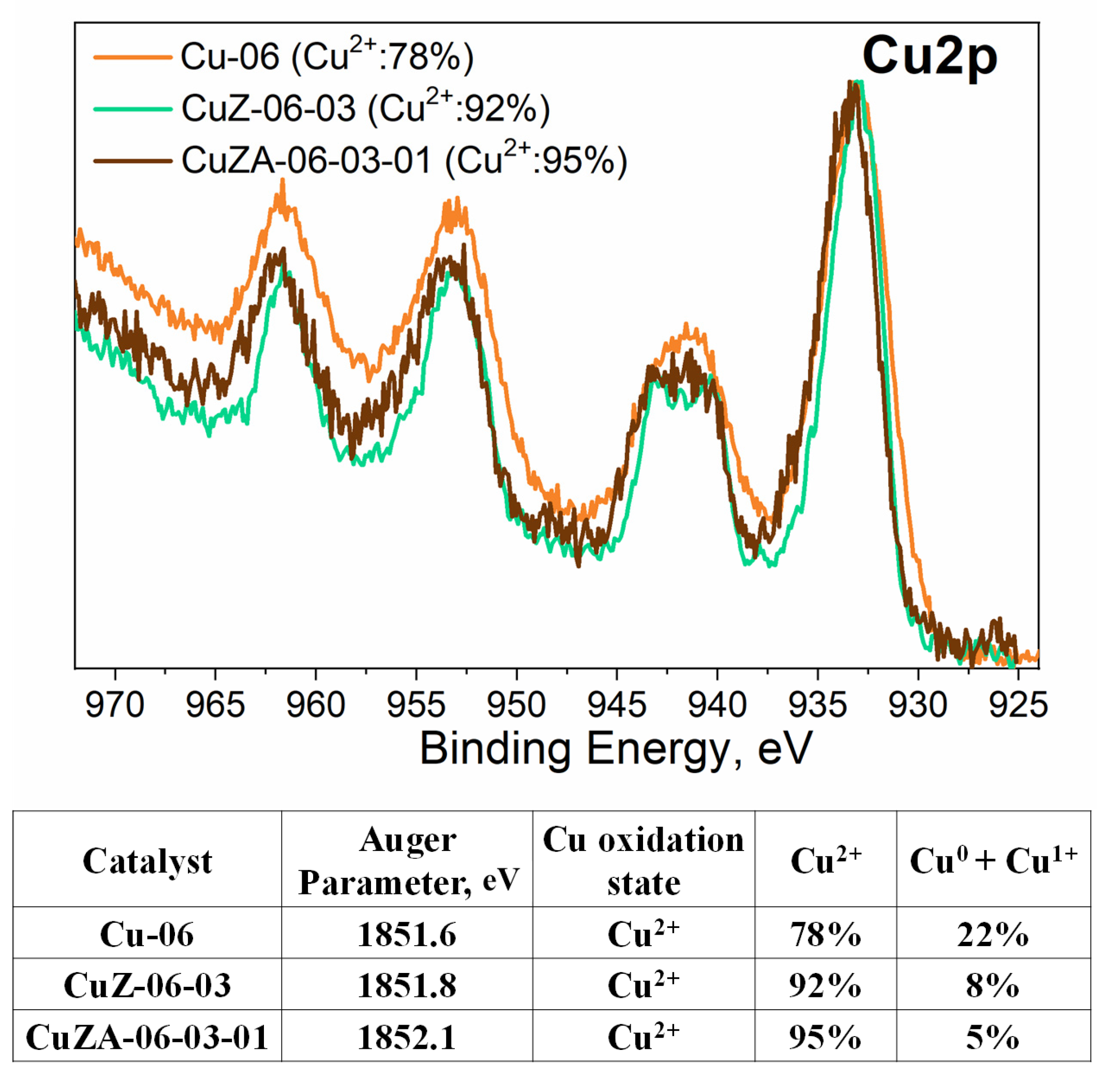
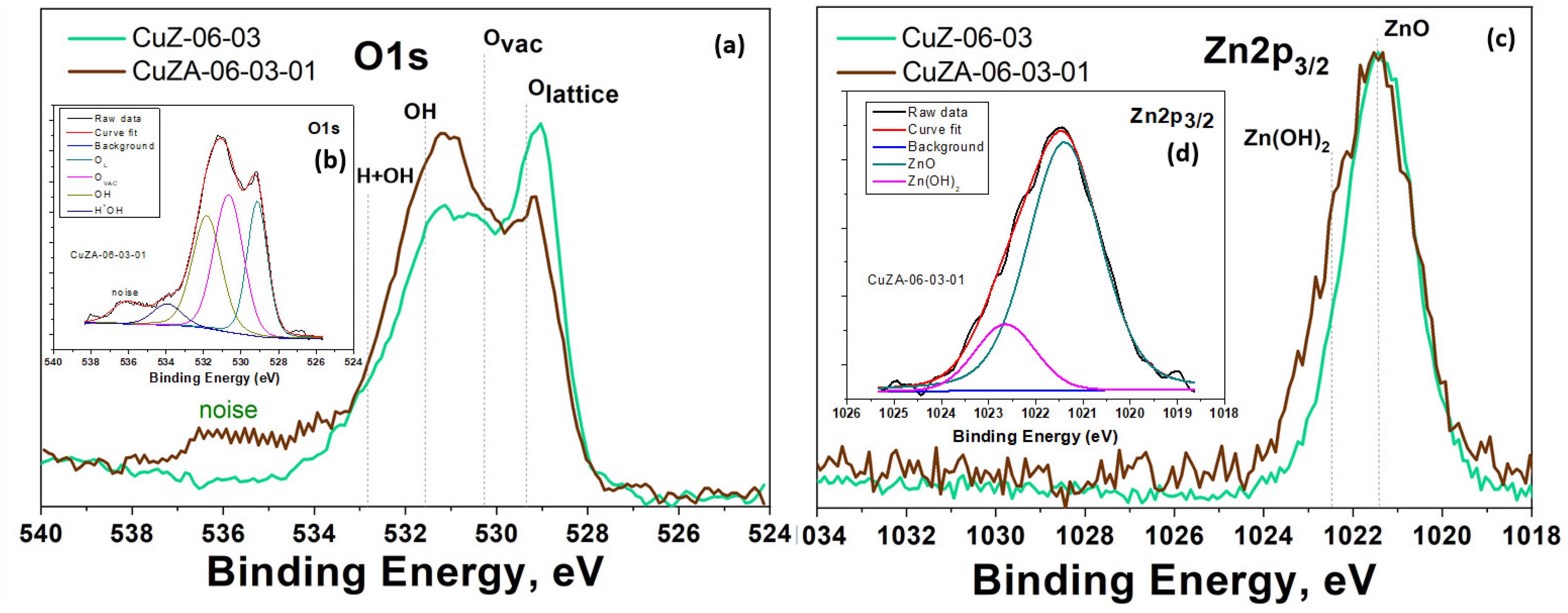
3.1. Physico-Chemical Characterization of Synthesized Powder Catalysts
3.2. Physico-Chemical Characterization of the Prepared Electrodes
3.3. Electrochemical Measurements
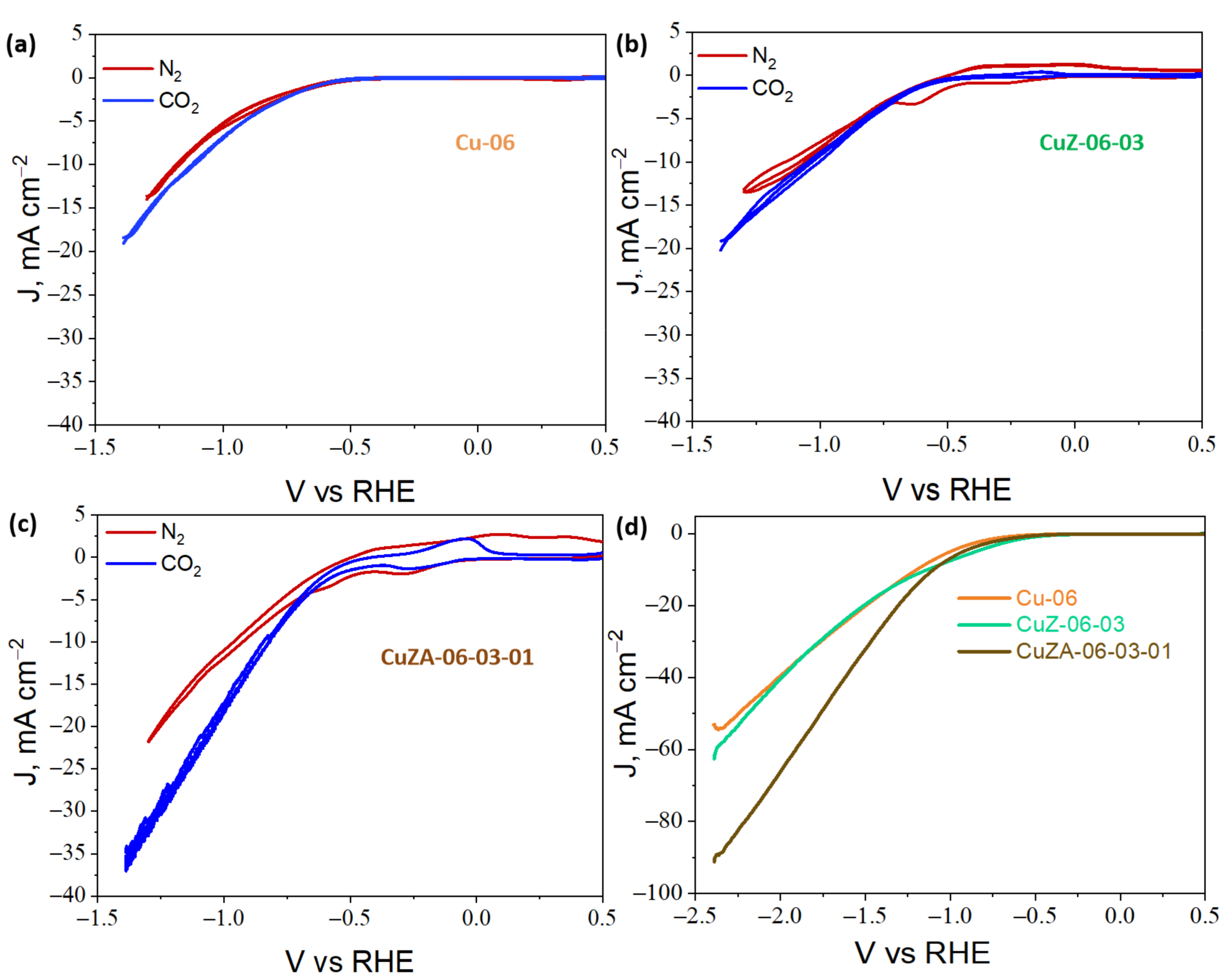
3.3.1. Cyclic Voltammetry Characterization of CuZnAl-Oxide Based Electrodes
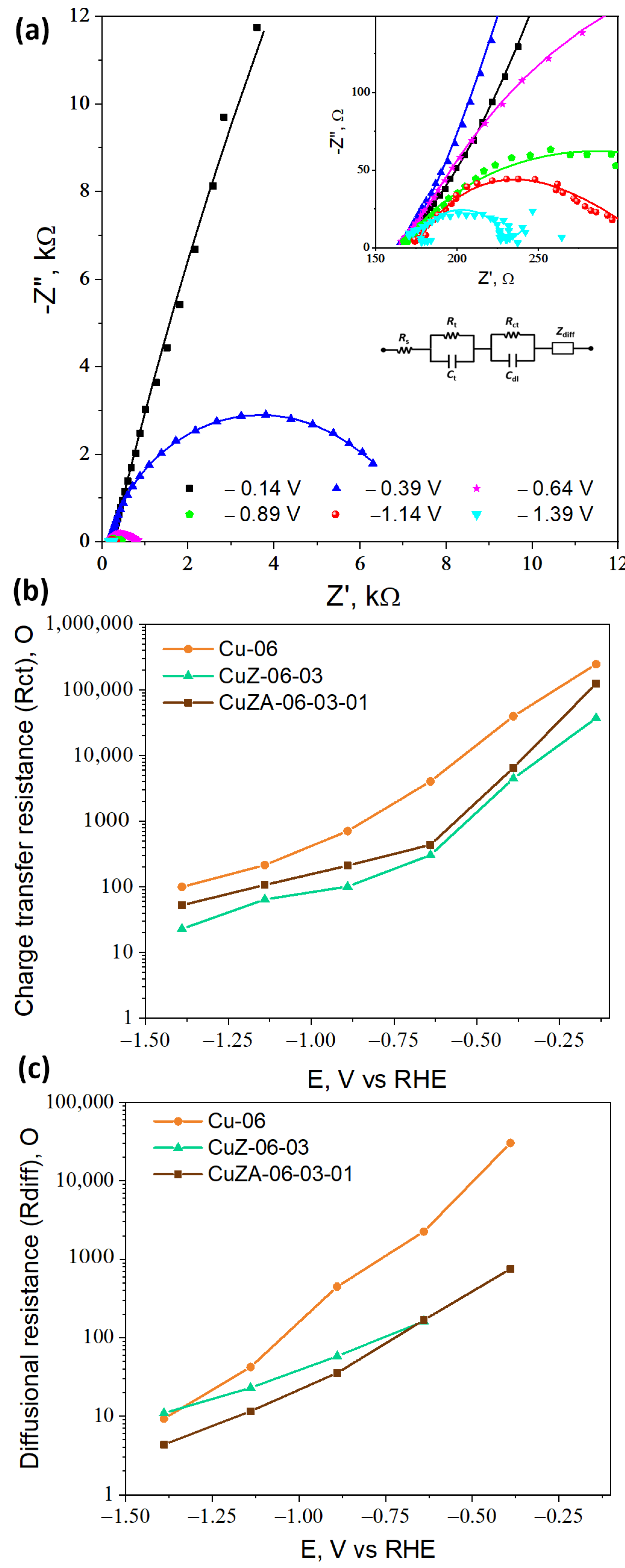
3.3.2. Electrochemical Impedance Spectroscopy
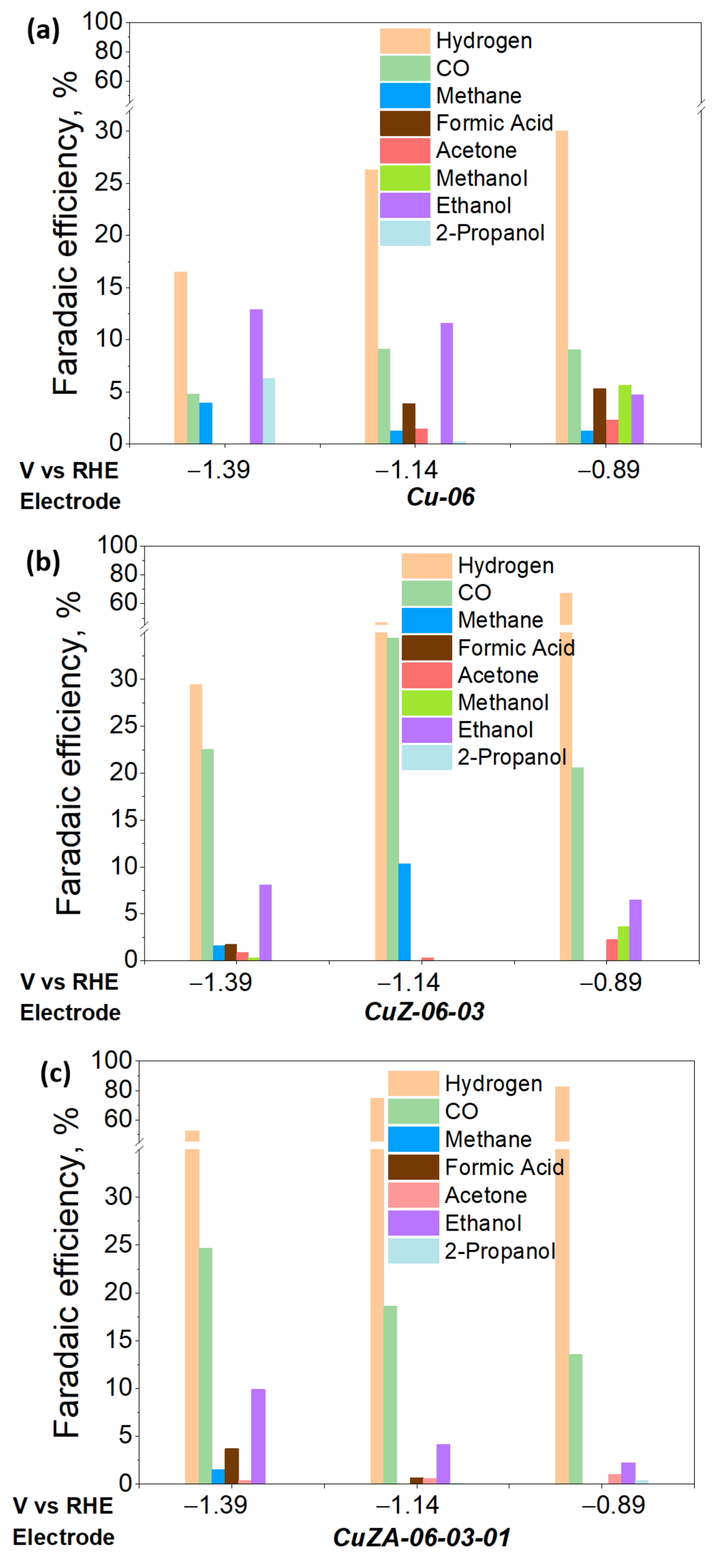
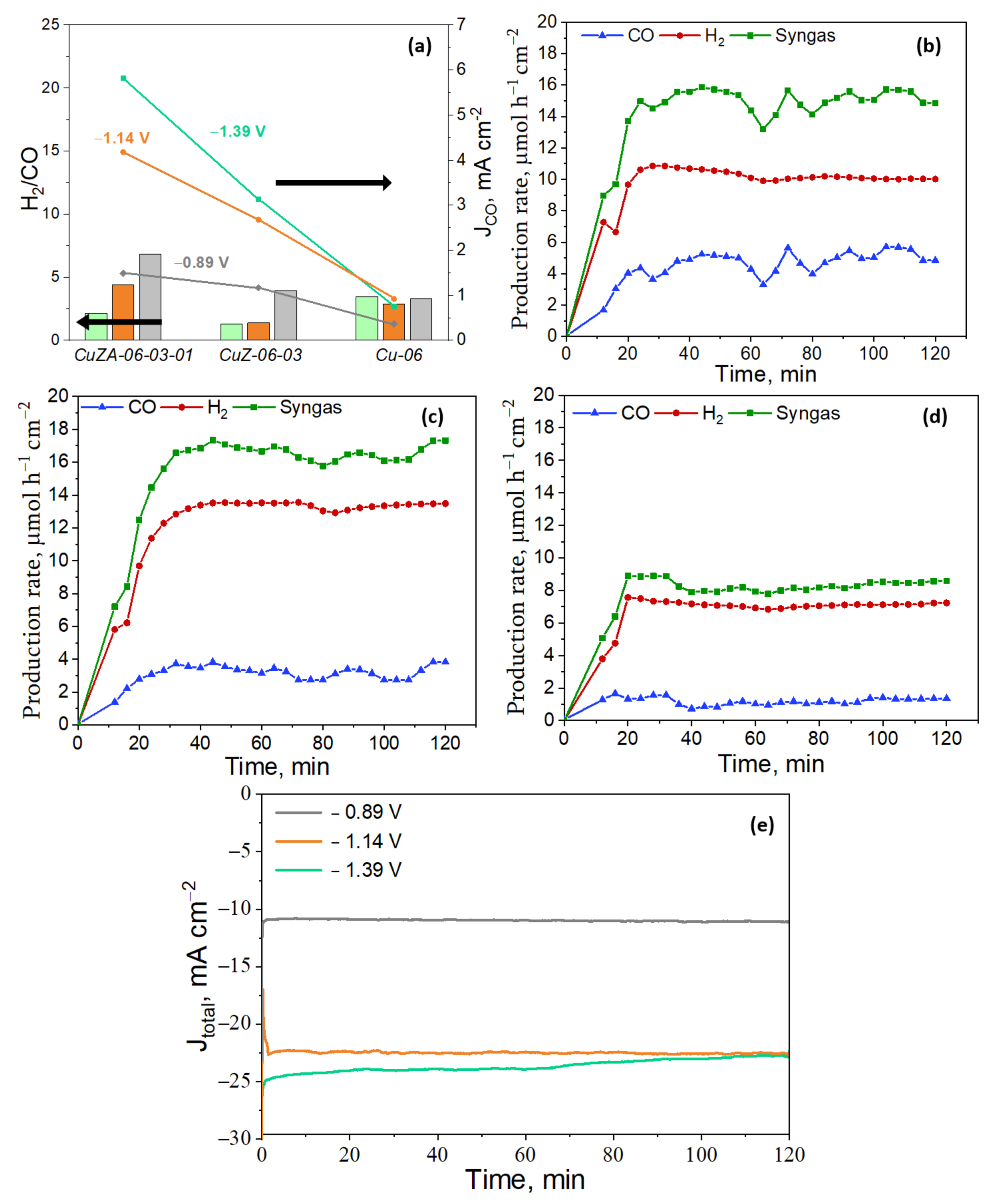
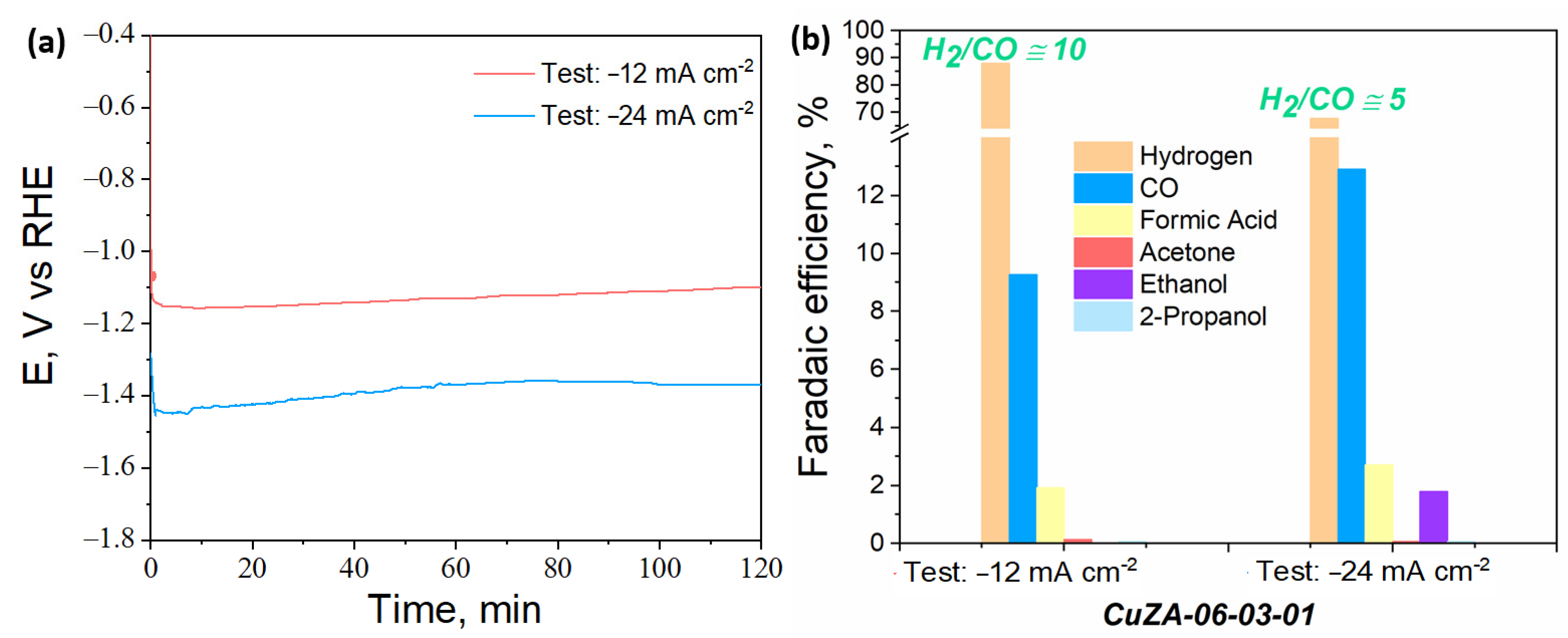
3.3.3. Electrochemical CO2 Reduction in the Aqueous Phase
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stocker, T.F.; Qin, D.; Plattner, G.-K.; Tignor, M.M.B.; Allen, S.K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P.M. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of IPCC the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Tolod, K.R.; Hernández, S.; Russo, N. Recent Advances in the BiVO4 Photocatalyst for Sun-Driven Water Oxidation: Top-Performing Photoanodes and Scale-Up Challenges. Catalysts 2017, 7, 13. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, M.; Jia, M.; Liu, S.; Qiu, J.; Sun, Z. Electrochemical CO2 reduction to C2+ species: Heterogeneous electrocatalysts, reaction pathways, and optimization strategies. Mater. Today Energy 2018, 10, 280–301. [Google Scholar] [CrossRef]
- Guzmán, H.; Russo, N.; Hernández, S. CO2 valorisation towards alcohols by Cu-based electrocatalysts: Challenges and perspectives. Green Chem. 2021, 23, 1896–1920. [Google Scholar] [CrossRef]
- Guzmán, H.; Farkhondehfal, M.A.; Rodulfo Tolod, K.; Russo, N.; Hernández, S. Photo/electrocatalytic hydrogen exploitation for CO2 reduction toward solar fuels production. In Solar Hydrogen Production Processes, Systems and Technologies; Elsevier Inc.: Amsterdam, The Netherlands, 2019; p. 560. ISBN 978-0-12-814853-2. [Google Scholar]
