Biomaterial Enhanced Regeneration Design Research for Skin and Load Bearing Applications

Abstract
:1. Introduction
1.1. Scope of Field
1.2. Scope of the Review
1.3. Principles
1.3.1. Design Hierarchy
- Biocompatibility is dependent on time. An implant can be biocompatible for short-term applications, but not long-term ones. In addition, an implant may trigger a “bad” (or inappropriate) response in the short-term in order to elicit a “good” (appropriate) response in the long-term [4].
- An “appropriate response” is much better than an “inert response”, but still does not provide much guidance on what is “appropriate”.
1.3.2. Emphasis
1.3.3. Responsibility for Design Studies
1.4. Design Options
1.4.1. Materials
1.4.2. Manufacturing Techniques
1.4.3. Properties
Process Modifications
Types of Modifications
Stability
1.4.4. Incorporation into the Biomaterial
Drug Delivery
Cell Incorporation
2. Strategies
2.1. General Design Strategies
2.1.1. Degradable/Regenerative Scaffolds
2.1.2. Synthetic Graft
2.1.3. Devices Adjacent to the Defect or Wound
2.2. Specific Strategies
2.2.1. Skin Scaffolds
Bioactivity
Scaffold Materials
System Design
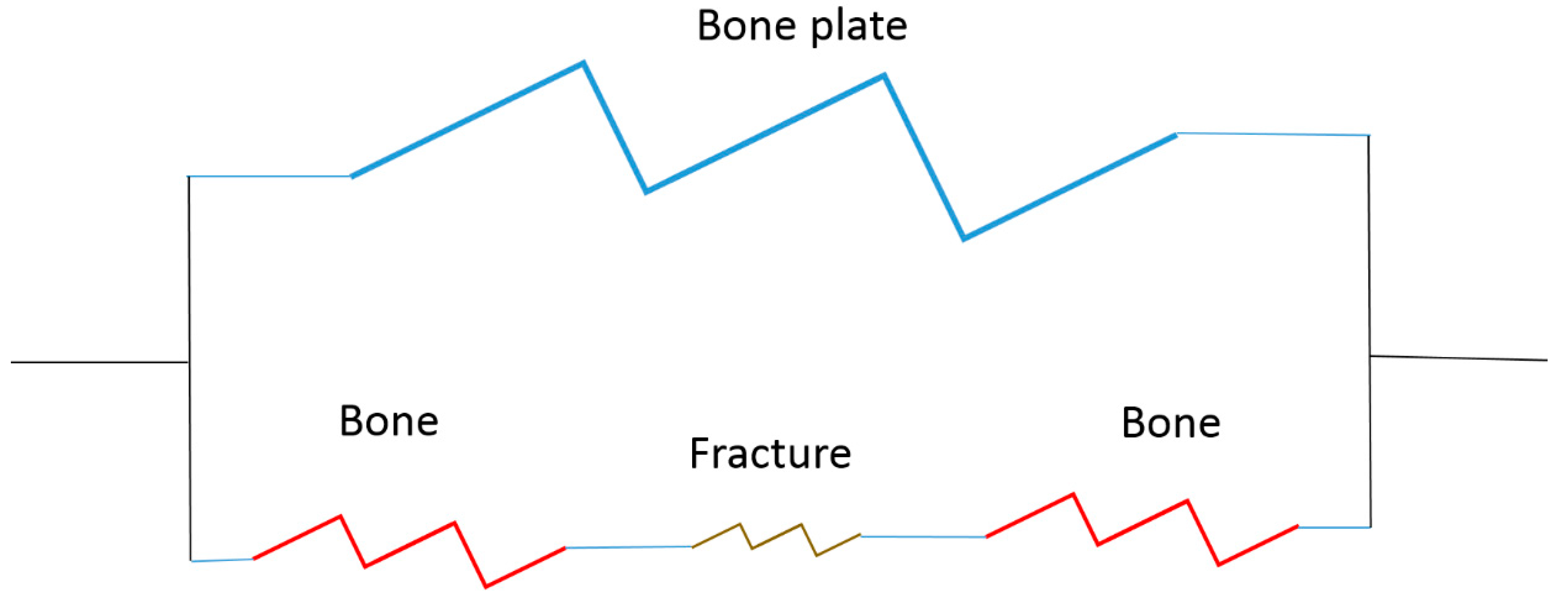
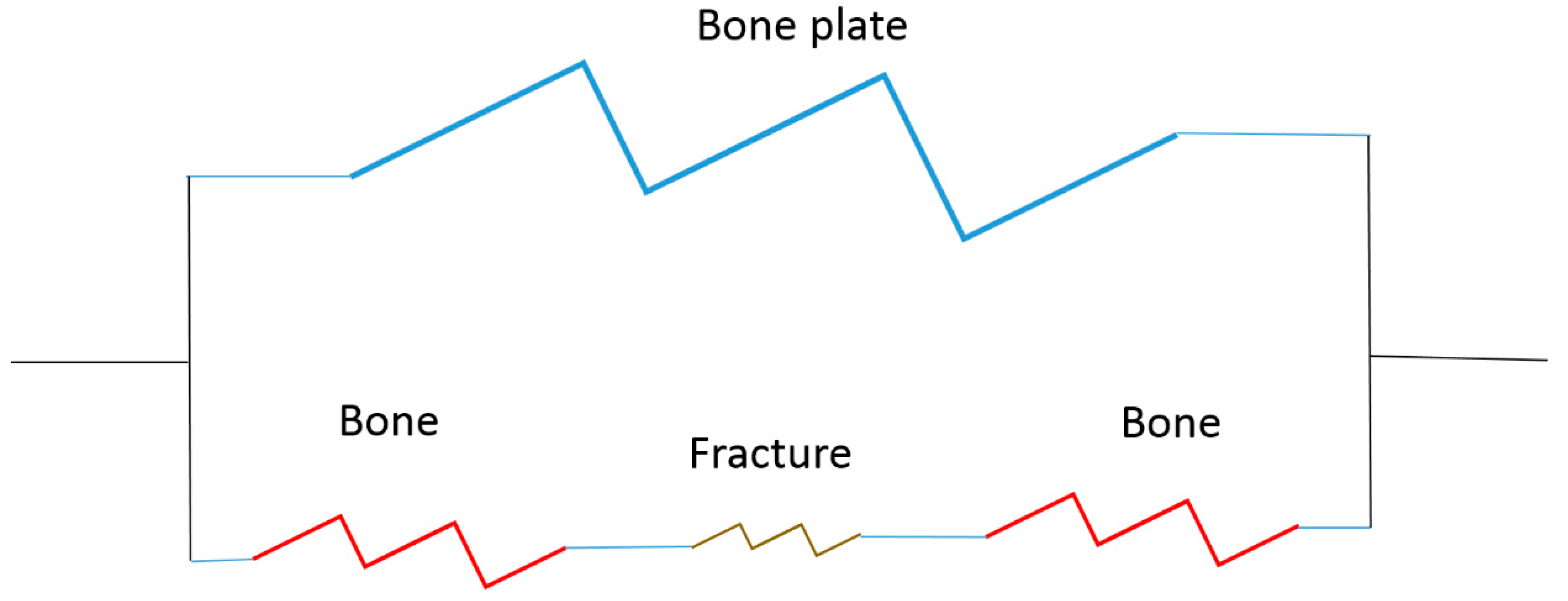
2.2.2. Load Bearing Systems
- How to stabilize the fracture site (bone to bone interfaces) to allow healing;
- To control loading to the fracture site to stimulate healing.
