Immunological and Toxicological Considerations for the Design of Liposomes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Toxicological Evaluation of Liposomes and Their Building Blocks
2.1. Toxicity of Cationic Liposomes
2.2. Interactions with the Mononuclear Phagocyte System
2.3. Effect of Cholesterol Content on the Toxicity of Liposome
3. Activation of the Immune System
3.1. Proinflammatory Cytokine Modulation
3.2. Recognition of Liposomes via Complement Activation
3.3. Hypersensitivity Reactions
3.4. Anti-PEG Response
3.5. Accelerated Blood Clearance
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Szoka, F., Jr.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural MODIFICATION BY surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252, IN26–IN27. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Huang, H.C.; Mallidi, S.; Obaid, G.; Sears, B.; Tangutoori, S.; Hasan, T. 23-Advancing photodynamic therapy with biochemically tuned liposomal nanotechnologies. In Applications of Nanoscience in Photomedicine; Hamblin, M.R., Avci, P., Eds.; Chandos Publishing: Oxford, UK, 2015; pp. 487–510. [Google Scholar] [CrossRef]
- Chrai, S.S.; Murari, R.; Ahmad, I. Liposomes (a review)-Part one: Manufacturing issues. Biopharm.-Appl. Technol. Biopharm. Dev. 2001, 14, 10–14. [Google Scholar]
- Dua, J.; Rana, A.; Bhandari, A. Liposome: Methods of preparation and applications. Int. J. Pharm. Stud. Res. 2012, 3, 14–20. [Google Scholar]
- Powers, D.; Nosoudi, N. Liposomes; from synthesis to targeting macrophages. Biomed. Res. 2019, 30, 288. [Google Scholar] [CrossRef][Green Version]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Fries, A.C.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Remote loading of preencapsulated drugs into stealth liposomes. Proc. Natl. Acad. Sci. USA 2014, 111, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Li, B.V. FDA Bioequivalence Standards; Springer: New York, NY, USA, 2014. [Google Scholar]
- Fahr, A.; Hoogevest, P.V.; May, S.; Bergstrand, N.; Leigh, M.L.S. Transfer of lipophilic drugs between liposomal membranes and biological interfaces: Consequences for drug delivery. Eur. J. Pharm. Sci. 2005, 26, 251–265. [Google Scholar] [CrossRef]
- Bhakay, A.; Rahman, M.; Dave, R.N.; Bilgili, E. Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation(-)Processing Aspects and Challenges. Pharmaceutics 2018, 10, 86. [Google Scholar] [CrossRef]
- Bressler, N.M. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: Two-year results of 2 randomized clinical trials-tap report 2. Arch. Ophthalmol. 2001, 119, 198–207. [Google Scholar]
- Schmidt-Erfurth, U.; Hasan, T.; Gragoudas, E.; Michaud, N.; Flotte, T.J.; Birngruber, R. Vascular targeting in photodynamic occlusion of subretinal vessels. Ophthalmology 1994, 101, 1953–1961. [Google Scholar] [CrossRef]
- Richter, A.M.; Cerruti-Sola, S.; Sternberg, E.D.; Dolphin, D.; Levy, J.G. Biodistribution of tritiated benzoporphyrin derivative (3H-BPD-MA), a new potent photosensitizer, in normal and tumor-bearing mice. J. Photochem. Photobiol. B Biol. 1990, 5, 231–244. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood-brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharm. Rev. 1999, 51, 691–743. [Google Scholar] [PubMed]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.H.; Lin, Y.Y.; Hwang, J.J.; Wang, H.E.; Tseng, Y.L.; Wang, S.J.; Liu, R.S.; Lin, W.J.; Yang, C.S.; Ting, G. Improvement of biodistribution and therapeutic index via increase of polyethylene glycol on drug-carrying liposomes in an HT-29/luc xenografted mouse model. Anticancer Res. 2009, 29, 2111–2120. [Google Scholar] [PubMed]
- Rahman, A.M.; Yusuf, S.W.; Ewer, M.S. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomed. 2007, 2, 567–583. [Google Scholar]
- Xing, M.; Yan, F.; Yu, S.; Shen, P. Efficacy and Cardiotoxicity of Liposomal Doxorubicin-Based Chemotherapy in Advanced Breast Cancer: A Meta-Analysis of Ten Randomized Controlled Trials. PLoS ONE 2015, 10, e0133569. [Google Scholar] [CrossRef]
- Kanter, P.M.; Bullard, G.A.; Pilkiewicz, F.G.; Mayer, L.D.; Cullis, P.R.; Pavelic, Z.P. Preclinical toxicology study of liposome encapsulated doxorubicin (TLC D-99): Comparison with doxorubicin and empty liposomes in mice and dogs. In Vivo 1993, 7, 85–95. [Google Scholar]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a Highly Active Nanoliposomal Irinotecan Using a Novel Intraliposomal Stabilization Strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Daemen, T.; Hofstede, G.; Ten Kate, M.T.; Bakker-Woudenberg, I.A.J.M.; Scherphof, G.L. Liposomal doxorubicin-induced toxicity: Depletion and impairment of phagocytic activity of liver macrophages. Int. J. Cancer 1995, 61, 716–721. [Google Scholar] [CrossRef]
- Filion, M.C.; Phillips, N.C. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim. Biophys. Acta 1997, 1329, 345–356. [Google Scholar] [CrossRef]
- Takano, S.; Aramaki, Y.; Tsuchiya, S. Physicochemical properties of liposomes affecting apoptosis induced by cationic liposomes in macrophages. Pharm. Res. 2003, 20, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, Y.; Takano, S.; Tsuchiya, S. Induction of apoptosis in macrophages by cationic liposomes. FEBS Lett. 1999, 460, 472–476. [Google Scholar] [CrossRef]
- Watson, D.S.; Endsley, A.N.; Huang, L. Design considerations for liposomal vaccines: Influence of formulation parameters on antibody and cell-mediated immune responses to liposome associated antigens. Vaccine 2012, 30, 2256–2272. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control Release 2006, 112, 15–25. [Google Scholar] [CrossRef]
