Role of Aspartic and Polyaspartic Acid on the Synthesis and Hydrolysis of Brushite




Abstract
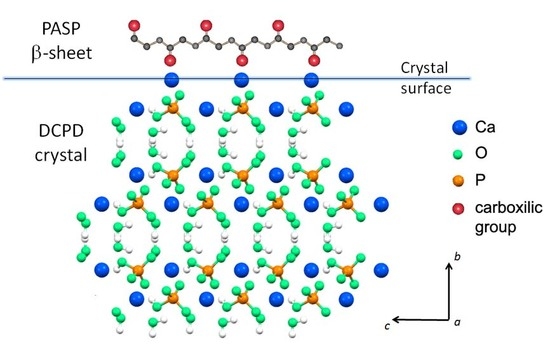
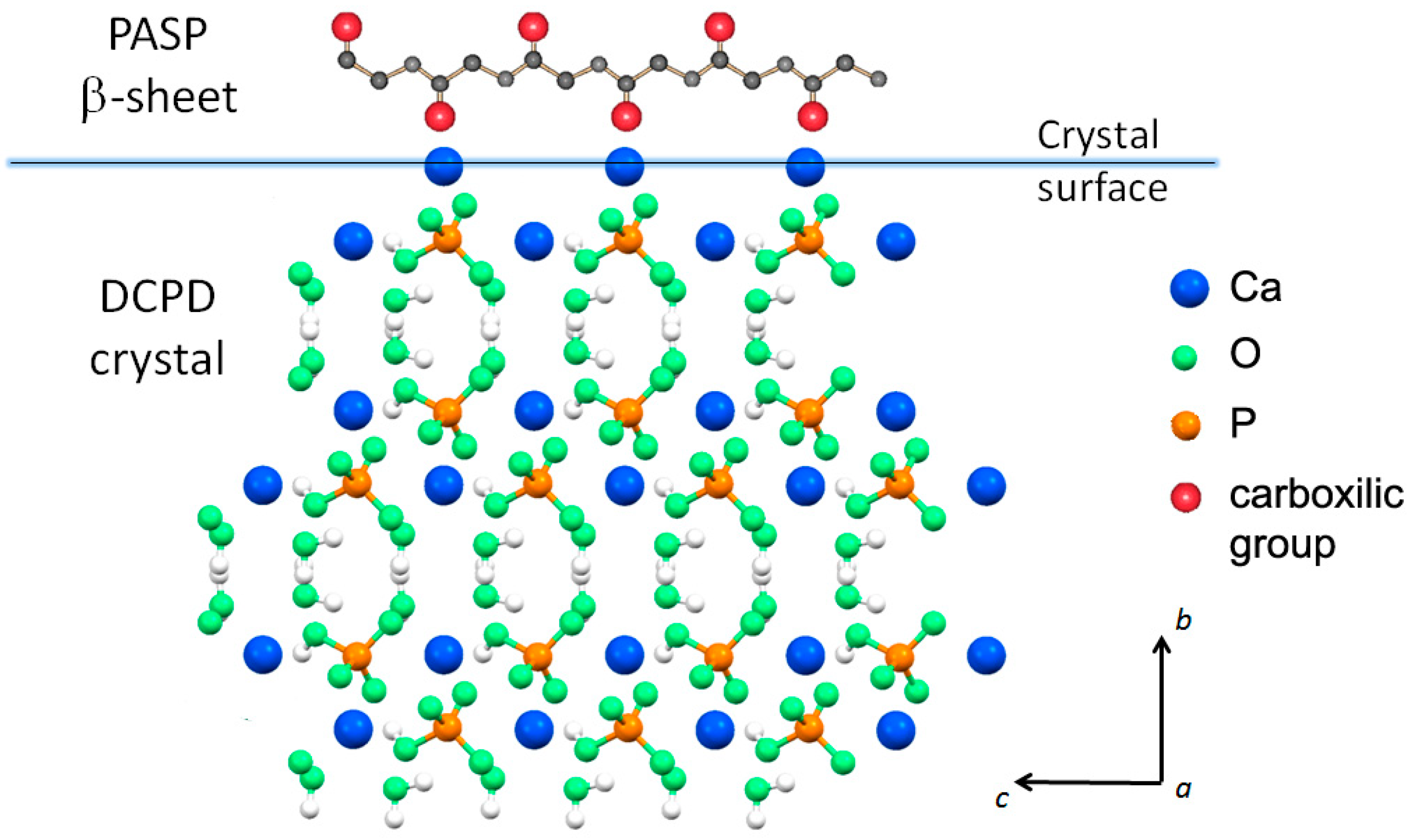
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Hydrolysis Procedures
2.2. Characterization
3. Results
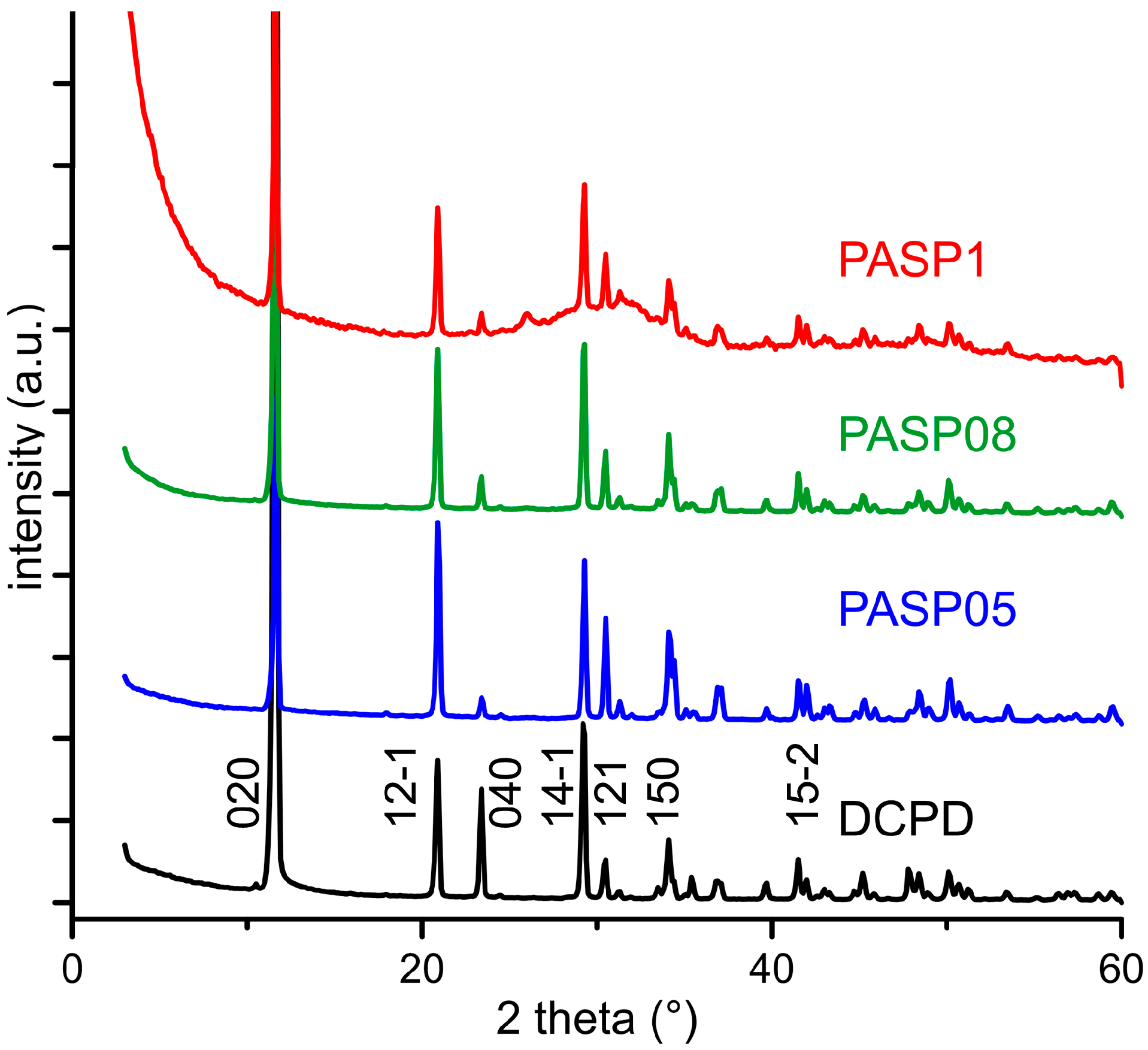
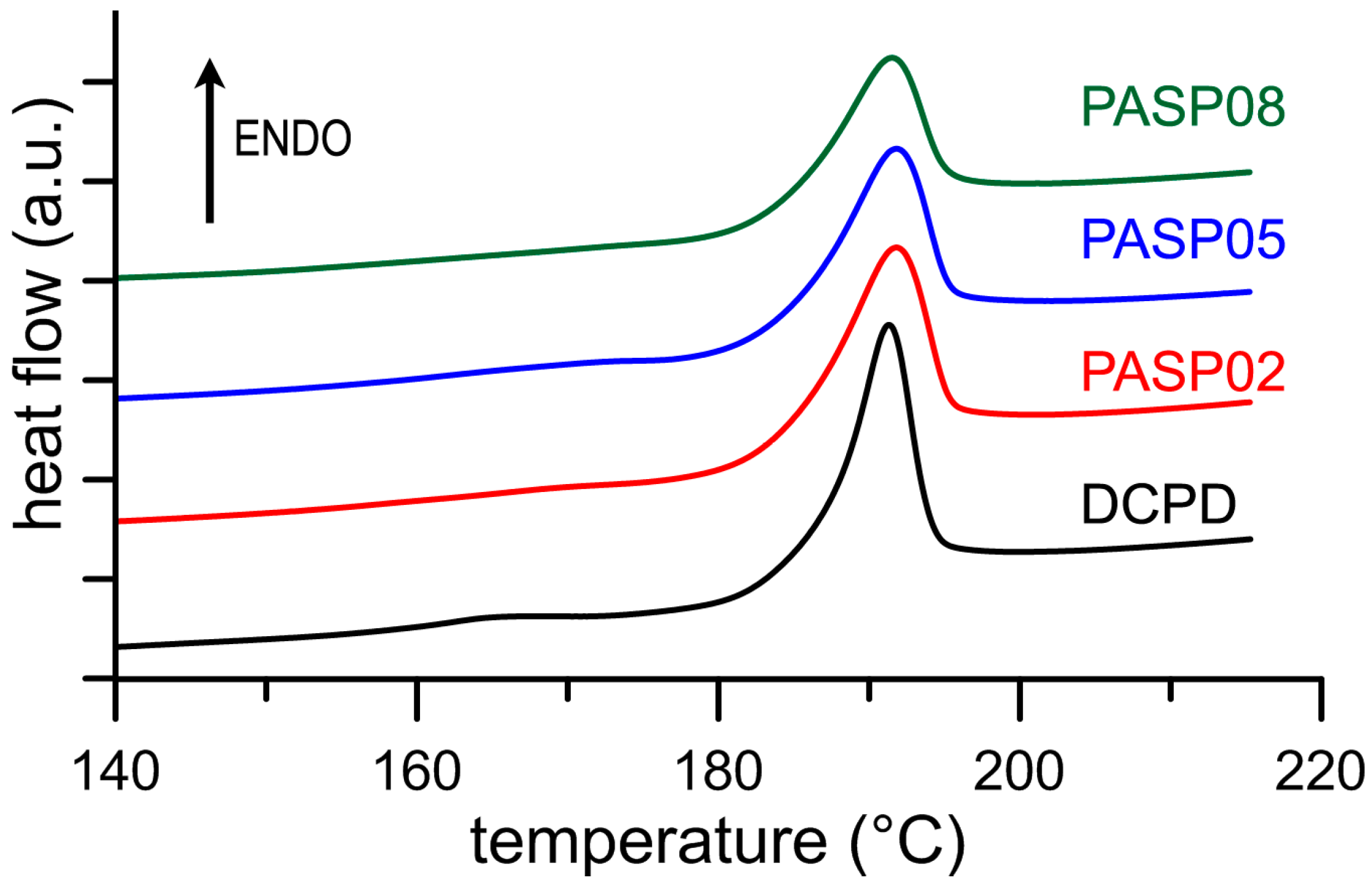
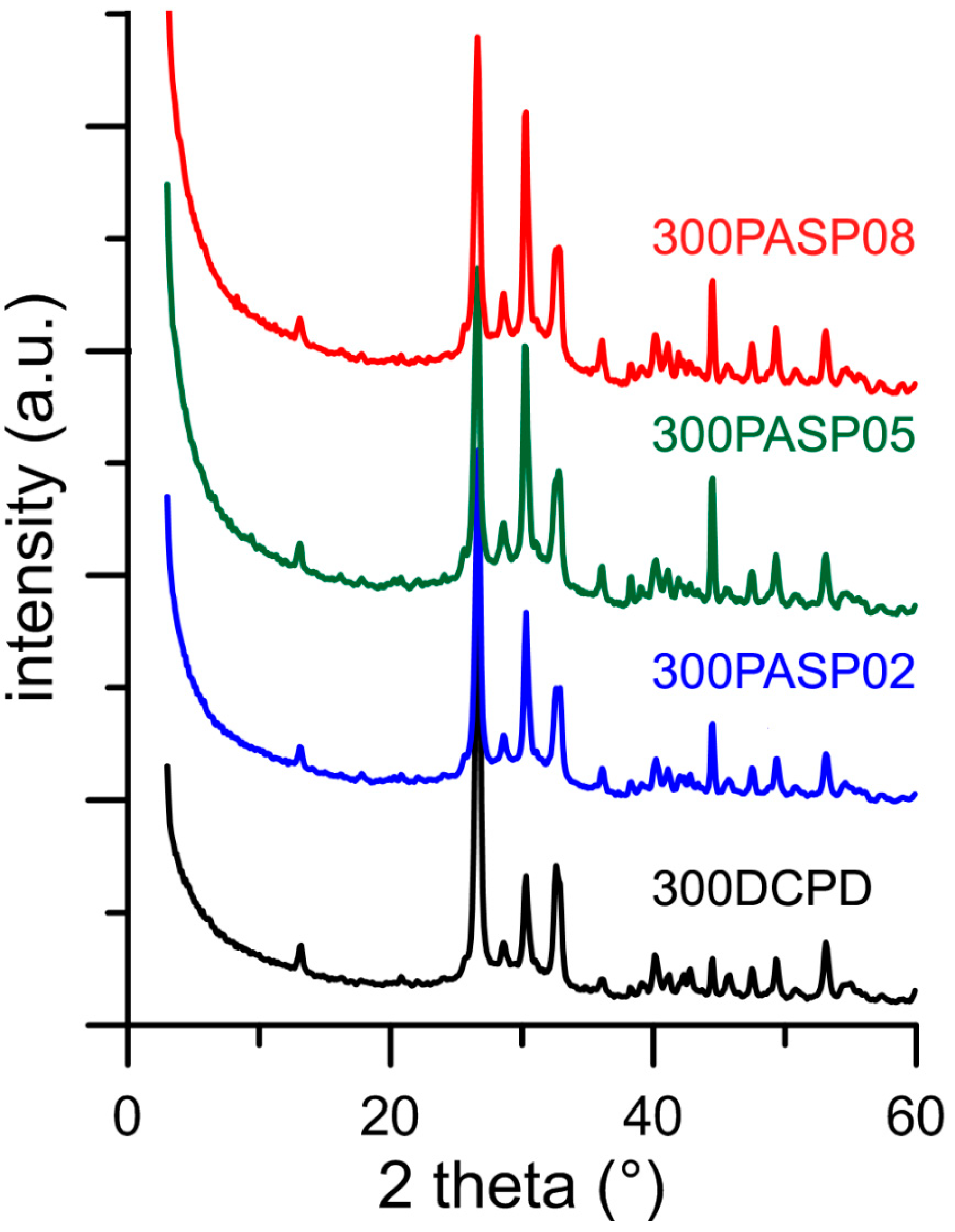
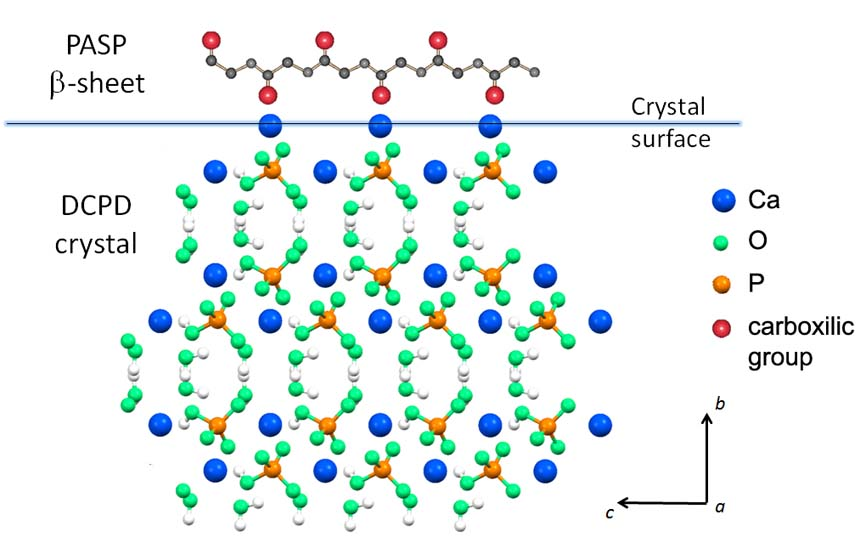
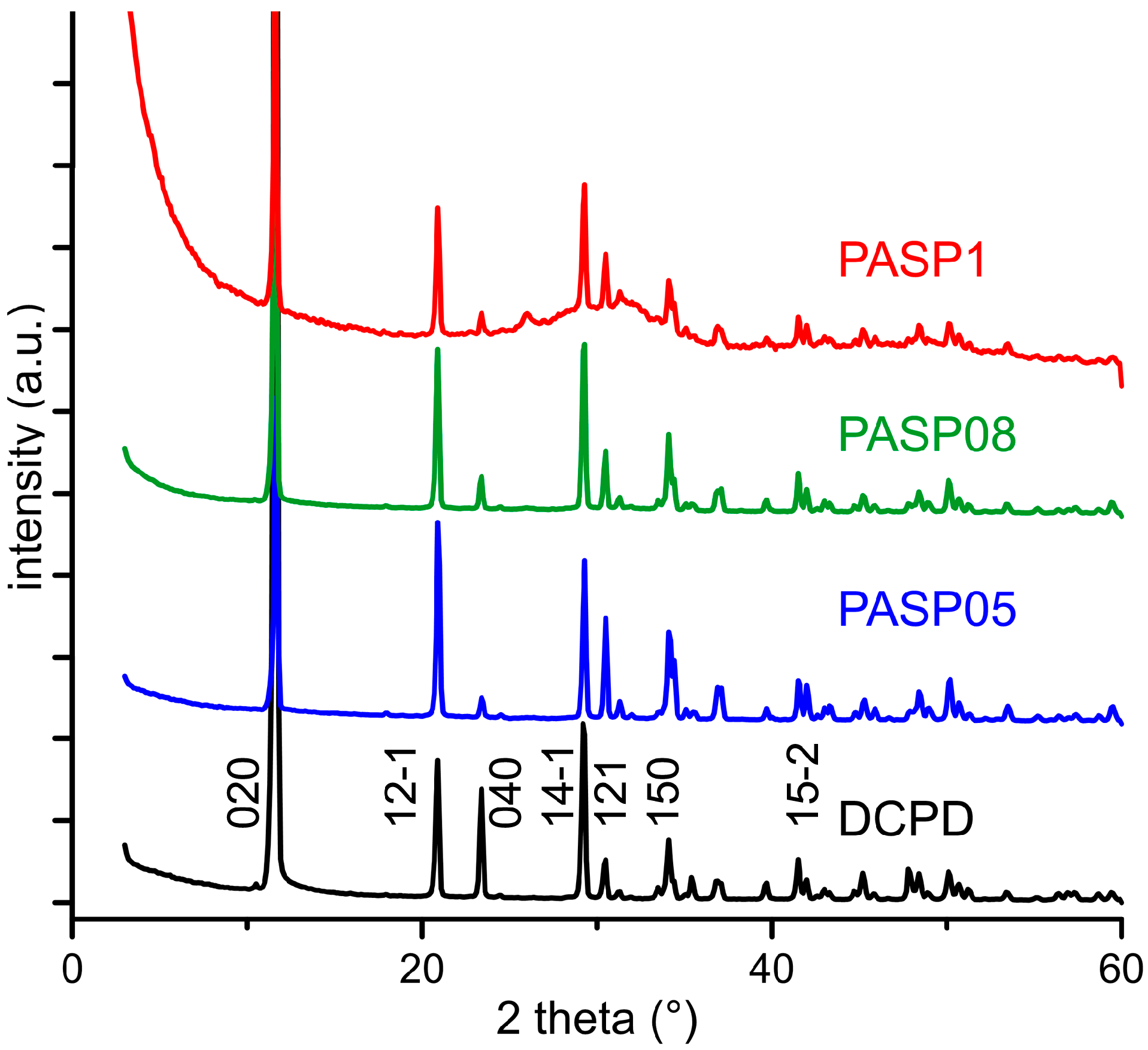
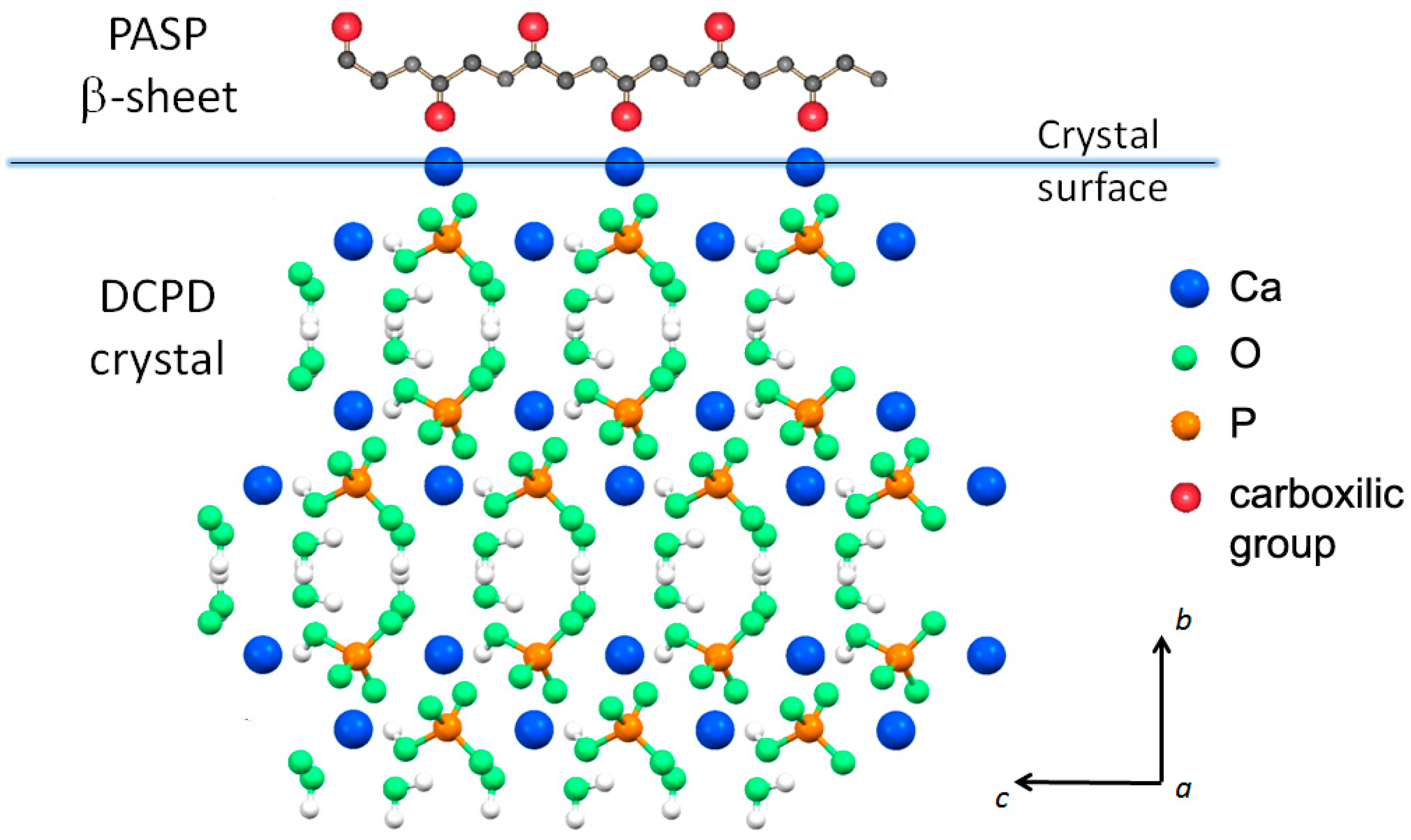
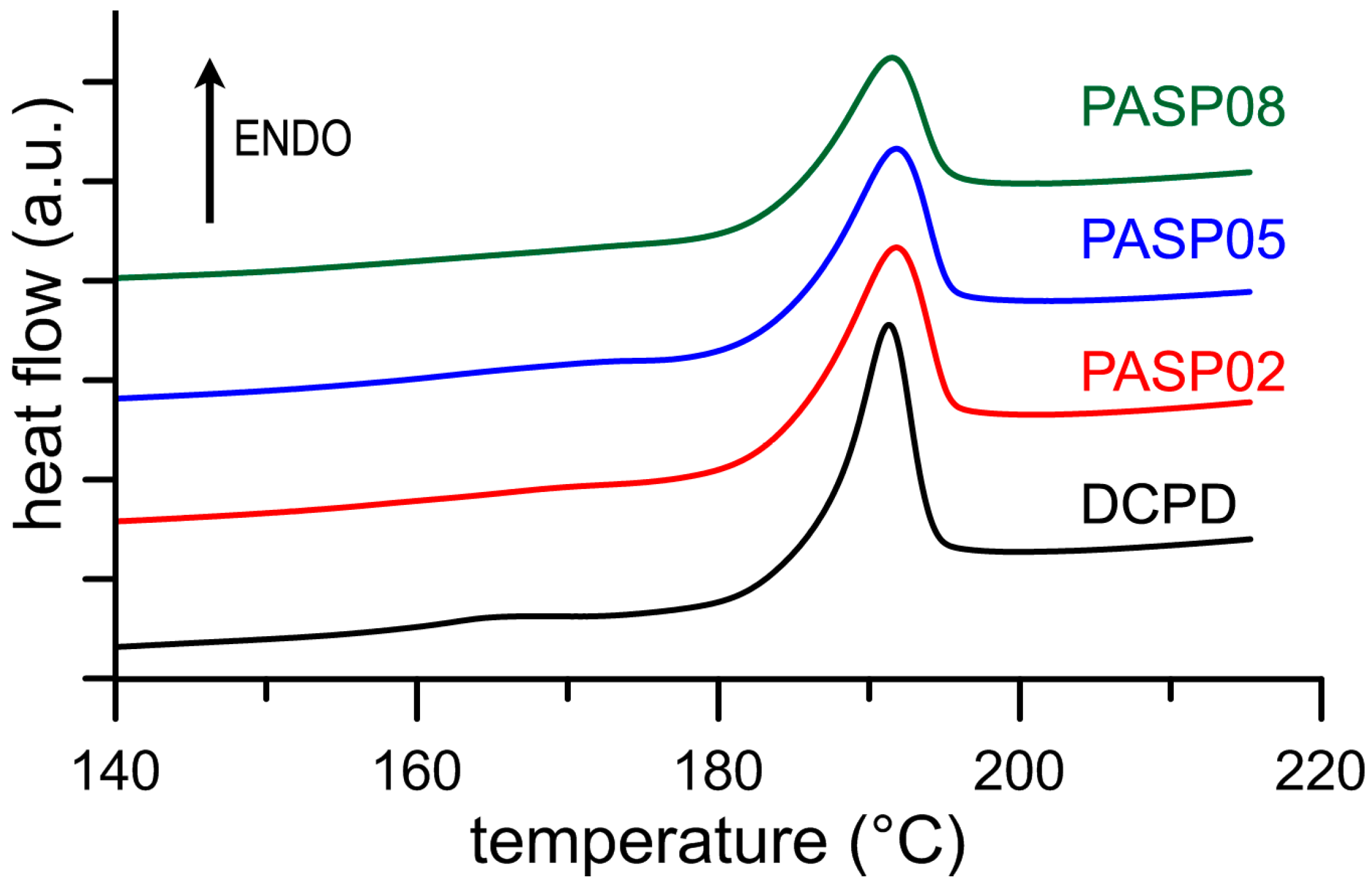
3.1. Synthesis of DCPD in the Presence of PASP
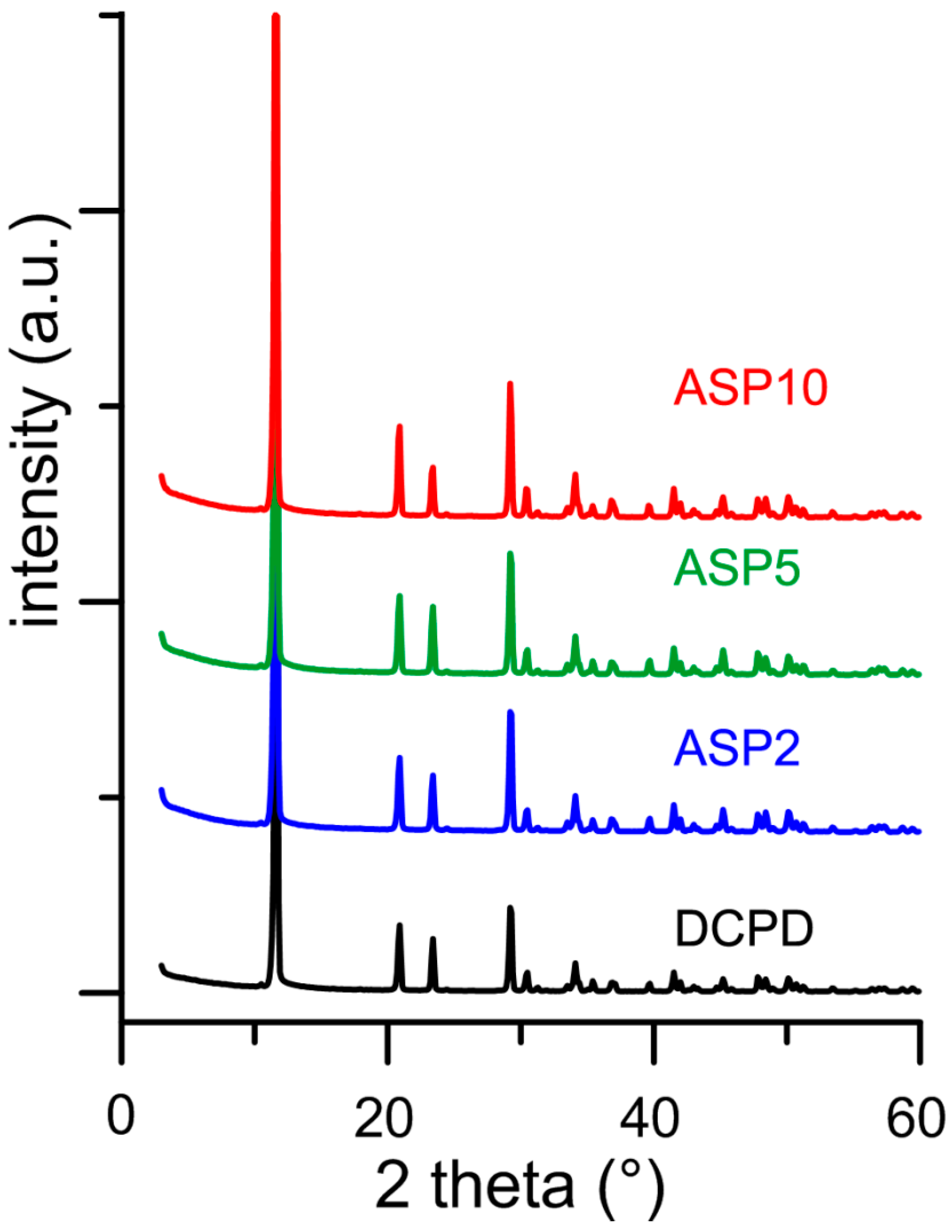
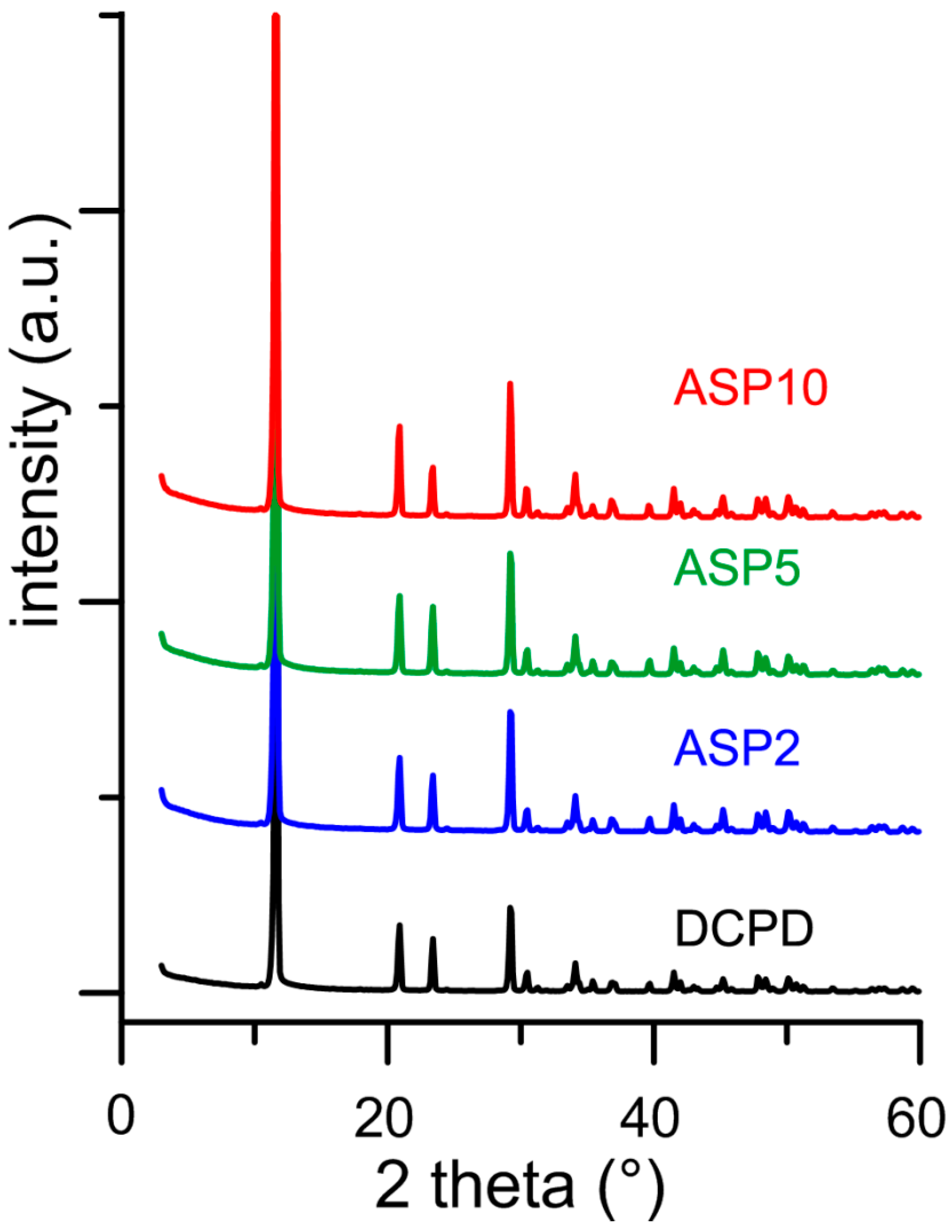
3.2. Synthesis of DCPD in the Presence of ASP
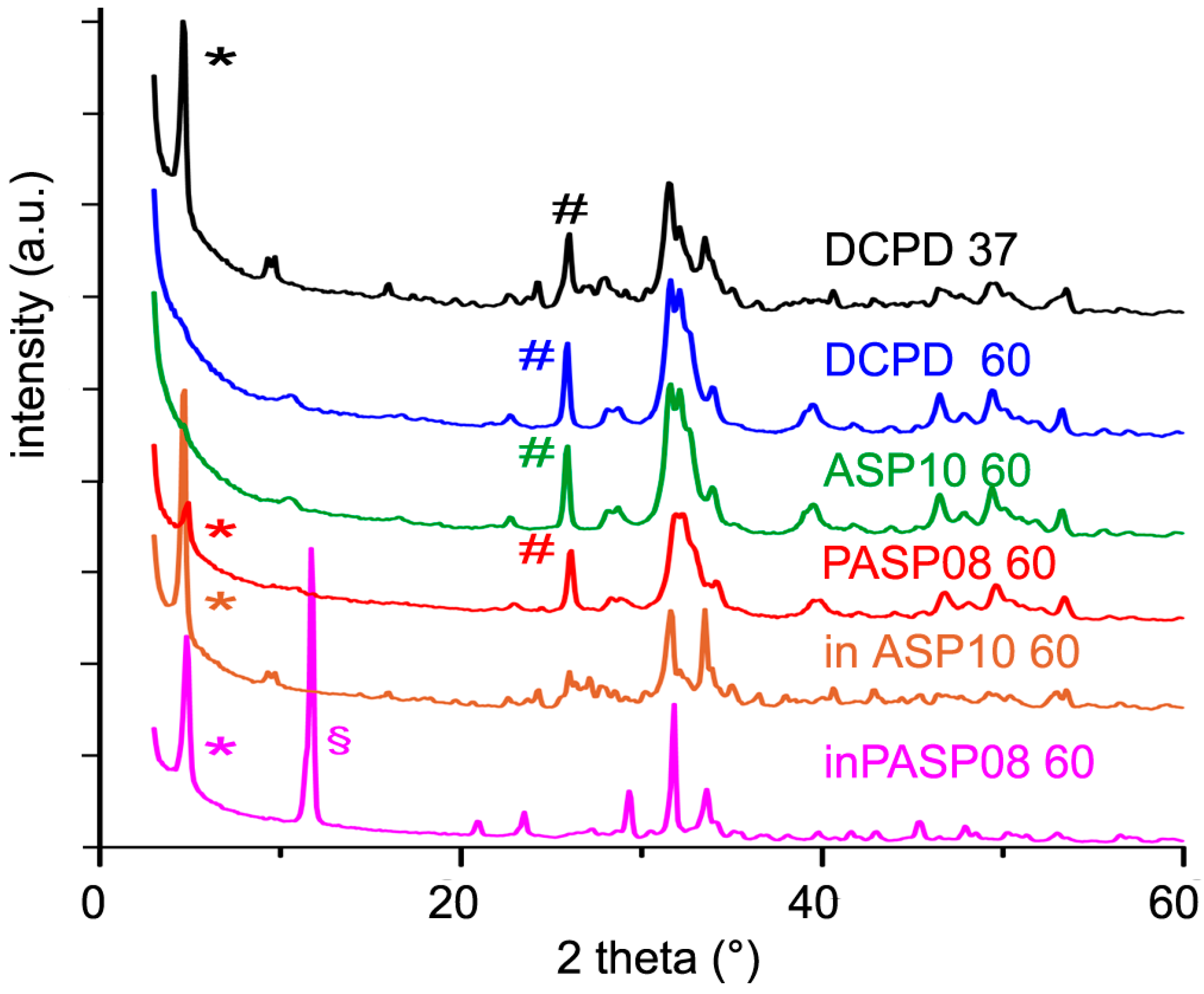
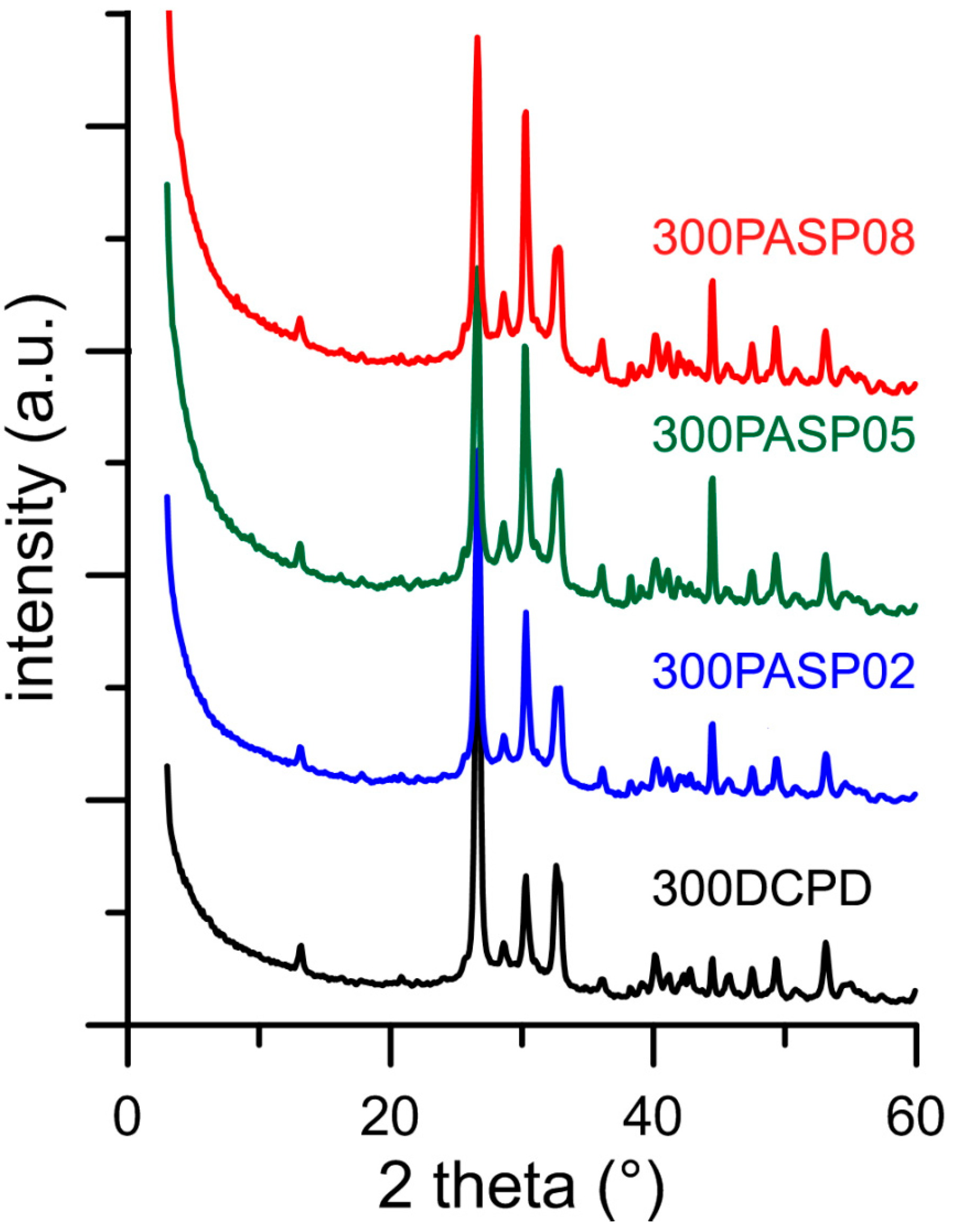
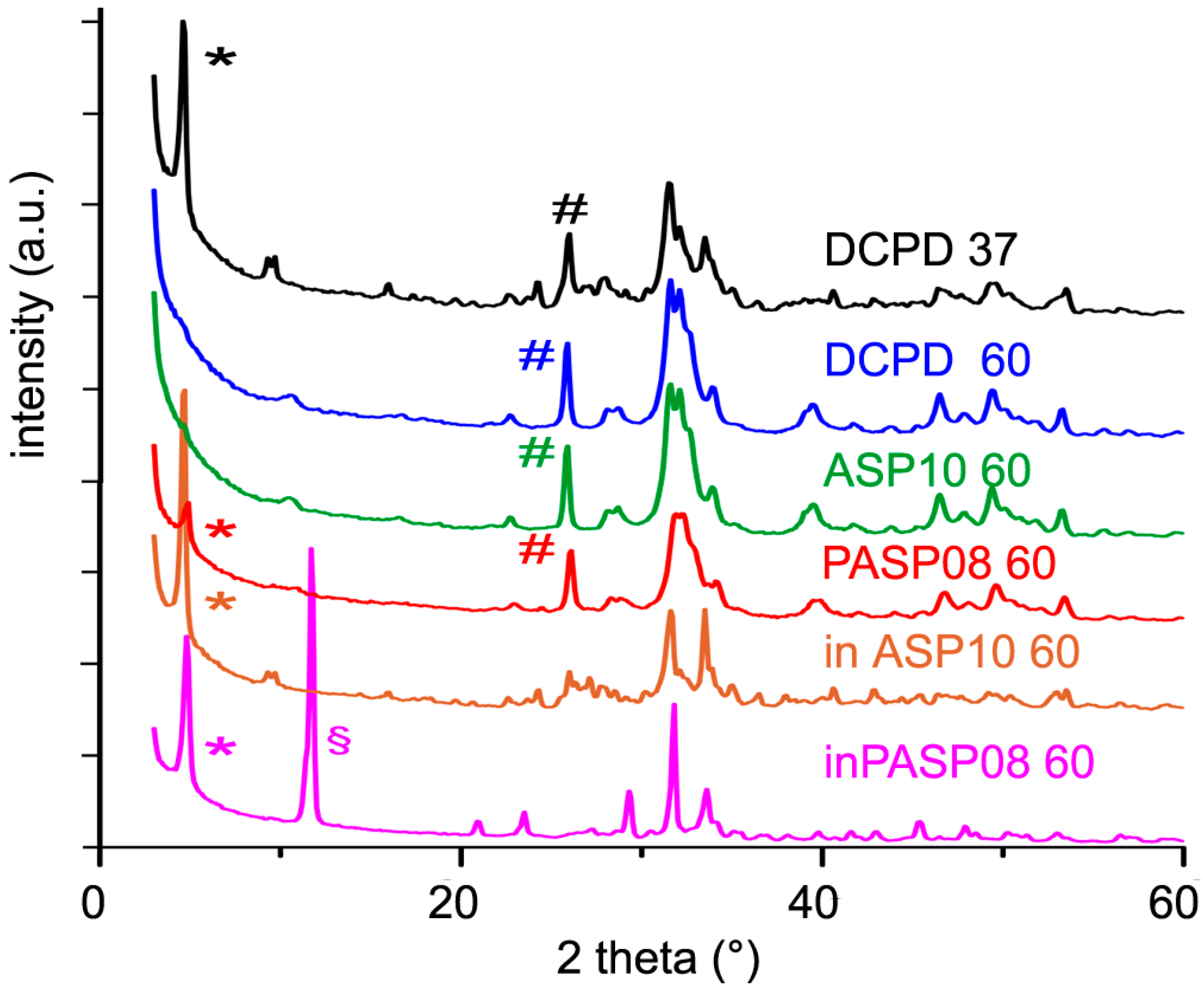