Load-Bearing Implant Design
Strategy Example
3. Current Clinical Devices
3.1. Current Biomaterial Enhanced Regeneration Techniques for Skin
3.1.1. Grafts (Skin Substitutes)
Natural Grafts
Synthetic Grafts
3.1.2. Limitations of Commercially Available Skin Substitutes
3.1.3. Future Directions
Scaffolds
Stem Cells
Additive Manufacturing
Skin Substitutes versus Scaffolds
Whole-Organ Decellularization
Overall Strategies
3.2. Current Biomaterial Enhanced Regeneration Techniques for Load Bearing Applications
3.2.1. Stability
3.2.2. Healing
3.2.3. Limitations of Commercially Available Load Bearing Systems
Mechanical
Healing
3.2.4. Future Directions
Funding
Acknowledgements
Conflicts of Interest
References
- Hench, L.; Ethridge, C. Biomaterials—The interfacial problem. Adv. Biomed. Eng. 1974, 5, 35–150. [Google Scholar]
- Vert, M. Terminology for biorelated polymers and applications. Pure Appl. Chem. 2012, 84, 377–410. [Google Scholar] [CrossRef]
- Williams, D.F. The Williams Dictionary of Biomaterials; Liverpool University Press: Liverpool, UK, 1999. [Google Scholar]
- Feldman, D.; Barker, T.; Bowman, J.; Blum, B.; Kilpadi, D.; Redden, R. Biomaterial enhanced regeneration for skin wounds. In Biomaterials and Bioengineering Handbook; Wise, D., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 807–842. [Google Scholar]
- Black, J. Biological Performance of Materials: Fundamentals of Biocompatibility; Marcel Dekker, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Williams, D.F. On the mechanism of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D. Quantification and modeling of biological processes for tissue engineering and regenerative medicine. Biomed. J. Sci. Tech. Res. 2019, in press. [Google Scholar]
- Feldman, D.; Czuwala, P.; Kelpke, S.; Pandit, A.; Wilson, D. A biocompatibility hierarchy: Justification for biomaterial enhanced regeneration. In Encyclopedic Handbook of Biomaterials and Bioengineering; Wise, D., Ed.; Marcel Dekker: New York, NY, USA, 1995; pp. 223–268. [Google Scholar]
- Yannas, I.V.; Burke, J.F.; Gordon, P.L.; Huang, C.; Rubenstein, R. Design of an artificial skin. II. Control of chemical composition. J. Biomed. Mater. Res. 1981, 14, 107–131. [Google Scholar] [CrossRef]
- Yannas, I.V.; Lee, E.; Orgill, D.P.; Skrabut, E.M.; Murphy, S.F. Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin. Proc. Natl. Acad. Sci. USA 1989, 86, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.; Feldman, D. Tissue adhesives for growth factor delivery. In Biomaterials and Bioengineering Handbook; Wise, D., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 261–312. [Google Scholar]
- Feldman, D. The importance of engineering design constraints to justify a study, particularly in an applied bioengineering journal. J. Biomed. Imaging Bioeng. 2017, 1, 5–6. [Google Scholar]
- Feldman, D. An Engineering Approach to the Scientific Method. Signif. Bioeng. Biosci. 2017, 1, 6. [Google Scholar] [CrossRef]
- Sharma, C.; Gautam, S.; Dinda, A.K.; Mishra, N.C. Cartilage tissue engineering: Current scenario and challenges. Adv. Mater. Lett. 2011, 2, 90–99. [Google Scholar] [CrossRef]
- Sharma, C.; Dinda, A.K.; Potdar, P.D.; Chou, C.F.; Mishra, N.C. Fabrication and characterization of novel nano-biocomposite scaffold of chitosan–gelatin–alginate–hydroxyapatite for bone tissue engineering. Mater. Sci. Eng. 2016, 64, 416–427. [Google Scholar] [CrossRef]
- Grimm, M.J. Orthopedic Biomaterials. In Biomedical Engineering and Design Handbook; Kutz, M., Ed.; McGraw-Hill: New York, NY, USA, 2009; pp. 421–444. [Google Scholar]
- Tenenhaus, M.; Rennekamoff, H.O. Current Concepts in Tissue Engineering: Skin Wound Healing. Plast. Reconstruct. Surg. 2016, 138 (Suppl. 3), 42S–50S. [Google Scholar] [CrossRef] [PubMed]
- Vig, K.; Chaudhari, A.; Tripathi, S.; Dixit, S.; Sahu, R.; Pillai, S.; Dennis, V.A.; Singh, S.R. Advances in Skin Regeneration Using Tissue Engineering. Int. J. Mol. Sci. 2017, 18, 789. [Google Scholar] [CrossRef] [PubMed]
- Tatara, A.M.; Mikos, A.G. Tissue Engineering in Orthopaedics. J. Bone Joint Surg. 2016, 98, 1132–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishero, B.A.; Kohli, N.; Das, A.; Christophel, J.J.; Cui, Q. Current Concepts of Bone Tissue Engineering for Craniofacial Bone Defect Repair. J. Bone Joint Surg. Am. 2016, 98, 1132–1139. [Google Scholar]