- Landesman-Milo, D.; Peer, D. Altering the immune response with lipid-based nanoparticles. J. Control Release 2012, 161, 600–608. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Miranda, D.; Lovell, J.F. Mechanisms of light-induced liposome permeabilization. Bioeng. Transl. Med. 2016, 1, 267–276. [Google Scholar] [CrossRef]
- Luo, D.; Li, N.; Carter, K.A.; Lin, C.; Geng, J.; Shao, S.; Huang, W.-C.; Qin, Y.; Atilla-Gokcumen, G.E.; Lovell, J.F. Rapid Light-Triggered Drug Release in Liposomes Containing Small Amounts of Unsaturated and Porphyrin-Phospholipids. Small (Weinheim an der Bergstrasse Germany) 2016, 12, 3039–3047. [Google Scholar] [CrossRef]
- Kohli, A.G.; Kierstead, P.H.; Venditto, V.J.; Walsh, C.L.; Szoka, F.C. Designer lipids for drug delivery: From heads to tails. J. Control Release Off. J. Control. Release Soc. 2014, 190, 274–287. [Google Scholar] [CrossRef]
- Tsui, F.C.; Ojcius, D.M.; Hubbell, W.L. The intrinsic pKa values for phosphatidylserine and phosphatidylethanolamine in phosphatidylcholine host bilayers. Biophys J. 1986, 49, 459–468. [Google Scholar] [CrossRef]
- Inglut, C.T.; Baglo, Y.; Liang, B.J.; Cheema, Y.; Stabile, J.; Woodworth, G.F.; Huang, H.C. Systematic Evaluation of Light-Activatable Biohybrids for Anti-Glioma Photodynamic Therapy. J. Clin. Med. 2019, 8, 1269. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.F.; Jin, C.S.; Huynh, E.; Jin, H.; Kim, C.; Rubinstein, J.L.; Chan, W.C.W.; Cao, W.; Wang, L.V.; Zheng, G. Porphysome nanovesicles generated by porphyrin bilayers for use as multimodal biophotonic contrast agents. Nat. Mater. 2011, 10, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Irby, D.; Du, C.; Li, F. Lipid-Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef]
- Signorell, R.D.; Luciani, P.; Brambilla, D.; Leroux, J.C. Pharmacokinetics of lipid-drug conjugates loaded into liposomes. Eur. J. Pharm. Biopharm. 2018, 128, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Baglo, Y.; Liang, B.J.; Robey, R.W.; Ambudkar, S.V.; Gottesman, M.M.; Huang, H.-C. Porphyrin-lipid assemblies and nanovesicles overcome ABC transporter-mediated photodynamic therapy resistance in cancer cells. Cancer Lett. 2019, 457, 110–118. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Nunes, S.S.; Fernandes, R.S.; Cavalcante, C.H.; da Costa César, I.; Leite, E.A.; Lopes, S.C.A.; Ferretti, A.; Rubello, D.; Townsend, D.M.; de Oliveira, M.C.; et al. Influence of PEG coating on the biodistribution and tumor accumulation of pH-sensitive liposomes. Drug Deliv. Transl. Res. 2019, 9, 123–130. [Google Scholar] [CrossRef]
- Swenson, C.E.; Bolcsak, L.E.; Batist, G.; Guthrie, T.H.J.; Tkaczuk, K.H.; Boxenbaum, H.; Welles, L.; Chow, S.-C.; Bhamra, R.; Chaikin, P. Pharmacokinetics of doxorubicin administered i.v. as Myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anti-Cancer Drugs 2003, 14, 239–246. [Google Scholar] [CrossRef]
- Hamilton, A.; Biganzoli, L.; Coleman, R.; Mauriac, L.; Hennebert, P.; Awada, A.; Nooij, M.; Beex, L.; Piccart, M.; Van Hoorebeeck, I.; et al. EORTC 10968: A phase I clinical and pharmacokinetic study of polyethylene glycol liposomal doxorubicin (Caelyx®, Doxil) at a 6-week interval in patients with metastatic breast cancer. Ann. Oncol. 2002, 13, 910–918. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Taraballi, F.; Toledano Furman, N.E.; Sherman, M.B.; Parodi, A.; Salvatore, F.; Tasciotti, E. Effects of the protein corona on liposome-liposome and liposome-cell interactions. Int. J. Nanomed. 2016, 11, 3049–3063. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomed. (Lond.) 2016, 11, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Barbero, F.; Russo, L.; Vitali, M.; Piella, J.; Salvo, I.; Borrajo, M.L.; Busquets-Fité, M.; Grandori, R.; Bastús, N.G.; Casals, E.; et al. Formation of the Protein Corona: The Interface between Nanoparticles and the Immune System. Semin. Immunol. 2017, 34, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, G. Liposome-protein corona in a physiological environment: Challenges and opportunities for targeted delivery of nanomedicines. Nanomedicine 2015, 11, 543–557. [Google Scholar] [CrossRef]
- Hadjidemetriou, M.; Al-Ahmady, Z.; Mazza, M.; Collins, R.F.; Dawson, K.; Kostarelos, K. In Vivo Biomolecule Corona around Blood-Circulating, Clinically Used and Antibody-Targeted Lipid Bilayer Nanoscale Vesicles. ACS Nano 2015, 9, 8142–8156. [Google Scholar] [CrossRef] [PubMed]
- Gessner, A.; Lieske, A.; Paulke, B.-R.; Müller, R.H. Functional groups on polystyrene model nanoparticles: Influence on protein adsorption. J. Biomed. Mater. Res. Part A 2003, 65, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Gessner, A.; Waicz, R.; Lieske, A.; Paulke, B.R.; Mäder, K.; Müller, R.H. Nanoparticles with decreasing surface hydrophobicities: Influence on plasma protein adsorption. Int. J. Pharm. 2000, 196, 245–249. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Poste, G.; Schaeffer, B.E. Fusion of mammalian cells by unilamellar lipid vesicles: Influence of lipid surface charge, fluidity and cholesterol. Biochim. Biophys. Acta (BBA)-Biomembr. 1973, 323, 23–42. [Google Scholar] [CrossRef]
- Pagano, R.E.; Huang, L.; Wey, C. Interaction of phospholipid vesicles with cultured mammalian cells. Nature 1974, 252, 166–167. [Google Scholar] [CrossRef]