3.3. Hydrolysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lowenstam, H.A.; Weiner, S. Transformation of amorphous calcium phosphate to crystalline dahllite in the radular teeth of chitons. Science 1985, 227, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Boanini, E. Calcium phosphates as delivery systems for bisphosphonates. J. Funct. Biomater. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.L.; Chen, X.R.; Bian, S.; Huang, J.; Zhang, T.L.; Wang, K. Identification of dicalcium phosphate dihydrate deposited during osteoblast mineralization in vitro. J. Inorg. Biochem. 2014, 131, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.C. Structure and Chemistry of the Apatites and Other Calcium Orthophosphates; Elsevier: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Fulmer, M.T.; Brown, P.W. Hydrolysis of dicalcium phosphate dihydrate to hydroxyapatite. J. Mater. Sci. Mater. Med. 1998, 9, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Štulajterová, R.; Medvecký, Ľ. Effect of calcium ions on transformation brushite to hydroxyapatite in aqueous solutions. Colloid Surf. A Physicochem. Eng. Asp. 2008, 316, 104–109. [Google Scholar] [CrossRef]
- Tas, A.C.; Bhaduri, S.B. Chemical processing of CaHPO4·2H2O: Its conversion to hydroxyapatite. J. Am. Ceram. Soc. 2004, 87, 2195–2200. [Google Scholar] [CrossRef]
- Obadia, L.; Rouillon, T.; Bujoli, B.; Daculsi, G.; Bouler, J.M. Calcium-deficient apatite synthesized by ammonia hydrolysis of dicalcium phosphate dihydrate: Influence of temperature, time, and pressure. J. Biomed. Mater. Res. Part B: Appl. Biomater. 2007, 80, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Tavafoghi, M.; Cerruti, M. The role of amino acids in hydroxyapatite mineralization. J. R. Soc. Interface 2016, 13, 20160462. [Google Scholar] [CrossRef] [PubMed]
- Jack, K.S.; Vizcarra, T.G.; Trau, M. Characterization and surface properties of amino-acid-modified carbonate-containing hydroxyapatite particles. Langmuir 2007, 23, 12233–12242. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-McQuire, R.; Chane-Ching, J.Y.; Vignaud, E.; Lebugle, A.; Mann, S. Synthesis and characterization of amino acid-functionalized hydroxyapatite nanorods. J. Mater. Chem. 2004, 14, 2277–2281. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E. Functionalized biomimetic calcium phosphates for bone tissue repair. J. Appl. Biomater. Funct. Mater. 2017, 15, e313–e325. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Boanini, E.; Gazzano, M.; Rubini, K.; Torricelli, P. Nanocrystalline hydroxyapatite-polyaspartate composites. Bio-Med. Mater. Eng. 2004, 14, 573–579. [Google Scholar]
- Sikirić, M.; Babić-Ivančić, V.; Milat, O.; Sarig, S.; Füredi-Milhofer, H. Factors influencing additive interactions with calcium hydrogenphosphate dihydrate crystals. Langmuir 2000, 16, 9261–9266. [Google Scholar] [CrossRef]
- Bigi, A.; Bracci, B.; Panzavolta, S.; Iliescu, M.; Plouet-Richard, M.; Werckmann, J.; Cam, D. Morphological and structural modifications of octacalcium phosphate induced by poly-l-aspartate. Cryst. Growth Des. 2004, 4, 141–146. [Google Scholar] [CrossRef]
- Gomez-Morales, J.; Delgado-Lopez, J.M.; Iafisco, M.; Hernandez-Hernandez, A.; Prat, M. Amino acidic control of calcium phosphate precipitation by using the vapor diffusion method in microdroplets. Cryst. Growth Des. 2011, 11, 4802–4809. [Google Scholar] [CrossRef]
- Landin, M.; Rowe, R.C.; York, P. Structural changes during the dehydration of dicalcium phosphate dihydrate. Eur. J. Pharm. Sci. 1994, 2, 245–252. [Google Scholar] [CrossRef]
- Xu, J.; Butler, I.S.; Gilson, D.F.R. FT-Raman and High-Pressure Infrared Spectroscopic Studies of CaHPO4_2H2O and CaHPO4. Spectrochim. Acta 1999, A55, 2801–2809. [Google Scholar] [CrossRef]
- Boanini, E.; Fini, M.; Gazzano, M.; Bigi, A. Hydroxyapatite nanocrystals modified with acidic amino acids. Eur. J. Inorg. Chem. 2006, 23, 4821–4826. [Google Scholar] [CrossRef]
- Fedotov, A.Y.; Komlev, V.S.; Teterina, A.Y.; Sirotinkin, V.P.; Shamrai, V.F.; Fadeeva, I.V.; Barinov, S.M. Preparation of octacalcium phosphate from calcium carbonate powder. Inorg. Mater. 2013, 49, 1148–1151. [Google Scholar] [CrossRef]