- Takato, T. Tissue Engineering in Bone: The Contention of a Hundred Schools of Thought. Artif. Organs 2016, 40, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Duranceau, L.; Genest, H.; Bortoluzzi, P.; Moulin, V.; Auger, F.A.; Germain, L. Successful grafting of a novel autologous tissue-engineered skin substitutes (dermis and epidermis) on twelve burn patients. J. Burn Care Res. 2014, 35, S121. [Google Scholar]
- Boyce, S.T.; Simpson, P.S.; Rieman, M.T.; Warner, P.M.; Yakuboff, K.P.; Bailey, J.K.; Nelson, J.K.; Fowler, L.A.; Kagan, R.J. Randomized, paired-site comparison of autologous engineered skin substitutes and split-thickness skin graft for closure of extensive, full-thickness burns. J. Burn Care Res. 2017, 38, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Varkey, M.; Ding, J.; Tredget, E.E. Advances in skin substitutes—Potential of tissue engineered skin for facilitating anti-fibrotic healing. J. Funct. Biomater. 2015, 6, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Boyce, S.T.; Supp, D.M. Biologic skin substitutes. In Skin Tissue Engineering and Regenerative Medicine; Albanna, M.Z., Holmes, J.H., Eds.; Academic Press: New York, NY, USA, 2016; pp. 211–238. [Google Scholar]
- Thomas-Virnig, C.L.; Allen-Hoffmann, B.L. A bioengineered human skin tissue for the treatment of infected wounds. Adv. Wound Care 2012, 1, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Boyce, S.T.; Kagan, R.J.; Greenhalgh, D.G.; Warner, P.; Yakuboff, K.P.; Palmieri, T.; Warden, G.D. Cultured skin substitutes reduce requirements for harvesting of skin autograft for closure of excised, full-thickness burns. J. Trauma Acute Care Surg. 2006, 60, 821–829. [Google Scholar]
- Kagan, R.J.; Peck, M.D.; Ahrenholz, D.H.; Hickerson, W.L.; Holmes, I.V.J.; Korentager, R.; Kraatz, J.; Pollock, K.; Kotoski, G. Surgical management of the burn wound and use of skin substitutes: An expert panel white paper. J. Burn Care Res. 2013, 34, e60–e79. [Google Scholar] [CrossRef] [PubMed]
- Heimbach, D.M.; Warden, G.D.; Luterman, A.; Jordan, M.H.; Ozobia, N.; Ryan, C.M.; Voigt, D.W.; Hickerson, W.L.; Saffle, J.R.; DeClement, F.A. Multicenter postapproval clinical trial of Integra dermal regeneration template for burn treatment. J. Burn Care Rehabil. 2003, 24, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D. Size vs. Chemistry in Biomaterial Pathology. Trans. Biomed. Eng. Soc. 2018, 4, 1–39. [Google Scholar]
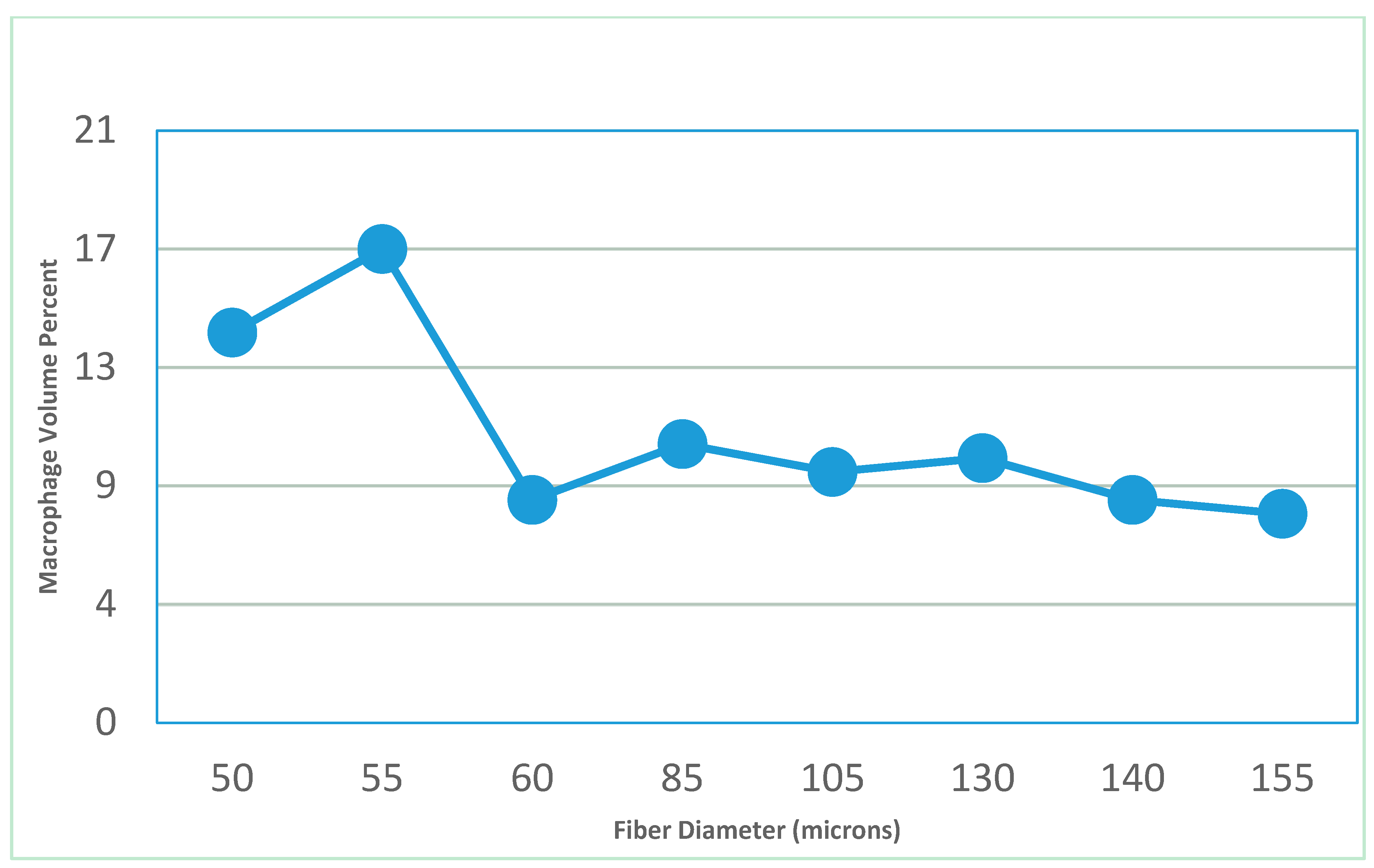
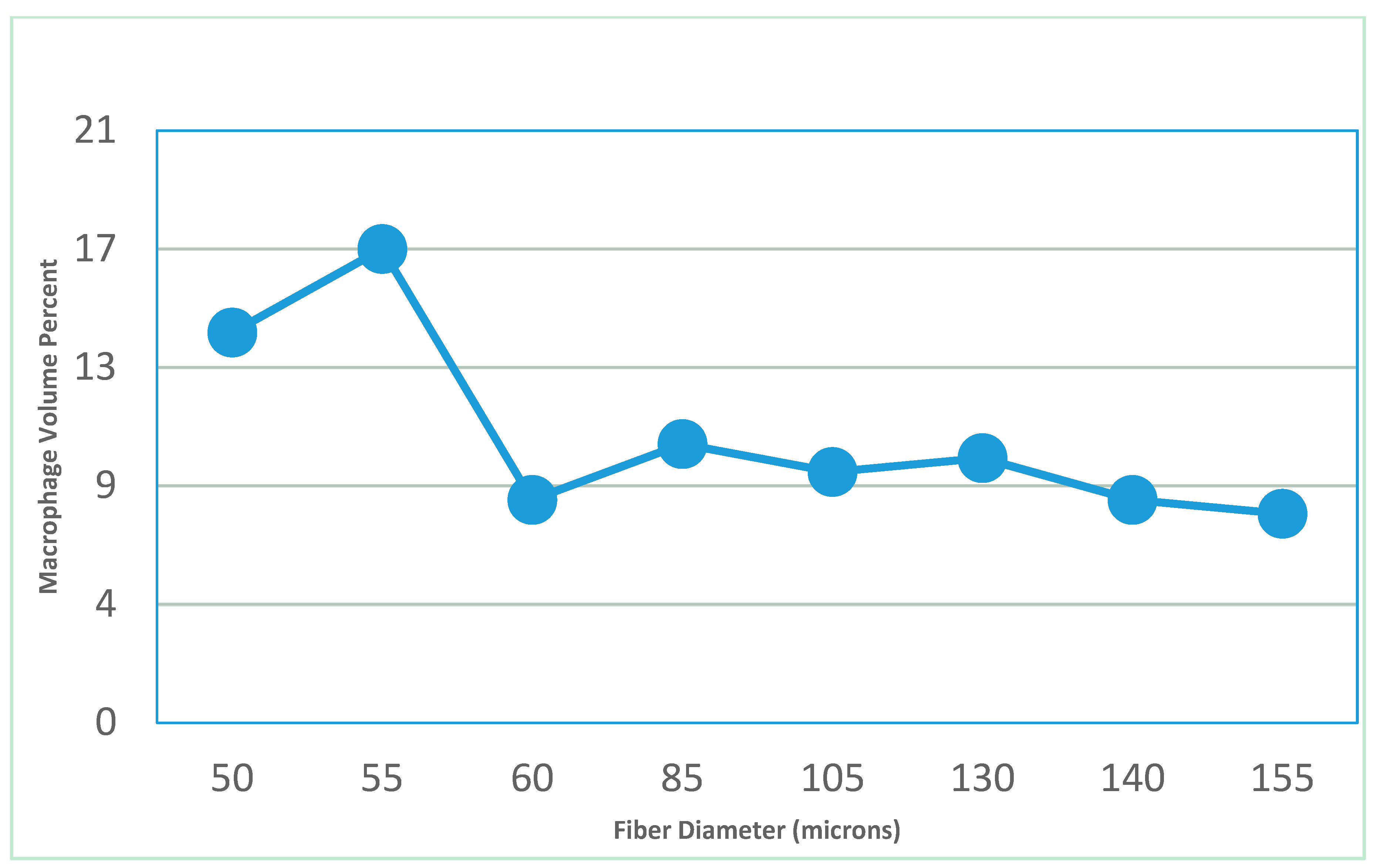
- Feldman, D.; Ferguson, D. The effect of fiber spacing and fiber diameter on soft tissue ingrowth for polyethylene terephthalate. J. Biomed. Imaging Bioeng. 2017, 1, 35–46. [Google Scholar]
- Feldman, D.; Myerson, A. Feasibility of a strategy to prevent gouty arthritis through limiting crystallization of monosodium urate. Int. J. Drug Res. Technol. 2017, 8, 4–21. [Google Scholar]
- Palsson, B. Coordination of Cellular-Fate Processes. In Tissue Engineering; Pearson Education: Upper Saddle River, NJ, USA, 2004; pp. 105–131. [Google Scholar]
- Sealy, M.; Ziye, L.; Chao, L.; Guo, Y.; White, B.; Barkey, M.; Jordan, B.; Brewer, L.; Feldman, D. A strategy to optimize recovery in orthopedic sports injuries. J. Bioanal. Biomed. 2017, 9, 144–151. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Rep. Reg. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- White, R.; McIntosh, C. A review of the literature on topical therapies for diabetic foot ulcers. Part 2: Advanced treatments. J. Wound Care 2009, 18, 335–341. [Google Scholar] [CrossRef]
- Zou, Z.; Sun, P.D. An improved recombinant mammalian cell expression system for human transforming growth factor-β2 and -β3 preparations. Protein Express. Purif. 2006, 50, 9–17. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, L.; Scott, P.G.; Tredget, E.E. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells 2007, 25, 2648–2659. [Google Scholar] [CrossRef]
- Nakagawa, H.; Akita, S.; Fukui, M.; Fujii, T.; Akino, K. Human mesenchymal stem cells successfully improve skin- substitute wound healing. Br. J. Dermatol. 2005, 153, 29–36. [Google Scholar] [CrossRef] [PubMed]