- Hernández, S.; Farkhondehfal, M.A.; Francesc, S.; Makkee, M.; Saracco, G.; Russo, N. Syngas production from electrochemical reduction of CO2: Current status and prospective implementation. Green Chem. 2017, 19, 2326–2346. [Google Scholar] [CrossRef]
- Ganesh, I. Conversion of carbon dioxide into methanol—A potential liquid fuel: Fundamental challenges and opportunities (a review). Renew. Sustain. Energy Rev. 2014, 31, 221–257. [Google Scholar] [CrossRef]
- Albo, J.; Irabien, A. Cu2O-loaded gas diffusion electrodes for the continuous electrochemical reduction of CO2 to methanol. J. Catal. 2016, 343, 232–239. [Google Scholar] [CrossRef]
- Gutiérrez-Guerra, N.; Moreno-López, L.; Serrano-Ruiz, J.C.; Valverde, J.L.; de Lucas-Consuegra, A. Gas phase electrocatalytic conversion of CO2 to syn-fuels on Cu based catalysts-electrodes. Appl. Catal. B Environ. 2016, 188, 272–282. [Google Scholar] [CrossRef]
- Fang, X.; Men, Y.; Wu, F.; Zhao, Q.; Singh, R.; Xiao, P.; Du, T.; Webley, P.A. Improved methanol yield and selectivity from CO2 hydrogenation using a novel Cu-ZnO-ZrO2 catalyst supported on Mg-Al layered double hydroxide (LDH). J. CO2 Util. 2019, 29, 57–64. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, X.D. Catalytic conversion of CO2 to value added fuels: Current status, challenges, and future directions. Chin. J. Catal. 2016, 37, 999–1015. [Google Scholar] [CrossRef]
- Jouny, M.; Luc, W.; Jiao, F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57, 2165–2177. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Raciti, D.; Braun, T.; Tackett, B.M.; Xu, H.; Cruz, M.; Wiley, B.J.; Moffat, T.P. High-Aspect-Ratio Ag Nanowire Mat Electrodes for Electrochemical CO Production from CO2. ACS Catal. 2021, 11, 11945–11959. [Google Scholar] [CrossRef]
- Nwabara, U.O.; Cofell, E.R.; Verma, S.; Negro, E.; Kenis, P.J.A. Durable Cathodes and Electrolyzers for the Efficient Aqueous Electrochemical Reduction of CO2. ChemSusChem 2020, 13, 855–875. [Google Scholar] [CrossRef] [PubMed]
