Abstract
Deep-sea eukaryotic microorganisms play a vital role in degrading organic matter and geochemically cycling elements in the deep ocean. However, the impact of sampling methods on detection of these microorganisms under high hydrostatic pressure remains uncertain. In this study, we compared a traditional water sampling method using a Niskin bottle, an in situ microbial filtration and fixation method (ISMIFF), and a multiple in situ nucleic acid collection (MISNAC) method to exhibit differences in the community structures that were collected at ~590–3100 m in the South China Sea (SCS). The classification and biodiversity indices of 18S rDNA Illumina sequencing reads from the V9 variation region revealed higher diversity for MISNAC DNA absorption column samples compared to others. Importantly, the relative abundance of Ciliophora (19.49%), Copepoda (23.31%), and Diplonemea (10.67%) was higher in MISNAC adsorption column samples, while Retaria (48.86%) were dominant in the MISNAC membrane samples. This indicates that MISNAC columns might collect more DNA in situ for the naked protists, while Retaria with a carbonate shell were more likely retained on the membrane. In conclusion, MISNAC is an effective method for DNA collection of deep-sea eukaryotic microorganisms and provides valuable materials for studying deep-sea microbial ecosystems.
1. Introduction
The deep ocean is a vast habitat for microbial eukaryotes that recycle nutrients from the overlying water column and consume prokaryotes to maintain a healthy marine ecosystem. These microorganisms are also exposed to the mounting anthropogenic pressures resulting from climate change, mining, and bottom-trawling activities [1]. Most of the microbial eukaryotes are difficult to cultivate [2], and the limited availability of samples renders investigations of the microbial eukaryotic community structures in the dark ocean sporadic. The development of metagenomics has remarkably renewed our understanding of the microbial world in the ocean, particularly for viruses and prokaryotes [3]. Using high-throughput sequencing of tagged 16S and 18S rRNA gene amplicons, ‘dark matter’ microbes have been revealed at different depths and locations in global oceans [2,4]. It is estimated that eukaryotic microbial richness in the deep sea might be at least three fold higher than in pelagic layers [5]. The study of deep-sea eukaryotic microorganisms is of significant importance for understanding the evolution of eukaryotes [5] and the adaptive mechanisms of microorganisms in extreme environments [6,7]. The major eukaryotic orders in euphotic and aphotic zones of global oceans are Dinophyceae and Eupelagonemidae, respectively [5]. However, the majority of the eukaryotic microbes in deep-sea zones are still unknown in terms of taxonomy and genomic features [8].
Some eukaryotic groups might be difficult to detect by sequencing tagged 18S rRNA gene amplicons, as primers introduce biases in the amplification process to some extent [9]. The selection of different primers results in different classification resolutions of eukaryotes [10]. Primer-independent detection of environmental prokaryotes using 16S rRNA gene Illumina reads has been applied to reveal community structures in deep-sea samples [11,12]. However, 18S rRNA gene Illumina reads (18S miTags) are rarely used for the study of eukaryotic microbial community structure in the deep ocean [4]. Additionally, eukaryotic microbial communities, as well as their morphology and metabolism in deep-sea waters, might be altered during the sampling and filtration steps that accompany environmental changes in pressure, temperature, optical density, and oxygen flux [13]. Particularly, when the samples are being recovered on board, rapidly decreasing hydrostatic pressure leads to the dissolution of gases inside cells of deep-sea eukaryotic microbes. This likely results in the explosion or partial damage of some eukaryotic naked microbes with a single-layered soft membrane. Therefore, it is hypothesized that the seawater samples obtained by conventional sampling devices cannot meet the requirement for research on eukaryotic microbial diversity and community structure in deep seawater environments [14]. Therefore, in order to deeply explore a complete eukaryotic microbial community in deep sea water and elaborate the role of microorganisms in the deep-sea nutrient cycle, a new deep-sea microbial sampling device (multiple in situ nucleic acid collection (MISNAC)) was developed for multiple in situ microbial filtration on membranes and nucleic acid collection using absorption columns [15], which may avoid DNA loss from damaged eukaryotic cells during sampling from the deep ocean. MISNAC has been evaluated in a study of prokaryotic microbial communities and time-course metabolism of the major deep-sea degraders from SAR202 [15,16]. In the mesopelagic and bathypelagic zones of the Mediterranean Sea, an in situ incubation device, MS-SID, was deployed to quantify phagotrophic activities of deep-sea protists, which could avoid sampling artifacts from sites with high levels of chemocline [17]. The ‘dark matter’ eukaryotic microbes that are likely lost during sampling can probably be recovered by MISNAC, as it can theoretically collect all the DNA of deep-sea eukaryotic microbes.