- Adams, D.H.; Joyce, G.; Richardson, V.J.; Ryman, B.E.; Wisniewski, H.M. Liposome toxicity in the mouse central nervous system. J. Neurol. Sci. 1977, 31, 173–179. [Google Scholar] [CrossRef]
- Shi, M.; Anantha, M.; Wehbe, M.; Bally, M.B.; Fortin, D.; Roy, L.-O.; Charest, G.; Richer, M.; Paquette, B.; Sanche, L. Liposomal formulations of carboplatin injected by convection-enhanced delivery increases the median survival time of F98 glioma bearing rats. J. Nanobiotechnol. 2018, 16, 77. [Google Scholar] [CrossRef]
- Fan, Y.; Sahdev, P.; Ochyl, L.J.; Akerberg, J.; Moon, J.J. Cationic liposome-hyaluronic acid hybrid nanoparticles for intranasal vaccination with subunit antigens. J. Control Release 2015, 208, 121–129. [Google Scholar] [CrossRef]
- Hernandez-Caselles, T.; Villalain, J.; Gomez-Fernandez, J.C. Influence of liposome charge and composition on their interaction with human blood serum proteins. Mol. Cell. Biochem. 1993, 120, 119–126. [Google Scholar] [CrossRef]
- Heath, T.D.; Lopez, N.G.; Papahadjopoulos, D. The effects of liposome size and surface charge on liposome-mediated delivery of methotrexate-gamma-aspartate to cells in vitro. Biochim. Biophys. Acta 1985, 820, 74–84. [Google Scholar] [CrossRef]
- Parnham, M.J.; Wetzig, H. Toxicity screening of liposomes. Chem. Phys. Lipids 1993, 64, 263–274. [Google Scholar] [CrossRef]
- Lappalainen, K.; Jääskeläinen, I.; Syrjänen, K.; Urtti, A.; Syrjänen, S. Comparison of Cell Proliferation and Toxicity Assays Using Two Cationic Liposomes. Pharm. Res. 1994, 11, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Lentz, B.R. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog. Lipid Res. 2003, 42, 423–438. [Google Scholar] [CrossRef]
- Zbinden, G.; Wunderli-Allenspach, H.; Grimm, L. Assessment of thrombogenic potential of liposomes. Toxicology 1989, 54, 273–280. [Google Scholar] [CrossRef]
- Ishiwata, H.; Suzuki, N.; Ando, S.; Kikuchi, H.; Kitagawa, T. Characteristics and biodistribution of cationic liposomes and their DNA complexes. J. Control Release 2000, 69, 139–148. [Google Scholar] [CrossRef]
- Office of Medical Products and Tobacco, Center for Drug Evaluation and Research. Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation; Guidance for Industry; Food and Drug Administration: Rockville, MD, USA, 2018. [Google Scholar]
- White, R.E.; Evans, D.C.; Hop, C.E.; Moore, D.J.; Prakash, C.; Surapaneni, S.; Tse, F.L. Radiolabeled mass-balance excretion and metabolism studies in laboratory animals: A commentary on why they are still necessary. Xenobiotica 2013, 43, 219–225. [Google Scholar] [CrossRef]
- Simoes, S.; Filipe, A.; Faneca, H.; Mano, M.; Penacho, N.; Duzgunes, N.; de Lima, M.P. Cationic liposomes for gene delivery. Expert Opin. Drug Deliv. 2005, 2, 237–254. [Google Scholar] [CrossRef]
- Balazs, D.A.; Godbey, W. Liposomes for use in gene delivery. J. Drug Deliv. 2011, 2011, 326497. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Shum, K.-T.; Burnett, J.C.; Rossi, J.J. Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges. Pharmaceuticals (Basel) 2013, 6, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Benter, I.F. Nonviral delivery of synthetic siRNAs in vivo. J. Clin. Investig. 2007, 117, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Szoka, F.C. Lipid-based Nanoparticles for Nucleic Acid Delivery. Pharm. Res. 2007, 24, 438–449. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef]
- Knudsen, K.B.; Northeved, H.; Kumar, P.E.; Permin, A.; Gjetting, T.; Andresen, T.L.; Larsen, S.; Wegener, K.M.; Lykkesfeldt, J.; Jantzen, K.; et al. In vivo toxicity of cationic micelles and liposomes. Nanomedicine 2015, 11, 467–477. [Google Scholar] [CrossRef]
- Hattori, Y.; Shimizu, S.; Ozaki, K.I.; Onishi, H. Effect of Cationic Lipid Type in Folate-PEG-Modified Cationic Liposomes on Folate Receptor-Mediated siRNA Transfection in Tumor Cells. Pharmaceutics 2019, 11, 181. [Google Scholar] [CrossRef]
- Zelphati, O.; Szoka, F.C. Intracellular Distribution and Mechanism of Delivery of Oligonucleotides Mediated by Cationic Lipids. Pharm. Res. 1996, 13, 1367–1372. [Google Scholar] [CrossRef]
- Romøren, K.; Thu, B.J.; Bols, N.C.; Evensen, Ø. Transfection efficiency and cytotoxicity of cationic liposomes in salmonid cell lines of hepatocyte and macrophage origin. Biochim. Biophys. Acta (BBA)-Biomembr. 2004, 1663, 127–134. [Google Scholar] [CrossRef]
- Cui, S.; Wang, Y.; Gong, Y.; Lin, X.; Zhao, Y.; Zhi, D.; Zhou, Q.; Zhang, S. Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol. Res. 2018, 7, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Lu, Y.; Xie, F.; Zou, H.; Fan, X.; Li, B.; Li, W.; Zhang, W.; Mei, L.; Feng, S.-S.; et al. Cationic liposomes induce cell necrosis through lysosomal dysfunction and late-stage autophagic flux inhibition. Nanomedicine 2016, 11, 3117–3137. [Google Scholar] [CrossRef] [PubMed]
- Soenen, S.J.H.; Brisson, A.R.; De Cuyper, M. Addressing the problem of cationic lipid-mediated toxicity: The magnetoliposome model. Biomaterials 2009, 30, 3691–3701. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Shao, B.; He, Z.; Ye, T.; Luo, M.; Sang, Y.; Liang, X.; Wang, W.; Luo, S.; Yang, S.; et al. Cationic nanocarriers induce cell necrosis through impairment of Na+/K+-ATPase and cause subsequent inflammatory response. Cell Res. 2015, 25, 237–253. [Google Scholar] [CrossRef]
- Aramaki, Y.; Takano, S.; Tsuchiya, S. Cationic Liposomes Induce Macrophage Apoptosis through Mitochondrial Pathway. Arch. Biochem. Biophys. 2001, 392, 245–250. [Google Scholar] [CrossRef]