- McFarlin, K.; Gao, X.; Liu, Y.B.; Dulchavsky, D.S.; Kwon, D.; Arbab, A.S.; Bansal, M.; Li, Y.; Chopp, M.; Dulchavsky, S.A.; et al. Bone marrow-derived mesenchymal stromal cells accelerate wound healing in the rat. Wound Rep. Reg. 2006, 14, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Badiavas, E.V.; Falanga, V. Treatment of chronic wounds with bone marrow-derived cells. Arch. Dermatol. 2003, 139, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Boxberger, S.; Jørgensen, B.; Feldmann, S.; Ehninger, G.; Bornhäuser, M.; Werner, C. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 2004, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.; Vanguri, P.; Simonetti, D.; Young, R. Adult mesenchymal stem cells: Potential for muscle and tendon regeneration and use in gene therapy. J. Muscoloskelet. Neuronal Interact. 2002, 2, 309–320. [Google Scholar]
- Zuk, P.A.; Zhu, M.I.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell- based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Abe, R.; Fujita, Y.; Ando, S.; Inokuma, D.; Shimizu, H. Mesenchymal Stem Cells Are Recruited into Wounded Skin and Contribute to Wound Repair by Transdifferentiation into Multiple Skin Cell Type. J. Immunol. 2008, 80, 2581–2587. [Google Scholar] [CrossRef]
- Fu, X.; Han, B.; Cai, S.; Lei, Y.; Sun, T.; Sheng, Z. Migration of bone marrow-derived mesenchymal stem cells induced by tumor necrosis factor-alpha and its possible role in wound healing. Wound Rep. Reg. 2009, 17, 185–191. [Google Scholar] [CrossRef]
- Fathke, C.; Wilson, L.; Hutter, J.; Kapoor, V.; Smith, A.; Hocking, A.; Isik, F. Contribution of bone marrow-derived cells to skin: Collagen deposition and wound repair. Stem Cells 2004, 22, 812–822. [Google Scholar] [CrossRef]
- Caplan, A.I. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J. Cell. Phys. 2007, 213, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Neman, J.; Hambrecht, A.; Cadry, C.; Goodarzi, A.; Youssefzadeh, J.; Chen, M.Y.; Jandial, R. Clinical efficacy of stem cell mediated osteogenesis and bioceramics for bone tissue engineering. Adv. Exp. Med. Biol. 2012, 760, 174–187. [Google Scholar] [PubMed]
- Neman, J.; Hambrecht, A.; Cadry, C.; Jandial, R. Stem cell-mediated osteogenesis: Therapeutic potential for bone tissue engineering. Biologics 2012, 6, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Martina, M.; Hutmacher, D.W.; Hui, J.H.; Lee, E.H.; Lim, B. Identification of common pathways mediating differentiation of bone marrow- and adipose tissue-derived human mesenchymal stem cells into three mesenchymal lineages. Stem Cells 2007, 25, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, O.; Katsube, Y.; Hirose, M.; Ohgushi, H.; Ito, H. Comparison of osteogenic ability of rat mesenchymal stem cells from bone marrow, periosteum, and adipose tissue. Calcif. Tissue Int. 2008, 82, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Weinand, C.; Xu, J.W.; Peretti, G.M.; Bonassar, L.J.; Gill, T.J. Conditions affecting cell seeding onto three-dimensional scaffolds for cellular-based biodegradable implants. J. Biomed. Mater. Res. B Appl. Biomater. 2009, 91, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; McCauley, M. Mesenchymal stem cells, TGF-β3, and an albumin scaffold to promote full-thickness wound healing. Trans. Biomed. Eng. Soc. 2012, 44, 152. [Google Scholar]
- Baranoski, S.; Ayello, E.A. Wound Care Essentials: Practice Principles; Lippincott, Williams, and Wilkins: Philadelphia, PA, USA, 2004; pp. 62–70. [Google Scholar]
- US Food and Drug Administration. Guidance for industry: Chronic cutaneous ulcer and burn wounds—Developing products for treatment. Fed. Regist. 2006, 65, 39912. [Google Scholar]
- Gibran, N.S.; Boyce, S.; Greenhalgh, D.G. Cutaneous wound healing. J. Burn Care Res. 2007, 28, 577–579. [Google Scholar] [CrossRef]
- McCullars, J.; Moore, S.; Jennings, A.; Feldman, D. Local versus systemic delivery of endothelial progenitor cells for a tissue scaffold. Trans. Soc. Biomater. 2006, 31, 588. [Google Scholar]
- Winter, G.W. Epidermal regeneration studied in the domestic pig. In Epidermal Wound Healing; Maibach, H.I., Rovee, D.T., Eds.; Year Book Publishers: Chicago, IL, USA, 1972; pp. 71–112. [Google Scholar]
- von Recum, A.; Park, J. Permanent percutaneous devices. CRC Crit. Rev. Bioeng. 1981, 11, 37. [Google Scholar]