In order to deeply understand the dynamic changes in eukaryotic microbial community composition and in the deep SCS, we employed MISNAC to collect in situ DNA at different time points from microbes in the deep water of the continental slope with a water depth of 1000 m in the SCS. At a nearby site, we also obtained samples with Niskin bottles for comparison. Based on the analysis of 18S miTags, the eukaryotic microbial communities of the MISNAC samples on a DNA absorption column were found to contain high proportions of Ciliophora, Copepoda, and Diplonemea, which were significantly higher than those in Niskin and MISNAC in situ preserved and lysis samples. Our result indicates that some eukaryotic microorganisms in deep-sea water columns have been missed or underestimated in terms of their abundance in previous studies due to sampling artifacts.
2. Materials and Methods
2.1. Sampling Using MISNAC Device
In two research cruises carried out in the SCS at depths of 591–3102 m in July 2018 and March 2022 (Figure 1 and Table S1), we deployed a deep-sea lander with a mounted MISNAC apparatus and two 10-L Niskin bottles. Ten minutes after the lander touched down on the sea floor, the MISNAC device began to collect samples automatically using the individual sets of filtration chambers and columns. During each cruise, the filtration chambers of MISNAC contained a polycarbonate membrane with a 142 mm diameter and a 0.22 μm pore size (Millipore, Bedford, MA, USA), each of which was connected to a DNA/RNA co-collection silica resin adsorption column (Tiangen, Beijing, China) for nucleic acid collection. The inlet water was prefiltered by a filter with a 1 mm pore size. After microbial filtration for 3 h (about 80 L of water), the filtration chambers were injected with 200 mL RNALater solution (TaKaRa, Dalian, China), cell lysis buffer, or nothing [15]. The microbes that had been lysed for 30 min on the membranes were then flooded with 500 mL lysis buffer and precipitated with 95% ethanol for nucleic acid extraction by the column. The columns and chambers were returned on board with the lander after 24 h. During each lander deployment, MISNAC collected six time-course membranes and column samples. At ~1000 m depth, some of the MISNAC membranes were not soaked with the cell lysis buffer and did not have a DNA collection column connected to the filtration chamber.
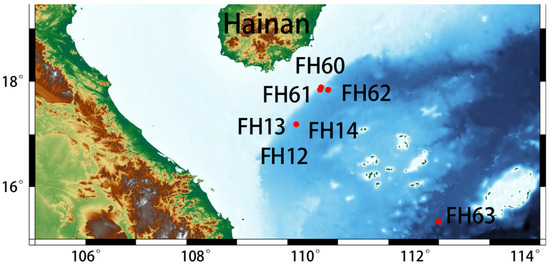
Figure 1.
Sampling sites in the South China Sea. FH12–14 were sampled in the summer of 2018; FH60–63 were sampled in the spring of 2022. The details of the deployment sites and samples collected by a deep-sea lander are shown in Table S1.