- Dinh, C.T.; García de Arquer, F.P.; Sinton, D.; Sargent, E.H. High rate, selective, and stable electroreduction of CO2 to CO in basic and neutral media. ACS Energy Lett. 2018, 3, 2835–2840. [Google Scholar] [CrossRef]
- Birdja, Y.Y.; Pérez-Gallent, E.; Figueiredo, M.C.; Göttle, A.J.; Calle-Vallejo, F.; Koper, M.T.M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4, 732–745. [Google Scholar] [CrossRef]
- Velasco-Vélez, J.J.; Jones, T.; Gao, D.; Carbonio, E.; Arrigo, R.; Hsu, C.J.; Huang, Y.C.; Dong, C.L.; Chen, J.M.; Lee, J.F.; et al. The Role of the Copper Oxidation State in the Electrocatalytic Reduction of CO2 into Valuable Hydrocarbons. ACS Sustain. Chem. Eng. 2019, 7, 1485–1492. [Google Scholar] [CrossRef]
- Qin, T.; Qian, Y.; Zhang, F.; Lin, B.L. Chloride-derived copper electrode for efficient electrochemical reduction of CO2 to ethylene. Chin. Chem. Lett. 2019, 30, 314–318. [Google Scholar] [CrossRef]
- Vasileff, A.; Xu, C.; Jiao, Y.; Zheng, Y.; Qiao, S.Z. Surface and Interface Engineering in Copper-Based Bimetallic Materials for Selective CO2 Electroreduction. Chem 2018, 4, 1809–1831. [Google Scholar] [CrossRef]
- Kim, D.; Resasco, J.; Yu, Y.; Asiri, A.M.; Yang, P. Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold-copper bimetallic nanoparticles. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Lu, Q.; Forest, R.V.; Moore, A.; Jiao, F. Electrodeposited Zn Dendrites with Enhanced CO Selectivity for Electrocatalytic CO2 Reduction. ACS Catal. 2015, 5, 4586–4591. [Google Scholar] [CrossRef]
- Meyer, B.; Marx, D. First-principles study of CO adsorption on ZnO surfaces. J. Phys. Condens. Matter 2003, 15, L89. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Hahn, C.; Hatsukade, T.; Kim, Y.G.; Vailionis, A.; Baricuatro, J.H.; Higgins, D.C.; Nitopi, S.A.; Soriaga, M.P.; Jaramillo, T.F. Engineering Cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons. Proc. Natl. Acad. Sci. USA 2017, 114, 5918–5923. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, H.; Salomone, F.; Batuecas, E.; Tommasi, T.; Russo, N.; Bensaid, S.; Hernández, S. How to make sustainable CO2 conversion to Methanol: Thermocatalytic versus electrocatalytic technology. Chem. Eng. J. 2021, 417, 127973. [Google Scholar] [CrossRef]