- Boyce, S.T.; Foreman, T.J.; English, K.B. Skin wound closure in athymic mice with cultured human cells, biopolymers, and growth factors. Surgery 1991, 110, 866. [Google Scholar] [PubMed]
- Feldman, D.; Sierra, D. Tissue adhesives in wound healing. In Encyclopedic Handbook of Biomaterials and Bioengineering; Marcel Dekker: New York, NY, USA, 1995. [Google Scholar]
- Sierra, D. Fibrin sealant adhesive systems: A review of their chemistry, material properties and clinical applications. J. Biomater. Appl. 1993, 7, 309–352. [Google Scholar] [CrossRef] [PubMed]
- Greisler, H.; Kim, D. Aspects of biodegradable vascular prosthesis. In Vascular Graft Update; Kahalic, H., Kantrowitz, A., Sung, P., Eds.; ASTM: Fairfield, PA, USA, 1986; pp. 197–218. [Google Scholar]
- Estridge, T.D. The Use of Oxygen, Growth Factors and Implants to Effect Cellular Activity In Vitro. Ph.D. Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1991. [Google Scholar]
- Estridge, T.; Feldman, D. The use of oxygen for optimal fibroblast activation. Trans. FASEB 1991, 75, 7230. [Google Scholar]
- Pandit, A. The Effect of Oxygen Treatment and Dressing Oxygen Permeability on Wound Healing. Master’s Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1991. [Google Scholar]
- Pandit, A.; Feldman, D. The effect of oxygen permeability on full-thickness skin defects. Wound Heal. Regen. 1994, 2, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Feldman, D. Acidic fibroblast growth factor (a-FGF) delivery through a fibrin matrix with oxygen treatments for full thickness defects. Wound Repair Regen. 1993, 1, 132. [Google Scholar]
- Andino, R. The Use of a Low Frequency PEMF in the Treatment of Full Thickness Skin Defects in the Rabbit Model. Master’s Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1991. [Google Scholar]
- Andino, R.; Feldman, D. Pulsating electromagnetic fields used to treat full thickness defects in the rabbit model. Trans. FASEB 1991, 5, 5. [Google Scholar]
- Kelpke, S. Pulsed Electromagnetic Field to Accelerate Wound Healing. Master’s Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1994. [Google Scholar]
- Kelpke, S.; Feldman, D. Polyurethane dressing in combination with pulsed electromagnetic fields to accelerate wound healing. Trans. Soc. Biomater. 1993, 19, 56. [Google Scholar]
- Jennings, A.; Chen, D.; Feldman, D. Upregulation of chemokine (C-C motif) ligand 20 in adult epidermal keratinocytes in direct current electric fields. Arch. Dermatol. Res. 2009, 302, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Jennings, A.; Chen, D.; Feldman, D. Transcriptional response of dermal fibroblasts in direct current electric fields. Bioelectromagnetics 2008, 29, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Jennings, A.; Feldman, D. Electric Field-induced gene expression in skin cells. Wound Repair Regen. 2006, 14, A52. [Google Scholar]
- Estridge, T.; Feldman, D.; Pandit, A.; Andino, R. The effect of wound matrices on the healing of full thickness defects in the rabbit model. In Proceedings of the 19th Annual Meeting of the Society for Biomaterials, Birmingham, AL, USA, 28 April–2 May 1993. [Google Scholar]
- Ashar, R. The Use of FGF-1 and TGF-β in the Treatment of Full-Thickness Skin Defects in the Rabbit Model; UAB: Birmingham, AL, USA, 1993. [Google Scholar]
- Pandit, A.; Ashar, R.; Feldman, D. The effect of TGF-β delivered through a collagen scaffold in stimulation of healing in full-thickness defects. J. Investig. Surg. 1999, 12, 89–100. [Google Scholar]
- Pandit, A.; Ashar, R.; Feldman, D.; Thompson, J. Investigation of acidic fibroblast growth factor delivered through a collagen scaffold for the treatment of full-thickness skin defects, in a rabbit model. Plast. Reconstruct. Surg. 1998, 101, 766–775. [Google Scholar] [CrossRef]
- Pandit, A.; Ashar, R.; Feldman, D. Acidic fibroblast growth factor and transforming growth factor beta in stimulation of healing in full thickness skin defects. In Proceedings of the Annual Meeting of the Wound Healing Society and the European Tissue Repair Society, Amsterdam, The Netherlands, 22–25 August 1993. [Google Scholar]
- Feldman, D. Adhesion and hemostasis in surgery. In Encyclopedia of Materials: Science and Technology; Williams, D., Ed.; Elsevier Science Ltd.: London, UK, 2002; pp. 38–43. [Google Scholar]