Proteins and ions attached to the adsorption columns were first removed by centrifugation at 4000 rpm; then, the nucleic acids on the adsorption columns were eluted by centrifugation at 4000 rpm with 1 mL of deionized distilled water. The concentration of the collected nucleic acid sample was measured by a Qubit 2.0 fluorometer (Life, Carlsbad, CA, USA). The water samples collected by the Niskin bottles were filtered through a 0.22 μm polycarbonate membrane (Millipore, Bedford, MA, USA). The collected nucleic acids and Niskin samples were immediately preserved at −80 °C for storage. In addition, the microbes on the membranes in the filtration chambers filled with RNALater and lysis buffer were transferred to a −80 °C refrigerator in the shipboard laboratory. The total DNA of the microbes on the polycarbonate membranes was extracted with a Mo Bio Powersoil DNA extraction kit (Qiagen, Carlsbad, CA, USA). The concentration of extracted DNA was determined using a Qubit 2.0 fluorometer (Life, Carlsbad, CA, USA).
In 2018 and 2022, MISNAC was used to collect microbial samples in body sizes spanning from 0.22 μm to 1 mm. Two samples preserved with RNALater were regarded as in situ microbial filtration and fixation samples (ISMIFF-1 and ISMIFF-2, respectively), and one unfixed sample was designated ISMIFF-N; the membranes with lysed microbes in the MISNAC filtration chambers were labeled as M-IN-Mn (n = 1–25); the time-course DNA samples collected from the absorption columns at different sites were denoted by M-IN-Cn (n = 1–16). In total, 40 MISNAC and 3 ISMIFF work units were completed at the deep-sea in situ sampling sites with a depth of ~590–3100 m (Figure 1 and Table S1). One Niskin sample was obtained as a control.
2.2. Construction and Sequencing of Metagenomic Library
The nucleic acid samples collected during the scientific expeditions in the SCS were incubated with RNAse (TaKaRa, Dalian, China) with a final concentration of 0.1 ng/μL at 37 °C for 10 min to remove RNA fragments from the nucleic acid samples. Due to the low concentration of the obtained DNA, VAHTS DNA clean beads (Vazyme, Nanjing, China) were used to concentrate the DNA sample after RNA removal. Then, ~100 ng DNA was used as a template for the construction of a metagenomic high-throughput library. First, the genomic DNA was ultrasonically broken into ~350 bp fragments by a Covaris M220 instrument (Covaris, Woburn, MA, USA). Secondly, the metagenomic high-throughput library was constructed using a VAHTS Universal DNA Library Prep Kit (Vazyme, Nanjing, China) for Illumina according to the instructions. The constructed metagenomic library was analyzed using an Illumina Novaseq 6000 platform (Illumina, San Diego, CA, USA) for paired-end 2 × 150 bp sequencing.
2.3. Data Processing and Analysis
The original metagenome data were processed by Fastp (v0.20.0) [18] to delete low-quality reads (base mass values < 20 accounted for more than 20%), those shorter than 50 bp, and linker sequences (‘-f 5 -F 5 -q 20 -u 20 -g -W 5 -3 -l 50’). Redundant reads were cleaned by Fastuniq [19] with default parameters after quality control. Subsequently, 18S miTags were identified by rRNA_HMM [14], and those >100 bp were extracted by an in-house script. Among the miTags, those belonging to the V9 region were further identified by HMMER 3 [20], then recruited by the in-house script. The miTags that shared 100% similarity were clustered into amplicon sequence variant (ASVs) using DADA2 (v1.8) (--p-trim-left 0, --p-trunc-len 120) [21]. The longest read in an ASV was then selected as the representative read. QIIME2 [22] was applied to reveal the eukaryotic community structure by searching the reads in the ASVs of 18S V9 miTags against the SILVA132 database of the integrated PyNAST and Ribosomal Database Project (RDP v2.2) classifier [23,24]. Principal coordinate analysis (PCoA) was performed using a Bray–Curtis dissimilarity matrix calculated based on the eukaryotic community structures at the order level.
The representative reads of the ASVs that showed differences in relative abundance between sample groups were used for the reconstruction of a maximum likelihood (ML) phylogenetic tree. Reference 18S rRNA gene sequences of the closest relatives were downloaded from the NCBI. All the sequences were aligned using MAFFT [25] (v.7.407). IQ-TREE (v1.6.12) [26] was used to build the 18S rRNA phylogenetic tree (GTRGAMMA model) with bootstrap values based on 1000 replicates. The phylogenetic tree was visualized by iTOL [27].