- Baltes, C.; Vukojević, S.; Schüth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Allahyari, S.; Haghighi, M.; Ebadi, A.; Hosseinzadeh, S. Ultrasound assisted co-precipitation of nanostructured CuO–ZnO–Al2O3 over HZSM-5: Effect of precursor and irradiation power on nanocatalyst properties and catalytic performance for direct syngas to DME. Ultrason. Sonochem. 2014, 21, 663–673. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V., Cu and Zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Hall, A.S.; Yoon, Y.; Wuttig, A.; Surendranath, Y. Mesostructure-Induced Selectivity in CO2 Reduction Catalysis. J. Am. Chem. Soc. 2015, 137, 14834–14837. [Google Scholar] [CrossRef]
- Witoon, T.; Kachaban, N.; Donphai, W.; Kidkhunthod, P.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J. Tuning of catalytic CO2 hydrogenation by changing composition of CuO-ZnO-ZrO2 catalysts. Energy Convers. Manag. 2016, 118, 21–31. [Google Scholar] [CrossRef]
- Xiao, J.; Mao, D.; Guo, X.; Yu, J. Effect of TiO2, ZrO2, and TiO2-ZrO2 on the performance of CuO-ZnO catalyst for CO2 hydrogenation to methanol. Appl. Surf. Sci. 2015, 338, 146–153. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, S.; Pastor, E.; Lázaro, M.J. Noble metal-free catalysts supported on carbon for CO2 electrochemical reduction. J. CO2 Util. 2017, 18, 41–52. [Google Scholar] [CrossRef]
- Chi, D.; Yang, H.; Du, Y.; Lv, T.; Sui, G.; Wang, H.; Lu, J. Morphology-controlled CuO nanoparticles for electroreduction of CO2 to ethanol. RSC Adv. 2014, 4, 37329–37332. [Google Scholar] [CrossRef]
- Albo, J.; Sáez, A.; Solla-Gullón, J.; Montiel, V.; Irabien, A. Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl. Catal. B Environ. 2015, 176–177, 709–717. [Google Scholar] [CrossRef]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2018, 95, 197–206. [Google Scholar] [CrossRef]