- Feldman, D.; Barker, T.; Blum, B.; Kilpadi, D.; Redden, R. Tissue assessment of skin substitutes. In Biomaterials and Bioengineering Handbook; Wise, D., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 773–780. [Google Scholar]
- Feldman, D. Wound healing applications of fibrin sealants. In Surgical Tissue Adhesives and Sealants; Sierra, D., Saltz, R., Eds.; Technomic: Lancaster, PA, USA, 1996; pp. 99–108. [Google Scholar]
- Saltz, R.; Sierra, D.; Feldman, D.; Saltz, M.; Dimick, A.; Vasconez, L. Experimental and clinical applications of fibrin glue. Plast. Reconstruct. Surg. 1991, 88, 1005–1015. [Google Scholar] [CrossRef]
- Lee, H.; Reddy, M.; Geurs, N.; Palcanis, K.; Lemons, J.; Rahemtulla, F.; Ho, K.; Chen, D.J.; Davis, C.; Feldman, D. Efficacy of platelet-rich plasma on wound healing in rabbits. J. Peridontol. 2008, 25, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Barker, T.; Fuller, G.; Klinger, M.; Feldman, D.; Hagood, S. Modification of fibrinogen with polyethylene glycol and its effects on fibrin clot characteristics. JBMR 2001, 56, 529–535. [Google Scholar] [CrossRef]
- Pandit, A.; Feldman, D.; Caufield, J.; Thompson, J. Stimulation of angiogenesis by FGF-1 delivered through a modified fibrin scaffold. Growth Factors 1998, 15, 113–123. [Google Scholar] [CrossRef]
- Pandit, A.; Caufield, J.; Feldman, D. In vivo wound healing response to a modified degradable fibrin scaffold. J. Biomater. Appl. 1997, 12, 222–236. [Google Scholar] [CrossRef]
- Pandit, A.; Feldman, D.; Listinsky, K.; Thompson, J. The effect on wound healing by a modified fibrin scaffold delivering acidic fibroblast growth factor (FGF-1). J. Bioact. Compat. Polym. 1997, 12, 99. [Google Scholar] [CrossRef]
- Pandit, A.; Wilson, D.; Feldman, D.; Thompson, J. Fibrin scaffold as an effective vehicle for the delivery of acidic fibroblast growth factor (FGF-1). J. Biomater. Appl. 2000, 14, 229–242. [Google Scholar] [CrossRef]
- Overby, R.; Feldman, D. Investigating how molecular weight of a PEG cross-linker affects the stability of an albumin tissue scaffold system. Trans. Soc. Biomater. 2003, 29, 294. [Google Scholar]
- Overby, R.; Feldman, D. Compositional effects on albumin system properties. Trans. Biomed. Eng. Soc. 2002, 20. [Google Scholar]
- Range, S.; Barker, A.; Feldman, D. Biofeedback-controlled protein delivery from a fibrin matrix. Trans. Soc. Biomater. 2002, 28, 147. [Google Scholar]
- Blum, B.; Huang, S.; Eberhardt, A.; Overby, R.; Feldman, D. Effect of composition and porosity on mechanical strength of an adhesive albumin. Trans. Soc. Biomater. 2002, 28, 334. [Google Scholar]
- Range, S.; Barker, T.; Blum, B.; Kilpadi, D.; Overby, R.; Feldman, D. Optimizing tissue adhesive scaffolds for skin wounds. Wound Repair Regen. 2001, 9, 143. [Google Scholar]
- Blum, B.; Huang, S.; Eberhardt, A.; Feldman, D. Effect of material composition on shear strength of an adhesive albumin. Trans. World Biomater. Congr. 2000, 6, 1067. [Google Scholar]
- Feldman, D.; Barker, T.; Blum, B.; Kilpadi, D.; Redden, R. Fibrin as a tissue adhesive and scaffold for meshed skin grafts in burn patients. Trans. Soc. Biomater. 1999, 25, 160. [Google Scholar]
- Blum, B.; Huang, S.; Barker, T.; Barker, S.; Kilpadi, D.; Redden, R.; Feldman, D. In vivo evaluation of an adhesive albumin used for incision closure. Trans. Soc. Biomater. 1999, 25, 43. [Google Scholar]
- Sierra, D. The Evaluation of Fibrin Glue Adhesive Strength. Master’s Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1991. [Google Scholar]
- Sierra, D.; Feldman, D.; Saltz, R. A method to determine the shear adhesive strength of fibrin sealants. Appl. Biomater. 1992, 3, 147–151. [Google Scholar] [CrossRef]
- Ronfard, V.; Broly, H.; Mitchell, V.; Galizia, J.P.; Hochart, D.; Chambon, E.; Pellerin, P.; Huart, J.J. Use of human keratinocytes cultured on fibrin glue in the treatment of burn wounds. Burns 1991, 17, 181–184. [Google Scholar] [CrossRef]
- Dahlstrom, K.K.; Weis-Fogh, U.S.; Medgyesi, S.; Rostgaard, J.; Sorenson, H. The use of autologous fibrin adhesive in skin transplantation. Plast. Reconstr. 1992, 89, 968–972. [Google Scholar] [CrossRef]
- Brown, D.M.; Barton, B.R.; Young, V.L.; Pruitt, B.A. Decreased wound contraction with fibrin glue-treated skin grafts. Arch. Surg. 1992, 127, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.; Redden, R.; Blum, B.; Osborne, S. Porous fibrin as a degradable adhesive and drug delivery system. Trans. Soc. Biomater. 1997, 23, 185. [Google Scholar]