- Shamrai, V.F.; Karpikhin, A.E.; Fedotov, A.Y.; Sirotinkin, V.P.; Barinov, S.M.; Komlev, V.S. Structural changes during the hydrolysis of dicalcium phosphate dihydrate to octacalcium phosphate and hydroxyapatite. Inorg. Mater. 2015, 51, 355–361. [Google Scholar] [CrossRef]
- Fowler, B.O.; Marković, M.; Brown, W.E. Octacalcium phosphate. 3. Infrared and raman vibrational spectra. Chem. Mater. 1993, 5, 1417–1423. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Bracci, B.; Falini, G.; Rubini, K. Interaction of acidic poly-amino acids with octacalcium phosphate. J. Inorg. Biochem. 2003, 95, 291–296. [Google Scholar] [CrossRef]
- Ginebra, M.P.; Canal, C.; Espanol, M.; Pastorino, D.; Montufar, E.B. Calcium phosphate cements as drug delivery materials. Adv. Drug. Deliv. Rev. 2012, 64, 1090–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, W.; Schnitzler, V.; Tancret, F.; Bouler, J.M. Calcium phosphate cements for bone substitution: Chemistry, handling and mechanical properties. Acta Biomater. 2014, 10, 1035–1049. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mass (g) | ΔH (J/g) | Zeta Potential (mV) | PASP Content (wt%) |