- Fan, L.; Xia, C.; Yang, F.; Wang, J.; Wang, H.; Lu, Y. Strategies in catalysts and electrolyzer design for electrochemical CO2 reduction toward C2 products. Sci. Adv. 2020, 6, eaay3111. [Google Scholar] [CrossRef]
- Hansen, H.A.; Shi, C.; Lausche, A.C.; Peterson, A.A.; Nørskov, J.K. Bifunctional alloys for the electroreduction of CO2 and CO. Phys. Chem. Chem. Phys. 2016, 18, 9194–9201. [Google Scholar] [CrossRef] [PubMed]
- Kutz, R.B.; Chen, Q.; Yang, H.; Sajjad, S.D.; Liu, Z.; Masel, I.R. Sustainion Imidazolium-Functionalized Polymers for Carbon Dioxide Electrolysis. Energy Technol. 2017, 5, 929–936. [Google Scholar] [CrossRef]
- Zeng, J.; Bejtka, K.; Ju, W.; Castellino, M.; Chiodoni, A.; Sacco, A.; Farkhondehfal, M.A.; Hernández, S.; Rentsch, D.; Battaglia, C.; et al. Advanced Cu-Sn foam for selectively converting CO2 to CO in aqueous solution. Appl. Catal. B Environ. 2018, 236, 475–482. [Google Scholar] [CrossRef]
- Sacco, A. Electrochemical impedance spectroscopy: Fundamentals and application in dye-sensitized solar cells. Renew. Sustain. Energy Rev. 2017, 79, 814–829. [Google Scholar] [CrossRef]
- Ren, D. The effects of currents and potentials on the selectivities of copper toward carbon dioxide electroreduction. Nat. Commun. 2018. [Google Scholar] [CrossRef]
- Todorova, T.K.; Schreiber, M.W.; Fontecave, M. Mechanistic Understanding of CO2 Reduction Reaction (CO2RR) Toward Multicarbon Products by Heterogeneous Copper-Based Catalysts. ACS Catal. 2020, 10, 1754–1768. [Google Scholar] [CrossRef]
- Gao, D.; Arán-Ais, R.M.; Jeon, H.S.; Roldan Cuenya, B. Rational catalyst and electrolyte design for CO2 electroreduction towards multicarbon products. Nat. Catal. 2019, 2, 198–210. [Google Scholar] [CrossRef]