3. Result
A total of 9,711,048 18S miTag data were identified in the metagenomic reads, among which 1,008,820 belonged to the V9 region of the 18S rRNA gene. We used the V9 miTag reads to analyze the composition of eukaryotic communities in the SCS deep-sea sites. The results showed that unclassified eukaryotic groups accounted for 13.6 ± 11.9% of the communities. The SAR superphylum dominated all the eukaryotic communities (48.1 ± 21.4%), while Opisthokonta (18.4 ± 16.3%), as another dominant phylum, was specialized in MISNAC column samples (Figure 2a). The SAR consisted of Alveolata and Rhizaria phyla, which could be further classified as Retaria (Rhizaria), Ciliophora (Alveolata), and Protalveolata (Alveolata); Opisthokonta was highly represented by Metazoa (largely Copepoda) (Figure 2b). Compared with the MISNAC column samples, Retaria and unknown Eukaryota were abundantly identified in MISNAC membrane samples. Regarding Alveolata, about 73% could not be further classified into Ciliophora. Notably, we identified about ninefold higher relative abundances of Ciliophora in the MISNAC columns (19.49%) compared with the other samples, including the MISNAC membranes (2.13%). Within Ciliophora, taxonomic classification also identified Oligotrichia, Scuticociliatia, and an unknown order that differed significantly between the MISNAC column samples and the others (Spearman Test, p < 0.0001) (Figure 2c). Among Metazoa, we identified much more Copepoda in the column samples (23.31%) compared with the membrane samples (2.49%). Calanoida, Harpacticoida, and Cyclopoida were the major orders in Copepoda (Figure 2d), which all showed a significant difference (Spearman test, p < 0.0001) between the MISNAC column samples and the others. This was also the case for Diplonemea of Discicristata (Discoba superphylum) (10.67% versus 3.28%) (Figure 2e). In contrast, the Retaria class (Rhizaria) members, including the orders of Collodaria, RAD, and Spumellaria, were prevalent in the MISNAC membrane samples (48.86%) rather than the column samples (9.62%) (Figure 2f), which was another significant difference (Spearman test; p < 0.0001). As an exception in Rhizaria, the species in the Cercozoa class were enriched in the column samples instead of the membrane samples (1.78% versus 0.27%). Regarding biodiversity, the Shannon indices of eukaryotic communities in the MISNAC column samples (on average, 4.42) were significantly higher than those in the other samples (on average, 4.17) (Spearman test; p < 0.0001). This was also true for Chao1 between the two groups of samples (Table S2).
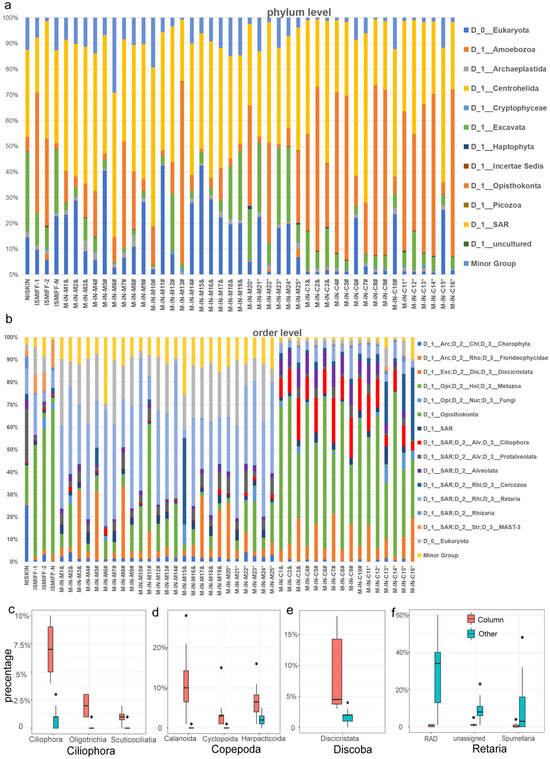
Figure 2.