- Iwaoka, S.; Nakamura, T.; Takano, S.; Tsuchiya, S.; Aramaki, Y. Cationic liposomes induce apoptosis through p38 MAP kinase–caspase-8–Bid pathway in macrophage-like RAW264.7 cells. J. Leukoc. Biol. 2006, 79, 184–191. [Google Scholar] [CrossRef]
- Chrai, S.S.; Murari, R.; Ahmad, I. Liposomes (a review): Part two: Drug delivery systems. BioPharm 2002, 15, 40–43, 49. [Google Scholar]
- Senior, J.H. Fate and behavior of liposomes in vivo: A review of controlling factors. Crit. Rev. Ther. Drug Carr. Syst. 1987, 3, 123–193. [Google Scholar]
- Kimelberg, H.K.; Tracy, T.F.; Biddlecome, S.M.; Bourke, R.S. The Effect of Entrapment in Liposomes on the in Vivo Distribution of [3H]Methotrexate in a Primate. Cancer Res. 1976, 36, 2949–2957. [Google Scholar]
- Yona, S.; Gordon, S. From the Reticuloendothelial to Mononuclear Phagocyte System—The Unaccounted Years. Front. Immunol. 2015, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, A.; Bao, A.; Phillips, W.T.; Perez, R., 3rd; Goins, B.A. [(186)Re]Liposomal doxorubicin (Doxil): In vitro stability, pharmacokinetics, imaging and biodistribution in a head and neck squamous cell carcinoma xenograft model. Nucl. Med. Biol. 2009, 36, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Beyer, I.; Cao, H.; Persson, J.; Song, H.; Richter, M.; Feng, Q.; Yumul, R.; van Rensburg, R.; Li, Z.; Berenson, R.; et al. Coadministration of epithelial junction opener JO-1 improves the efficacy and safety of chemotherapeutic drugs. Clin. Cancer Res. 2012, 18, 3340–3351. [Google Scholar] [CrossRef]
- Arnold, R.D.; Mager, D.E.; Slack, J.E.; Straubinger, R.M. Effect of repetitive administration of Doxorubicin-containing liposomes on plasma pharmacokinetics and drug biodistribution in a rat brain tumor model. Clin. Cancer Res. 2005, 11, 8856–8865. [Google Scholar] [CrossRef]
- Lu, W.L.; Qi, X.R.; Zhang, Q.; Li, R.Y.; Wang, G.L.; Zhang, R.J.; Wei, S.L. A pegylated liposomal platform: Pharmacokinetics, pharmacodynamics, and toxicity in mice using doxorubicin as a model drug. J. Pharm. Sci. 2004, 95, 381–389. [Google Scholar] [CrossRef]
- Luo, R.; Li, Y.; He, M.; Zhang, H.; Yuan, H.; Johnson, M.; Palmisano, M.; Zhou, S.; Sun, D. Distinct biodistribution of doxorubicin and the altered dispositions mediated by different liposomal formulations. Int. J. Pharm. 2017, 519, 1–10. [Google Scholar] [CrossRef]
- Allen, T.M.; Murray, L.; MacKeigan, S.; Shah, M. Chronic liposome administration in mice: Effects on reticuloendothelial function and tissue distribution. J. Pharmacol. Exp. Ther. 1984, 229, 267. [Google Scholar]
- Ellens, H.; Mayhew, E.; Rustum, Y.M. Reversible depression of the reticuloendothelial system by liposomes. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1982, 714, 479–485. [Google Scholar] [CrossRef]
- Van Rooijen, N.; Sanders, A. Kupffer cell depletion by liposome-delivered drugs: Comparative activity of intracellular clodronate, propamidine, and ethylenediaminetetraacetic acid. Hepatology 1996, 23, 1239–1243. [Google Scholar] [CrossRef]
- Storm, G.; ten Kate, M.T.; Working, P.K.; Bakker-Woudenberg, I.A. Doxorubicin entrapped in sterically stabilized liposomes: Effects on bacterial blood clearance capacity of the mononuclear phagocyte system. Clin. Cancer Res. 1998, 4, 111–115. [Google Scholar] [PubMed]
- Rooijen, N.V.; Kesteren-Hendrikx, E.V. “In Vivo” Depletion of Macrophages by Liposome-Mediated “Suicide”. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2003; Volume 373, pp. 3–16. [Google Scholar]
- Daemen, T.; Regts, J.; Meesters, M.; Ten Kate, M.T.; Bakker-Woudenberg, I.A.J.M.; Scherphof, G.L. Toxicity of doxorubicin entrapped within long-circulating liposomes. J. Control Release 1997, 44, 1–9. [Google Scholar] [CrossRef]
- La-Beck, N.M.; Liu, X.; Wood, L.M. Harnessing Liposome Interactions with the Immune System for the Next Breakthrough in Cancer Drug Delivery. Front. Pharmacol. 2019, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Leal, Y.; Lucatelli Laurindo, M.F.; Osugui, L.; Luzardo Mdel, C.; Lopez-Requena, A.; Alonso, M.E.; Alvarez, C.; Popi, A.F.; Mariano, M.; Perez, R.; et al. Liposomes of phosphatidylcholine and cholesterol induce an M2-like macrophage phenotype reprogrammable to M1 pattern with the involvement of B-1 cells. Immunobiology 2014, 219, 403–415. [Google Scholar] [CrossRef]
- Rajan, R.; Sabnani, M.K.; Mavinkurve, V.; Shmeeda, H.; Mansouri, H.; Bonkoungou, S.; Le, A.D.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome-induced immunosuppression and tumor growth is mediated by macrophages and mitigated by liposome-encapsulated alendronate. J. Control. Release Off. J. Control. Release Soc. 2018, 271, 139–148. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Miao, X.; Leng, X.; Zhang, Q. The Current State of Nanoparticle-Induced Macrophage Polarization and Reprogramming Research. Int. J. Mol. Sci. 2017, 18, 336. [Google Scholar] [CrossRef]
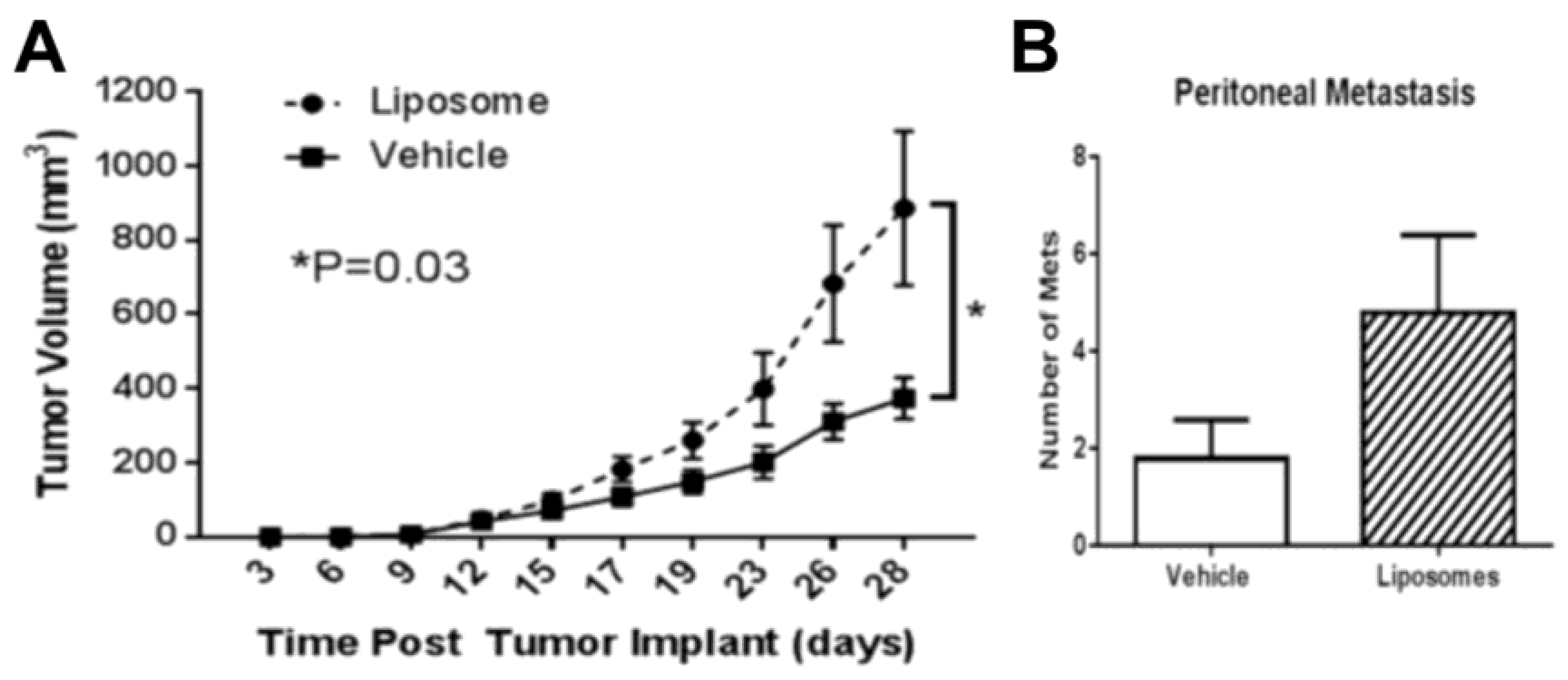
- Sabnani, M.K.; Rajan, R.; Rowland, B.; Mavinkurve, V.; Wood, L.M.; Gabizon, A.A.; La-Beck, N.M. Liposome promotion of tumor growth is associated with angiogenesis and inhibition of antitumor immune responses. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 259–262. [Google Scholar] [CrossRef]
- Batenjany, M.M.; Boni, L.T.; Guo, Y.; Neville, M.E.; Bansal, S.; Robb, R.J.; Popescu, M.C. The effect of cholesterol in a liposomal Muc1 vaccine. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1514, 280–290. [Google Scholar] [CrossRef]