- Flahiff, C. Mechanical Testing of Fibrin Adhesives for Blood Vessel Anastomosis. Master’s Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1990. [Google Scholar]
- Flahiff, C.; Feldman, D.; Saltz, R.; Huang, S. Mechanical testing of fibrin adhesives for blood vessel anastomosis. J. Biomater. Mater. Res. 1992, 26, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, C. Fibrin sealants for haemostasis and drug delivery. Lancet 1997, 349, 334. [Google Scholar] [CrossRef]
- Huang, S.; Kilpadi, D.; Feldman, D. A comparison of the shear strength of fibrin and albumin glues. Trans. Wound Heal. Soc. 1997, 7, 63. [Google Scholar]
- Slimane, S.B.; Guidoin, R.; Mourad, W.; Hebert, J.; King, M.W.; Sigot-Luizard, M.F. Polyester arterial grafts impregnated with cross-linked albumin: The rate of degradation of the coating in vivo. Eur. J. Surg. Res. 1998, 20, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Slimane, S.B.; Guidoin, R.; Marceau, D.; Merhi, Y.; King, M.W.; Sigot-Luizard, M.F. Characteristics of polyester arterial grafts coated with albumin: The role and importance of the cross-linking chemicals. Eur. J. Surg. Res. 1998, 20, 18–28. [Google Scholar] [CrossRef]
- Slimane, S.B.; Guidoin, R.; Merhi, Y.; King, M.W.; Domurado, D.; Sigot-Luizard, M.F. In vivo evaluation of polyester arterial grafts coated with albumin: The role and importance of cross-linking agents. Eur. J. Surg. Res. 1988, 20, 66–74. [Google Scholar] [CrossRef]
- Chafke, N.; Gasser, B.; Lindner, V.; Rouyer, N.; Rooke, R.; Kretz, J.G.; Nicolini, P.; Eisenmann, B. Albumin as a sealant for a polyester vascular prosthesis: Its impact on the healing sequence in humans. J. Cardiovasc. Surg. 1996, 37, 431–440. [Google Scholar]
- An, Y.H.; Stuart, G.W.; McDowell, S.J.; McDaniel, S.E.; Kang, Q.; Friedman, R.J. Prevention of bacterial adherence to implant surfaces with a crosslinked albumin coating in vivo. J. Orthop. Res. 1996, 14, 846–849. [Google Scholar] [CrossRef]
- An, H.; Bradley, J.; Powers, D.L.; Friedman, R.J. Preventing prosthetic infection using a crosslinked albumin coating in vivo. In Proceedings of the 5th World Biomaterials Conference, Toronto, ON, Canada, 29 May–2 June 1996. [Google Scholar]
- Weissleder, R.; Poss, K.; Wilkinson, R.; Zhou, C.; Bogdanov, A. Quantitation of slow drug release from an implantable and degradable gentamicin conjugate by in vivo magnetic resonance imaging. Antimicrob. Agents Chemother. 1995, 29, 839–845. [Google Scholar] [CrossRef]
- Osborne, S.; Pandit, A.; Feldman, D.; Thompson, J.A. The use of FGF-1 delivered through a porous scaffold to enhance meshed skin graft healing in a rabbit model. Wound Repair Regen. 1995, 3, 99. [Google Scholar]
- Sahni, A.; Odrljin, T.; Francis, C. Binding of basic fibroblast growth factor to fibrinogen and fibrin. J. Biol. Chem. 1997, 273, 7554–7559. [Google Scholar] [CrossRef]
- Pandit, A. Fibrin as a Matrix for FGF-1 Delivery In Vivo. Ph.D. Thesis, University of Alabama at Birmingham, Birmingham, AL, USA, 1998. [Google Scholar]
- France, R.; DeBloise, C.; Doillon, C. Extracellular matrix analogs as carriers for growth factors: in vitro fibroblast behavior. J. Biomed. Mater. Res. 1993, 27, 389–397. [Google Scholar]
- Sealy, M.; Guo, Y.; Caslaru, R.; Sharkins, J.; Feldman, D. Fatigue performance of biodegradable magnesium-calcium alloy processed by laser shock peening for orthopedic implants. Int. J. Fatigue 2016, 82, 428–436. [Google Scholar] [CrossRef]
- Staiger, M.P.; Pietak, A.M.; Huadmai, J.; Dias, G. Magnesium and its alloys as orthopedic biomaterials: A review. Biomaterials 2006, 27, 1728–1734. [Google Scholar] [CrossRef]
- Kirkland, N.T.; Birbilis, N. Magnesium Biomaterials: Design, Testing, and Best Practice; Springer International: Cham, Switzerland, 2014. [Google Scholar]
- Kaltenthaler, E.; Withfield, M.D.; Walters, S.J.; Akehurst, R.L.; Paisley, S. UK, USA and Canada: How do their pressure ulcer prevalence and incidence data compare? J. Wound Care 2001, 10, 530–535. [Google Scholar] [CrossRef]
- Braden, B. Costs of Pressure Ulcer Prevention. National Pressure Ulcer Advisory Panel. 2012. Available online: http://www.npuap.org (accessed on 7 January 2018).