- Hjorth, I.; Nord, M.; Rønning, M.; Yang, J.; Chen, D. Electrochemical reduction of CO2 to synthesis gas on CNT supported CuxZn1-xO catalysts. Catal. Today 2020, 357, 311–321. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Liu, X.; Schlexer, P.; Xiao, J.; Ji, Y.; Wang, L.; Sandberg, R.B.; Tang, M.; Brown, K.S.; Peng, H.; Ringe, S.; et al. pH effects on the electrochemical reduction of CO2 towards C2 products on stepped copper. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Back, S.; Kim, H.; Jung, Y. Selective heterogeneous CO2 electroreduction to methanol. ACS Catal. 2015, 5, 965–971. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Roldan Cuenya, B.; Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.S.; Sinev, I.; Scholten, F.; Divins, N.J.; Zegkinoglou, I.; Pielsticker, L.; Cuenya, B.R. Operando Evolution of the Structure and Oxidation State of Size-Controlled Zn Nanoparticles during CO2 Electroreduction. J. Am. Chem. Soc. 2018, 140, 9383–9386. [Google Scholar] [CrossRef]
- Ye, R.P.; Lin, L.; Li, Q.; Zhou, Z.; Wang, T.; Russell, C.K.; Adidharma, H.; Xu, Z.; Yao, Y.G.; Fan, M. Recent progress in improving the stability of copper-based catalysts for hydrogenation of carbon-oxygen bonds. Catal. Sci. Technol. 2018, 8, 3428–3449. [Google Scholar] [CrossRef]
- Resini, C.; Lucarelli, C.; Taillades-Jacquin, M.; Liew, K.E.; Gabellini, I.; Albonetti, S.; Wails, D.; Rozire, J.; Vaccari, A.; Jones, D. Pt-Sn/γ-Al2O3 and Pt-Sn-Na/γ-Al2O3 catalysts for hydrogen production by dehydrogenation of Jet A-1 fuel: Characterisation and preliminary activity tests. Int. J. Hydrog. Energy 2011, 36, 5972–5982. [Google Scholar] [CrossRef][Green Version]
- Abidli, A.; Hamoudi, S.; Belkacemi, K. Synthesis, characterization and insights into stable and well organized hexagonal mesoporous zinc-doped alumina as promising metathesis catalysts carrier. Dalton Trans. 2015, 44, 9823–9838. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Marcandalli, G.; Mints, V.A.; Koper, M.T.M. Competition between CO2 reduction and hydrogen evolution on a gold electrode under well-defined mass transport conditions. J. Am. Chem. Soc. 2020. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. The role of copper particle size in low pressure methanol synthesis via CO2 hydrogenation over Cu/ZnO catalysts. Catal. Sci. Technol. 2015, 5, 869–881. [Google Scholar] [CrossRef]