- Demel, R.A.; Kinsky, S.C.; Kinsky, C.B.; Van Deenen, L.L.M. Effects of temperature and cholesterol on the glucose permeability of liposomes prepared with natural and synthetic lecithins. Biochim. Biophys. Acta (BBA)-Biomembr. 1968, 150, 655–665. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Cowden, M.; Kimelberg, H. Role of cholesterol in membranes. Effects on phospholipid-protein interactions, membrane permeability and enzymatic activity. Biochim. Biophys. Acta 1973, 330, 8–26. [Google Scholar] [CrossRef]
- Briuglia, M.L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Kirby, C.; Clarke, J.; Gregoriadis, G. Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and In Vitro. Biochem. J. 1980, 186, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pinto, J.M.; Gonzalez-Rodriguez, M.L.; Rabasco, A.M. Effect of cholesterol and ethanol on dermal delivery from DPPC liposomes. Int. J. Pharm. 2005, 298, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smaby, J.M.; Momsen, M.M.; Brockman, H.L.; Brown, R.E. Phosphatidylcholine acyl unsaturation modulates the decrease in interfacial elasticity induced by cholesterol. Biophys. J. 1997, 73, 1492–1505. [Google Scholar] [CrossRef]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Nir, S.; Ohki, S. Permeability properties of phospholipid membranes: Effect of cholesterol and temperature. Biochim. Biophys. Acta (BBA)-Biomembr. 1972, 266, 561–583. [Google Scholar] [CrossRef]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [CrossRef]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef]
- Roerdink, F.H.; Regts, J.; Handel, T.; Sullivan, S.M.; Baldeschwieler, J.D.; Scherphof, G.L. Effect of cholesterol on the uptake and intracellular degradation of liposomes by liver and spleen; a combined biochemical and γ-ray perturbed angular correlation study. Biochim. Biophys. Acta (BBA)-Biomembr. 1989, 980, 234–240. [Google Scholar] [CrossRef]
- Kaur, R.; Henriksen-Lacey, M.; Wilkhu, J.; Devitt, A.; Christensen, D.; Perrie, Y. Effect of incorporating cholesterol into DDA:TDB liposomal adjuvants on bilayer properties, biodistribution, and immune responses. Mol. Pharm. 2014, 11, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Mori, M.; Yamamura, H.; Naito, S.; Kato, H.; Taneichi, M.; Tanaka, Y.; Komuro, K.; Uchida, T. Cholesterol inclusion in liposomes affects induction of antigen-specific IgG and IgE antibody production in mice by a surface-linked liposomal antigen. Bioconjug. Chem. 2002, 13, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, R.; Krishan, A.; Wodinsky, I.; Zubrod, C.G.; Lesko, L.J. Effect of cholesterol content on antitumor activity and toxicity of liposome-encapsulated 1-beta-D-arabinofuranosylcytosine In Vivo. Cancer Res. 1980, 40, 630–633. [Google Scholar] [PubMed]
- Ishida, T.; Kojima, H.; Harashima, H.; Kiwada, H. Biodistribution of liposomes and C3 fragments associated with liposomes: Evaluation of their relationship. Int. J. Pharm. 2000, 205, 183–193. [Google Scholar] [CrossRef]
- Cunningham, C.M.; Kingzette, M.; Richards, R.L.; Alving, C.R.; Lint, T.F.; Gewurz, H. Activation of Human Complement by Liposomes: A Model for Membrane Activation of the Alternative Pathway. J. Immunol. 1979, 122, 1237–1242. [Google Scholar]
- Chonn, A.; Cullis, P.R.; Devine, D.V. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J. Immunol. 1991, 146, 4234–4241. [Google Scholar]
- Remes, A.; Williams, D.F. Immune response in biocompatibility. Biomaterials 1992, 13, 731–743. [Google Scholar] [CrossRef]
- Lynn, A.D.; Blakney, A.K.; Kyriakides, T.R.; Bryant, S.J. Temporal progression of the host response to implanted poly(ethylene glycol)-based hydrogels. J. Biomed. Mater. Res. A 2011, 96, 621–631. [Google Scholar] [CrossRef]
- Szebeni, J.; Moghimi, S.M. Liposome triggering of innate immune responses: A perspective on benefits and adverse reactions. J. Liposome Res. 2009, 19, 85–90. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Aggarwal, P.; Hall, J.B.; McNeil, S.E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 2008, 5, 487–495. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Choi, E.-J.; Webster, T.J.; Kim, S.-H.; Khang, D. Effect of the protein corona on nanoparticles for modulating cytotoxicity and immunotoxicity. Int. J. Nanomed. 2014, 10, 97–113. [Google Scholar] [CrossRef]
- Kinsky, S.C.; Haxby, J.A.; Zopf, D.A.; Alving, C.R.; Kinsky, C.B. Complement-dependent damage to liposomes prepared from pure lipids and Forssman hapten. Biochemistry 1969, 8, 4149–4158. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ishida, T.; Kiwada, H. Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J. Control Release 2007, 119, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Alving, C.R.; Rao, M.; Steers, N.J.; Matyas, G.R.; Mayorov, A.V. Liposomes containing lipid A: An effective, safe, generic adjuvant system for synthetic vaccines. Expert Rev. Vaccines 2012, 11, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Alving, C.R. Liposomes as carriers of antigens and adjuvants. J. Immunol. Methods 1991, 140, 1–13. [Google Scholar] [CrossRef]
- Perrie, Y.; Crofts, F.; Devitt, A.; Griffiths, H.R.; Kastner, E.; Nadella, V. Designing liposomal adjuvants for the next generation of vaccines. Adv. Drug Deliv. Rev. 2016, 99, 85–96. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Laupeze, B.; Di Pasquale, A.; Hergli, N.; Collignon, C.; Garcon, N. Adjuvant system AS01: Helping to overcome the challenges of modern vaccines. Expert Rev. Vaccines 2017, 16, 55–63. [Google Scholar] [CrossRef]