- Weigand, A.; Beier, J.P.; Hess, A.; Gerber, T.; Arkudas, A.; Horch, R.E.; Boos, A.M. Acceleration of vascularized bone tissue-engineered constructs in a large animal model combining intrinsic and extrinsic vascularization. Tissue Eng. Part A 2015, 21, 1680–1694. [Google Scholar] [CrossRef]
- Meinel, L.; Karageorgiou, V.; Fajardo, R.; Snyder, B.; Shinde-Patil, V.; Zichner, L.; Kaplan, D.; Langer, R.; Vunjak-Novakovic, G. Bone tissue engineering using human mesenchymal stem cells: Effects of scaffold material and medium flow. Ann. Biomed. Eng. 2004, 32, 112–122. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Z.; Smith, C.; Sankar, J. Recent advances on the development of magnesium alloys for biodegradable implants. Acta Biomater. 2014, 10, 4561–4573. [Google Scholar] [CrossRef]
- Witte, F.; Hort, N.; Vogt, C.; Cohen, S.; Kainer, K.U.; Willumeit, R.; Feyerabend, F. Degradable biomaterials based on magnesium corrosion. Curr. Opin. Solid State Mater. Sci. 2008, 12, 63–72. [Google Scholar] [CrossRef]
- Xin, Y.; Hu, T.; Chu, P.K. In vitro studies of biomedical magnesium alloys in a simulated physiological environment: A review. Acta Biomater. 2011, 7, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, N.; Bondarenko, A.; Hewicker-Trautwein, M.; Angrisani, N.; Reifenrath, J.; Lucas, A.; Meyer-Lindenberg, A. Evaluation of the soft tissue biocompatibility of MgCa0.8 and surgical steel 316L in vivo: A comparative study in rabbits. BioMed. Eng. OnLine 2010, 9, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Parker, P.M. The 2009–2014 World Outlook for Medical and Surgical Bone Nails, Plates, and Screws and Other Internal Fixation Devices; ICON Group International, Inc.: San Diego, CA, USA, 2008; pp. 16–17. [Google Scholar]
- Elder, M. Market Research Report: Advanced Orthopedic Technologies, Implants and Regenerative Products; HLC052B; BCC Research: Wellesley, MA, USA, 2010. [Google Scholar]
- Anscomb, A. Orthopedic Biomaterials: World Market; KLI1399489; Kalorama Information: New York, NY, USA, 2007. [Google Scholar]


{kind=link}
{kind=link}
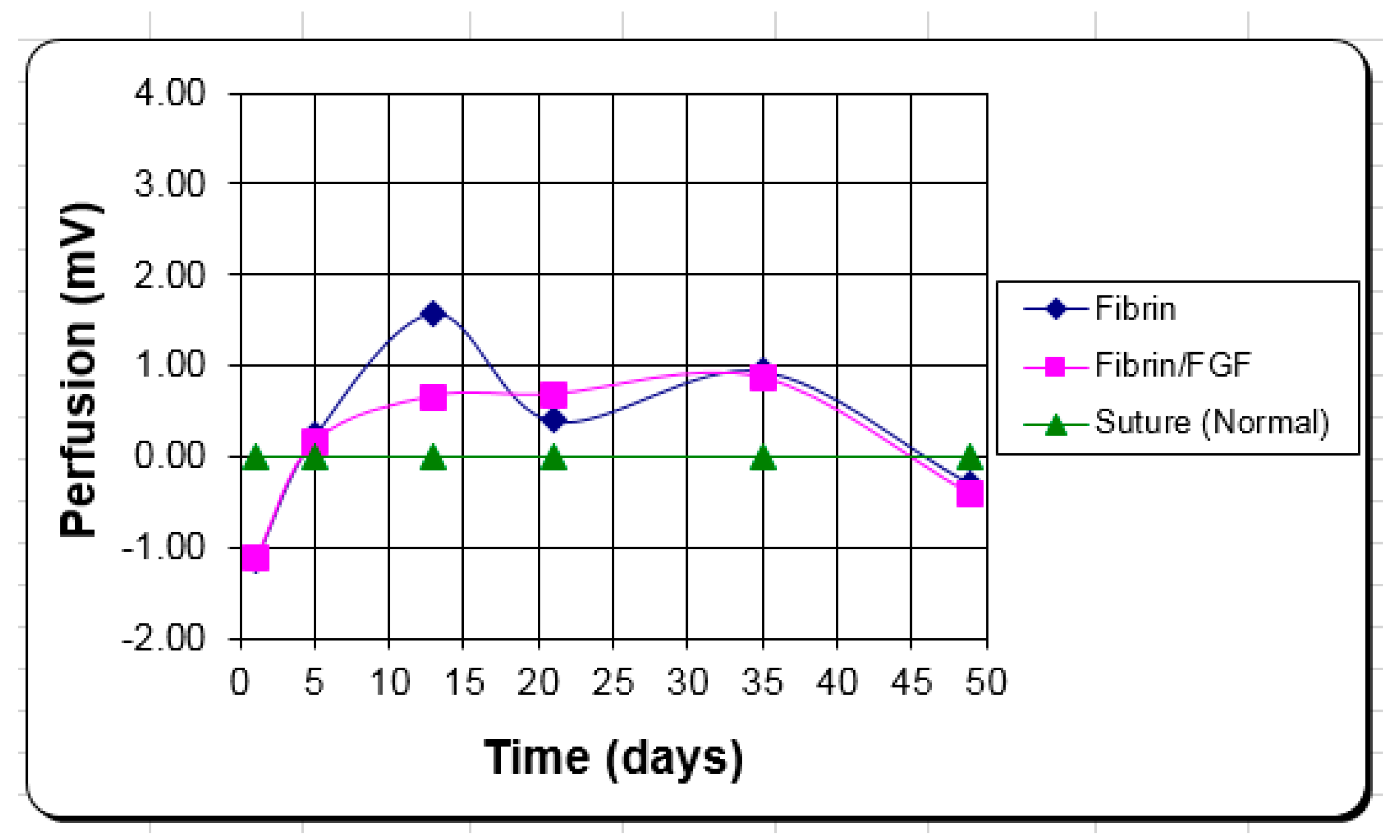{kind=link}
| Host | Implant |
|---|---|
| Regeneration | Degradable |
| Integration | Bioactive |
| Minimal Inflammation | Inert |
| Inert | - |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feldman, D.S. Biomaterial Enhanced Regeneration Design Research for Skin and Load Bearing Applications. J. Funct. Biomater. 2019, 10, 10. https://doi.org/10.3390/jfb10010010
Feldman DS. Biomaterial Enhanced Regeneration Design Research for Skin and Load Bearing Applications. Journal of Functional Biomaterials. 2019; 10(1):10. https://doi.org/10.3390/jfb10010010
Chicago/Turabian StyleFeldman, Dale S. 2019. "Biomaterial Enhanced Regeneration Design Research for Skin and Load Bearing Applications" Journal of Functional Biomaterials 10, no. 1: 10. https://doi.org/10.3390/jfb10010010
APA StyleFeldman, D. S. (2019). Biomaterial Enhanced Regeneration Design Research for Skin and Load Bearing Applications. Journal of Functional Biomaterials, 10(1), 10. https://doi.org/10.3390/jfb10010010