|---|---|---|---|---|
| DCPD | 1.146 | 400 | −12.0 | --- |
| PASP0.2 | 0.691 | 326 | −23.0 | 0.4 |
| PASP0.5 | 0.481 | 302 | −21.1 | 1.0 |
| PASP0.8 | 0.220 | 216 | −23.3 | 2.3 |
| ASP5 | 1.151 | 406 | −11.6 | --- |
| ASP10 | 1.134 | 400 | −12.5 | --- |
| T = 37 °C | 3 h | 6 h | 9 h | 15 h | 24 h | 48 h | 72 h |
|---|---|---|---|---|---|---|---|
| DCPD | D + O | D + O | D + O | D + O | D + O | O | O + H |
| PASP08 | D + O | D + O | D + O | D + O | D + O | D + O | D + O |
| inPASP08 | D | D | D | D | D | D | D + O |
| ASP10 | D + O | D + O | D + O | D + O | D + O | O | O + H |
| inASP10 | D | D | D | D | D | D | D + O |
| T = 60 °C | 3 h | 6 h | 9 h | 15 h | 24 h | 48 h | 72 h |
|---|---|---|---|---|---|---|---|
| DCPD | O + D + H | O + H | O + H | O + H | H + O | H | H |
| PASP08 | D + O + H | D + O + H | D + O + H | H + O + D | H + O + D | H + O + D | H + O |
| inPASP08 | D + OCP | D + O | D + O | D + O | O + D | O + D | O + D |
| ASP10 | O + D + H | O + H | O + H | O + H | H + O | H | H |
| inASP10 | D + O | O + D | O | O + D | O | O | O |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubini, K.; Boanini, E.; Bigi, A. Role of Aspartic and Polyaspartic Acid on the Synthesis and Hydrolysis of Brushite. J. Funct. Biomater. 2019, 10, 11. https://doi.org/10.3390/jfb10010011
Rubini K, Boanini E, Bigi A. Role of Aspartic and Polyaspartic Acid on the Synthesis and Hydrolysis of Brushite. Journal of Functional Biomaterials. 2019; 10(1):11. https://doi.org/10.3390/jfb10010011
Chicago/Turabian StyleRubini, Katia, Elisa Boanini, and Adriana Bigi. 2019. "Role of Aspartic and Polyaspartic Acid on the Synthesis and Hydrolysis of Brushite" Journal of Functional Biomaterials 10, no. 1: 11. https://doi.org/10.3390/jfb10010011
APA StyleRubini, K., Boanini, E., & Bigi, A. (2019). Role of Aspartic and Polyaspartic Acid on the Synthesis and Hydrolysis of Brushite. Journal of Functional Biomaterials, 10(1), 11. https://doi.org/10.3390/jfb10010011