- Badiee, A.; Jaafari, M.R.; Khamesipour, A.; Samiei, A.; Soroush, D.; Kheiri, M.T.; Barkhordari, F.; McMaster, W.R.; Mahboudi, F. The role of liposome charge on immune response generated in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63). Exp. Parasitol. 2009, 121, 362–369. [Google Scholar] [CrossRef]
- Badiee, A.; Khamesipour, A.; Samiei, A.; Soroush, D.; Shargh, V.H.; Kheiri, M.T.; Barkhordari, F.; Robert Mc Master, W.; Mahboudi, F.; Jaafari, M.R. The role of liposome size on the type of immune response induced in BALB/c mice against leishmaniasis: rgp63 as a model antigen. Exp. Parasitol. 2012, 132, 403–409. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Devitt, A.; Perrie, Y. The vesicle size of DDA:TDB liposomal adjuvants plays a role in the cell-mediated immune response but has no significant effect on antibody production. J. Control Release 2011, 154, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Ishida, T.; Inoue, A.; Mikami, J.; Muraguchi, M.; Ohmoto, Y.; Kiwada, H. HEPC-based liposomes trigger cytokine release from peripheral blood cells: Effects of liposomal size, dose and lipid composition. Int. J. Pharm. 2002, 236, 125–133. [Google Scholar] [CrossRef]
- Niwa, K.; Ikebuchi, K.; Fujihara, M.; Abe, H.; Wakamoto, S.; Ito, T.; Yamaguchi, M.; Sekiguchi, S. Inflammatory cytokine production in whole blood modified by liposome-encapsulated hemoglobin. Artif. Cells Blood Substit. Immobil. Biotechnol. 1998, 26, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef]
- van den Berg, R.H.; Faber-Krol, M.C.; Sim, R.B.; Daha, M.R. The First Subcomponent of Complement, C1q, Triggers the Production of IL-8, IL-6, and Monocyte Chemoattractant Peptide-1 by Human Umbilical Vein Endothelial Cells. J. Immunol. 1998, 161, 6924–6930. [Google Scholar]
- Wagner, H. Bacterial CpG DNA Activates Immune Cells to Signal Infectious Danger. In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 73, pp. 329–368. [Google Scholar]
- Ahmad-Nejad, P.; Hacker, H.; Rutz, M.; Bauer, S.; Vabulas, R.M.; Wagner, H. Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol. 2002, 32, 1958–1968. [Google Scholar] [CrossRef]
- Sellins, K.; Fradkin, L.; Liggitt, D.; Dow, S. Type I interferons potently suppress gene expression following gene delivery using liposome(-)DNA complexes. Mol. Ther. 2005, 12, 451–459. [Google Scholar] [CrossRef]
- Yasuda, S.; Yoshida, H.; Nishikawa, M.; Takakura, Y. Comparison of the type of liposome involving cytokine production induced by non-CpG Lipoplex in macrophages. Mol. Pharm. 2010, 7, 533–542. [Google Scholar] [CrossRef]
- Pusztai, L.; Mendoza, T.R.; Reuben, J.M.; Martinez, M.M.; Willey, J.S.; Lara, J.; Syed, A.; Fritsche, H.A.; Bruera, E.; Booser, D.; et al. Changes in plasma levels of inflammatory cytokines in response to paclitaxel chemotherapy. Cytokine 2004, 25, 94–102. [Google Scholar] [CrossRef]
- Szebeni, J. The interaction of liposomes with the complement system. Crit Rev. Ther. Drug Carr. Syst. 1998, 15, 57–88. [Google Scholar] [CrossRef]
- Cullis, P.R.; Chonn, A.; Semple, S.C. Interactions of liposomes and lipid-based carrier systems with blood proteins: Relation to clearance behaviour In Vivo. Adv. Drug Deliv. Rev. 1998, 32, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.J.; Brooks, D.E.; Norris-Jones, R.; Devine, D.V. C1q Binding to liposomes is surface charge dependent and is inhibited by peptides consisting of residues 14–26 of the human C1qA chain in a sequence independent manner. Biochim. Biophys. Acta (BBA)-Biomembr. 1999, 1418, 19–30. [Google Scholar] [CrossRef]
- Harashima, H.; Sakata, K.; Funato, K.; Kiwada, H. Enhanced Hepatic Uptake of Liposomes Through Complement Activation Depending on the Size of Liposomes. Pharm. Res. 1994, 11, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Solow, R.; Hu, V.W. Fluorescence analysis of size distribution and mode of dye release from carboxyfluorescein-loaded vesicles: Application to the study of complement-membrane interactions. Biochim. Biophys. Acta (BBA)-Biomembr. 1988, 945, 253–262. [Google Scholar] [CrossRef]
- Alving, C.R.; Richards, R.L.; Guirguis, A.A. Cholesterol-Dependent Human Complement Activation Resulting in Damage to Liposomal Model Membranes. J. Immunol. 1977, 118, 342–347. [Google Scholar]
- Szebeni, J.; Baranyi, L.; Savay, S.; Milosevits, J.; Bunger, R.; Laverman, P.; Metselaar, J.M.; Storm, G.; Chanan-Khan, A.; Liebes, L.; et al. Role of complement activation in hypersensitivity reactions to doxil and hynic PEG liposomes: Experimental and clinical studies. J. Liposome Res. 2002, 12, 165–172. [Google Scholar] [CrossRef]
- Chanan-Khan, A.; Szebeni, J.; Savay, S.; Liebes, L.; Rafique, N.M.; Alving, C.R.; Muggia, F.M. Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil): Possible role in hypersensitivity reactions. Ann. Oncol. 2003, 14, 1430–1437. [Google Scholar] [CrossRef]
- van den Hoven, J.M.; Nemes, R.; Metselaar, J.M.; Nuijen, B.; Beijnen, J.H.; Storm, G.; Szebeni, J. Complement activation by PEGylated liposomes containing prednisolone. Eur. J. Pharm. Sci. 2013, 49, 265–271. [Google Scholar] [CrossRef]
- Fülöp, T.; Kozma, G.T.; Vashegyi, I.; Mészáros, T.; Rosivall, L.; Urbanics, R.; Storm, G.; Metselaar, J.M.; Szebeni, J. Liposome-induced hypersensitivity reactions: Risk reduction by design of safe infusion protocols in pigs. J. Control Release 2019, 309, 333–338. [Google Scholar] [CrossRef]