- Phongamwong, T.; Chantaprasertporn, U.; Witoon, T.; Numpilai, T.; Poo-arporn, Y.; Limphirat, W.; Donphai, W.; Dittanet, P.; Chareonpanich, M.; Limtrakul, J. CO2 hydrogenation to methanol over CuO–ZnO–ZrO2–SiO2 catalysts: Effects of SiO2 contents. Chem. Eng. J. 2017, 316, 692–703. [Google Scholar] [CrossRef]
- Zeng, J.; Bejtka, K.; Di Martino, G.; Sacco, A.; Castellino, M.; Re Fiorentin, M.; Risplendi, F.; Farkhondehfal, M.A.; Hernández, S.; Cicero, G.; et al. Microwave-Assisted Synthesis of Copper-Based Electrocatalysts for Converting Carbon Dioxide to Tunable Syngas. ChemElectroChem 2020, 7, 229–238. [Google Scholar] [CrossRef]
- Qin, B.; Li, Y.; Fu, H.; Wang, H.; Chen, S.; Liu, Z.; Peng, F. Electrochemical reduction of CO2 into tunable syngas production by regulating crystal facets of earth-abundant Zn catalyst. ACS Appl. Mater. Interfaces 2018. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Rino, T.; Bejtka, K.; Castellino, M.; Sacco, A.; Farkhondehfal, M.A.; Chiodoni, A.; Drago, F.; Pirri, C.F. Coupled Copper–Zinc Catalysts for Electrochemical Reduction of Carbon Dioxide. ChemSusChem 2020, 13, 4128–4139. [Google Scholar] [CrossRef]
- Lv, J.J.; Jouny, M.; Luc, W.; Zhu, W.; Zhu, J.J.; Jiao, F. A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction. Adv. Mater. 2018, 30, 1803111. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Goddard, W.A.; Cheng, T.; Liu, Y. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6685–6688. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Precursor Concentration, M | Main Composition after Calcination | Electrode Name | ||
|---|---|---|---|---|---|
| Cu(NO3)2·3H2O | Zn(NO3)2·6H2O | Al(NO3)3·9 H2O | |||
| Cu/Zn/Al | 0.6 | 0.3 | 0.1 | CuO/ZnO/Al2O3 | CuZA-06-03-01 |
| Cu/Zn | 0.6 | 0.3 | - | CuO/ZnO | CuZ-06-03 |