- Szebeni, J.; Baranyi, L.; Savay, S.; Bodo, M.; Morse, D.S.; Basta, M.; Stahl, G.L.; Bunger, R.; Alving, C.R. Liposome-induced pulmonary hypertension: Properties and mechanism of a complement-mediated pseudoallergic reaction. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1319–H1328. [Google Scholar] [CrossRef]
- Ingen-Housz-Oro, S.; Pham-Ledard, A.; Brice, P.; Lebrun-Vignes, B.; Zehou, O.; Reitter, D.; Ram-Wolff, C.; Dupin, N.; Bagot, M.; Chosidow, O.; et al. Immediate hypersensitivity reaction to pegylated liposomal doxorubicin: Management and outcome in four patients. Eur. J. Derm. 2017, 27, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Arning, M.; Heer-Sonderhoff, A.H.; Wehmeier, A.; Schneider, W. Pulmonary toxicity during infusion of liposomal amphotericin B in two patients with acute leukemia. Eur. J. Clin. Microbiol. Infect. Dis. 1995, 14, 41–43. [Google Scholar] [CrossRef]
- Judge, A.; McClintock, K.; Phelps, J.R.; Maclachlan, I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Ringden, O.; Andstrom, E.; Remberger, M.; Svahn, B.M.; Tollemar, J. Allergic reactions and other rare side-effects of liposomal amphotericin. Lancet 1994, 344, 1156–1157. [Google Scholar] [CrossRef]
- Alberts, D.S.; Garcia, D.J. Safety Aspects of Pegylated Liposomal Doxorubicin in Patients with Cancer. Drugs 1997, 54, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Fontana, J.L.; Wassef, N.M.; Mongan, P.D.; Morse, D.S.; Dobbins, D.E.; Stahl, G.L.; Bunger, R.; Alving, C.R. Hemodynamic changes induced by liposomes and liposome-encapsulated hemoglobin in pigs: A model for pseudoallergic cardiopulmonary reactions to liposomes. Role of complement and inhibition by soluble CR1 and anti-C5a antibody. Circulation 1999, 99, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, D.; Hudson, L.; Weaver, L.J.; Craddock, P.; Jacob, H. Association of Complement Activation and Elevated Plasma-C5a with Adult Respiratory Distress Syndrome: Pathophysiological Relevance and Possible Prognostic Value. Lancet 1980, 315, 947–949. [Google Scholar] [CrossRef]
- Marceau, F.; Lundberg, C.; Hugli, T.E. Effects of the anaphylatoxins on circulation. Immunopharmacology 1987, 14, 67–84. [Google Scholar] [CrossRef]
- Kozma, G.T.; Mészáros, T.; Vashegyi, I.; Fülöp, T.; Örfi, E.; Dézsi, L.; Rosivall, L.; Bavli, Y.; Urbanics, R.; Mollnes, T.E.; et al. Pseudo-anaphylaxis to Polyethylene Glycol (PEG)-Coated Liposomes: Roles of Anti-PEG IgM and Complement Activation in a Porcine Model of Human Infusion Reactions. ACS Nano 2019, 13, 9315–9324. [Google Scholar] [CrossRef]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: Prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef]
- Weiss, R.B.; Donehower, R.C.; Wiernik, P.H.; Ohnuma, T.; Gralla, R.J.; Trump, D.L.; Baker, J.R., Jr.; Van Echo, D.A.; Von Hoff, D.D.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.C.; Tennant, N.M.; Sloane, D.E.; Hsu, F.I.; Barrett, N.A.; Hong, D.I.; Laidlaw, T.M.; Legere, H.J.; Nallamshetty, S.N.; Palis, R.I.; et al. Hypersensitivity reactions to chemotherapy: Outcomes and safety of rapid desensitization in 413 cases. J. Allergy Clin. Immunol. 2008, 122, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Bedőcs, P.; Rozsnyay, Z.; Weiszhár, Z.; Urbanics, R.; Rosivall, L.; Cohen, R.; Garbuzenko, O.; Báthori, G.; Tóth, M.; et al. Liposome-induced complement activation and related cardiopulmonary distress in pigs: Factors promoting reactogenicity of Doxil and AmBisome. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 176–184. [Google Scholar] [CrossRef] [PubMed]
- ALZA Pharmaceuticals; I. Doxil Package Insert: Mountain View, CA, USA, 2000.
- Szebeni, J.; Simberg, D.; González-Fernández, Á.; Barenholz, Y.; Dobrovolskaia, M.A. Roadmap and strategy for overcoming infusion reactions to nanomedicines. Nat. Nanotechnol. 2018, 13, 1100–1108. [Google Scholar] [CrossRef]
- Ishida, T.; Wang, X.; Shimizu, T.; Nawata, K.; Kiwada, H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J. Control Release 2007, 122, 349–355. [Google Scholar] [CrossRef]
- Verhoef, J.J.F.; Anchordoquy, T.J. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv. Transl. Res. 2013, 3, 499–503. [Google Scholar] [CrossRef]
- Ichihara, M.; Shimizu, T.; Imoto, A.; Hashiguchi, Y.; Uehara, Y.; Ishida, T.; Kiwada, H. Anti-PEG IgM Response against PEGylated Liposomes in Mice and Rats. Pharmaceutics 2010, 3, 1–11. [Google Scholar] [CrossRef]
- Mohamed, M.; Abu Lila, A.S.; Shimizu, T.; Alaaeldin, E.; Hussein, A.; Sarhan, H.A.; Szebeni, J.; Ishida, T. PEGylated liposomes: Immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710–724. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef]
- Mond, J.J.; Vos, Q.; Lees, A.; Snapper, C.M. T cell independent antigens. Curr. Opin. Immunol. 1995, 7, 349–354. [Google Scholar] [CrossRef]
- Boes, M. Role of natural and immune IgM antibodies in immune responses. Mol. Immunol. 2000, 37, 1141–1149. [Google Scholar] [CrossRef]
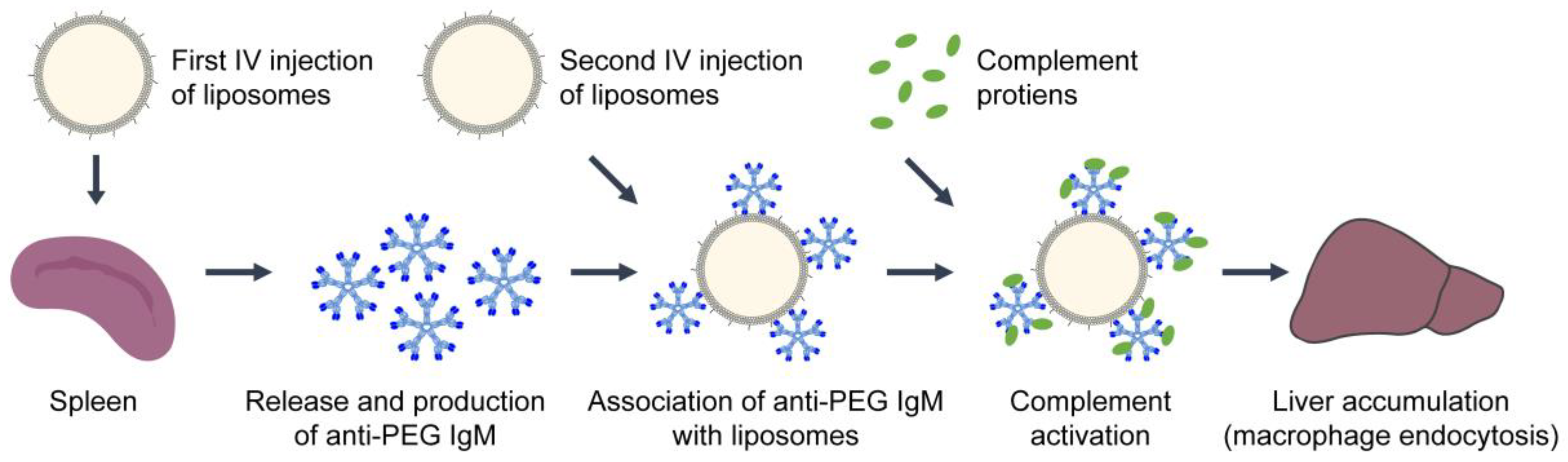
- Ishida, T.; Ichihara, M.; Wang, X.; Kiwada, H. Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J. Control Release 2006, 115, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Bucke, W.E.; Leitzke, S.; Diederichs, J.E.; Borner, K.; Hahn, H.; Ehlers, S.; Müller, R.H. Surface-Modified Amikacin-Liposomes: Organ Distribution and Interaction with Plasma Proteins. J. Drug Target. 1998, 5, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-L.; Wu, P.-Y.; Wu, M.-F.; Chern, J.-W.; Roffler, S.R. Accelerated Clearance of Polyethylene Glycol-Modified Proteins by Anti-Polyethylene Glycol IgM. Bioconjug. Chem. 1999, 10, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Hauet, T.; Eugene, M. A new approach in organ preservation: Potential role of new polymers. Kidney Int. 2008, 74, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Dams, E.T.M.; Laverman, P.; Oyen, W.J.G.; Storm, G.; Scherphof, G.L.; van der Meer, J.W.M.; Corstens, F.H.M.; Boerman, O.C. Accelerated Blood Clearance and Altered Biodistribution of Repeated Injections of Sterically Stabilized Liposomes. J. Pharmacol. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar]
- Laverman, P.; Carstens, M.G.; Boerman, O.C.; Dams, E.T.M.; Oyen, W.J.G.; van Rooijen, N.; Corstens, F.H.M.; Storm, G. Factors Affecting the Accelerated Blood Clearance of Polyethylene Glycol-Liposomes upon Repeated Injection. J. Pharmacol. Exp. Ther. 2001, 298, 607–612. [Google Scholar]
- Ishida, T.; Maeda, R.; Ichihara, M.; Irimura, K.; Kiwada, H. Accelerated clearance of PEGylated liposomes in rats after repeated injections. J. Control Release 2003, 88, 35–42. [Google Scholar] [CrossRef]
- Xu, H.; Ye, F.; Hu, M.; Yin, P.; Zhang, W.; Li, Y.; Yu, X.; Deng, Y. Influence of phospholipid types and animal models on the accelerated blood clearance phenomenon of PEGylated liposomes upon repeated injection. Drug Deliv. 2015, 22, 598–607. [Google Scholar] [CrossRef]
- Ishida, T.; Ichikawa, T.; Ichihara, M.; Sadzuka, Y.; Kiwada, H. Effect of the physicochemical properties of initially injected liposomes on the clearance of subsequently injected PEGylated liposomes in mice. J. Control Release 2004, 95, 403–412. [Google Scholar] [CrossRef]
- Semple, S.C.; Harasym, T.O.; Clow, K.A.; Ansell, S.M.; Klimuk, S.K.; Hope, M.J. Immunogenicity and Rapid Blood Clearance of Liposomes Containing Polyethylene Glycol-Lipid Conjugates and Nucleic Acid. J. Pharmacol. Exp. Ther. 2005, 312, 1020–1026. [Google Scholar] [CrossRef]
- Ishida, T.; Atobe, K.; Wang, X.; Kiwada, H. Accelerated blood clearance of PEGylated liposomes upon repeated injections: Effect of doxorubicin-encapsulation and high-dose first injection. J. Control Release 2006, 115, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kashima, S.; Kiwada, H. The contribution of phagocytic activity of liver macrophages to the accelerated blood clearance (ABC) phenomenon of PEGylated liposomes in rats. J. Control Release 2008, 126, 162–165. [Google Scholar] [CrossRef]
- Koide, H.; Asai, T.; Hatanaka, K.; Akai, S.; Ishii, T.; Kenjo, E.; Ishida, T.; Kiwada, H.; Tsukada, H.; Oku, N. T cell-independent B cell response is responsible for ABC phenomenon induced by repeated injection of PEGylated liposomes. Int. J. Pharm. 2010, 392, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ichihara, M.; Hyodo, K.; Yamamoto, E.; Ishida, T.; Kiwada, H.; Ishihara, H.; Kikuchi, H. Accelerated blood clearance of PEGylated liposomes containing doxorubicin upon repeated administration to dogs. Int. J. Pharm. 2012, 436, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Lopes, T.C.M.; Silva, D.F.; Costa, W.C.; Frézard, F.; Barichello, J.M.; Silva-Barcellos, N.M.; de Lima, W.G.; Rezende, S.A. Accelerated Blood Clearance (ABC) Phenomenon Favors the Accumulation of Tartar Emetic in Pegylated Liposomes in BALB/c Mice Liver. Pathol. Res. Int. 2018, 2018, 9076723. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.L.; Wang, H.H.; Wu, Y.F.; Wang, L.; Zhang, L.; Ye, X.; Peng, D.Y.; Chen, W.D. Activation of PXR-Cytochrome P450s axis: A Possible Reason for the Enhanced Accelerated Blood Clearance Phenomenon of PEGylated Liposomes In Vivo. Drug Metab. Dispos. 2019, 47, 785–793. [Google Scholar] [CrossRef]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef]
- Gabizon, A.; Isacson, R.; Rosengarten, O.; Tzemach, D.; Shmeeda, H.; Sapir, R. An open-label study to evaluate dose and cycle dependence of the pharmacokinetics of pegylated liposomal doxorubicin. Cancer Chemother. Pharm. 2008, 61, 695–702. [Google Scholar] [CrossRef]
- Yang, Q.; Jacobs, T.M.; McCallen, J.D.; Moore, D.T.; Huckaby, J.T.; Edelstein, J.N.; Lai, S.K. Analysis of Pre-existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population. Anal. Chem. 2016, 88, 11804–11812. [Google Scholar] [CrossRef]
- Wang, F.; Ye, X.; Wu, Y.; Wang, H.; Sheng, C.; Peng, D.; Chen, W. Time Interval of Two Injections and First-Dose Dependent of Accelerated Blood Clearance Phenomenon Induced by PEGylated Liposomal Gambogenic Acid: The Contribution of PEG-Specific IgM. J. Pharm. Sci. 2019, 108, 641–651. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.-C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. https://doi.org/10.3390/nano10020190
Inglut CT, Sorrin AJ, Kuruppu T, Vig S, Cicalo J, Ahmad H, Huang H-C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials. 2020; 10(2):190. https://doi.org/10.3390/nano10020190
Chicago/Turabian StyleInglut, Collin T., Aaron J. Sorrin, Thilinie Kuruppu, Shruti Vig, Julia Cicalo, Haroon Ahmad, and Huang-Chiao Huang. 2020. "Immunological and Toxicological Considerations for the Design of Liposomes" Nanomaterials 10, no. 2: 190. https://doi.org/10.3390/nano10020190
APA StyleInglut, C. T., Sorrin, A. J., Kuruppu, T., Vig, S., Cicalo, J., Ahmad, H., & Huang, H.-C. (2020). Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials, 10(2), 190. https://doi.org/10.3390/nano10020190