| Cu | 0.6 | - | - | CuO | Cu-06 |
| Catalyst | BET Surface Area, m2 g−1 | Total Pore Volume, cm³ g−1 | EDS, Atomic Ratio | Crystallite Size, nm | |
|---|---|---|---|---|---|
| (11-1) Facet of CuO | (100) Facet of ZnO | ||||
| Cu-06 | 18.4 | 0.11 | Cu/O 1:1 | 16.98 | - |
| CuZ-06-03 | 55.16 | 0.23 | Cu/Zn 60:40 | 7.08 | 7.44 |
| CuZA-06-03-01 | 101.65 | 0.37 | Cu/Zn/Al 60:30:10 | 8.49 | 11.40 |
| Sample | Molar Composition, % mol | ||
|---|---|---|---|
| Cu | Zn | Al | |
| CuZA-06-03-01 | 52.3 | 38.6 | 9.1 |
| CuZ-06-03 | 56.9 | 44.2 | - |
| Electrode | Rs, Ω | Rt, Ω | Ct, µF | Cdl, mF cm−2 | ECSA, cm2 |
|---|---|---|---|---|---|
| Cu-06 | 166.1 | 61.5 | 3.1 | 0.2 | 8.1 |
| CuZ-06-03 | 160.5 | 50.5 | 13.2 | 4.5 | 160.2 |
| CuZA-06-03-01 | 158.9 | 38.2 | 4.6 | 0.6 | 21.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzmán, H.; Roldán, D.; Sacco, A.; Castellino, M.; Fontana, M.; Russo, N.; Hernández, S. CuZnAl-Oxide Nanopyramidal Mesoporous Materials for the Electrocatalytic CO2 Reduction to Syngas: Tuning of H2/CO Ratio. Nanomaterials 2021, 11, 3052. https://doi.org/10.3390/nano11113052
Guzmán H, Roldán D, Sacco A, Castellino M, Fontana M, Russo N, Hernández S. CuZnAl-Oxide Nanopyramidal Mesoporous Materials for the Electrocatalytic CO2 Reduction to Syngas: Tuning of H2/CO Ratio. Nanomaterials. 2021; 11(11):3052. https://doi.org/10.3390/nano11113052
Chicago/Turabian StyleGuzmán, Hilmar, Daniela Roldán, Adriano Sacco, Micaela Castellino, Marco Fontana, Nunzio Russo, and Simelys Hernández. 2021. "CuZnAl-Oxide Nanopyramidal Mesoporous Materials for the Electrocatalytic CO2 Reduction to Syngas: Tuning of H2/CO Ratio" Nanomaterials 11, no. 11: 3052. https://doi.org/10.3390/nano11113052
APA StyleGuzmán, H., Roldán, D., Sacco, A., Castellino, M., Fontana, M., Russo, N., & Hernández, S. (2021). CuZnAl-Oxide Nanopyramidal Mesoporous Materials for the Electrocatalytic CO2 Reduction to Syngas: Tuning of H2/CO Ratio. Nanomaterials, 11(11), 3052. https://doi.org/10.3390/nano11113052