Eukaryotic microbial community structure. The samples were obtained by two different sampling tools: Niskin bottles and MISNAC. The ISMIFF-1 and -2 samples were not treated with cell lysis butter in MISNAC filtration chambers but were preserved with RNALater. The ISMIFF-N sample was maintained in a MISNAC filtration chamber without treatment before being brought on board. The community structures for the samples (see Table S1) from the MISNAC filtration membranes and columns after cell lysis are displayed separately. The eukaryotic microbial communities were classified using 18S V9 miTags from the metagenomes for these samples. The phylum (a) and order (b) classifications of ASVs were performed with reference to the SILVA v132 database. The minor group includes the phyla or orders with relative abundances <1%. The relative abundances of the taxa at the lower levels were compared between the MISNAC column samples and the others (c). The major lineages that showed a significant difference between the MISNAC column samples (Column) and the others (Spearman test, p < 0.0001) are shown for Copepoda (c), Discoba (d), Ciliophora (e), and Retaria (f). &: a sample from ~590 m depth; #: ~1100 m depth; *: ~3100 m depth.
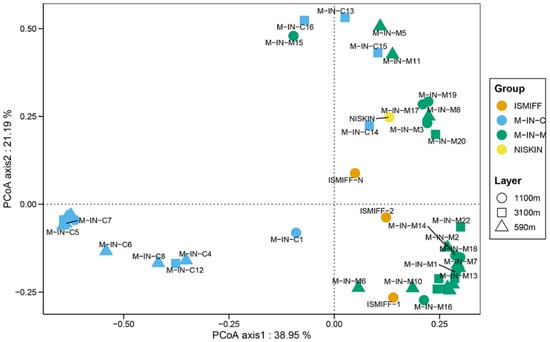
Our PCoA result showed that all the MISNAC membrane samples stayed away from the column samples (Figure 3), while the ISMIFF and NISKIN samples were relatively close to the MISNAC membrane samples. Moreover, the sampling depth is not a factor that separates the eukaryotic communities, regardless of the four 3100 m MISNAC column samples that were away from the others.
To further confirm the taxonomic status of the dominant ASVs in the MISNAC column samples, the representative 18S joint miTags for the abundant ASVs were recruited to construct ML phylogenetic trees (Figure 4). Seven Metazoa ASVs were mainly derived from Copepoda (total of the average percentages: 10.8%). ASV2 was not grouped with any references. ASV3 had a short phylogenetic distance to Ryocalanus squamatus from the Pacific Ocean and Spinocalanus aspinosus from the Atlantic Ocean. ASV4 was adjacent to Ergasilus tumidus. ASV1 showed almost no distance to Sinergasilus major (Figure 4a). The non-Copepoda Metazoa ASVs were affiliated with Margalopsis hartlaubii from the Raunefjord, Phoxichilidium femoratum and Spiophanes soederstroemi. The only solely abundant ASV from Discoba accounted for, on average, 6.8% of the eukaryotic communities and showed affinity to Diplonemea species isolated from ~6000 m depth (Figure 4b). There were three major Ciliophora ASVs that accounted for, on average, 0.4–1.7% of the eukaryotic communities of all the samples. In light of the phylogenetic distance, these ASVs are relatives of Dexiotricha and Spirostrombidium agathae. Two non-Ciliophora Alveolata ASVs were approximate to Fragilariopsis and Prorocentrum spinulentum based on the topology of the three (Figure 4c). The Retaria ASVs were mainly split into two clades in the tree (Figure 4d)—one consisting of Ehrenbergina glabra, Vanhoeffenella, and Nonionella and another composed of Spumellaria from the Pacific Ocean and Sticholonchida from the Lost City hydrothermal vent. ASV18 was independent of any references, suggesting its novel taxonomic status.
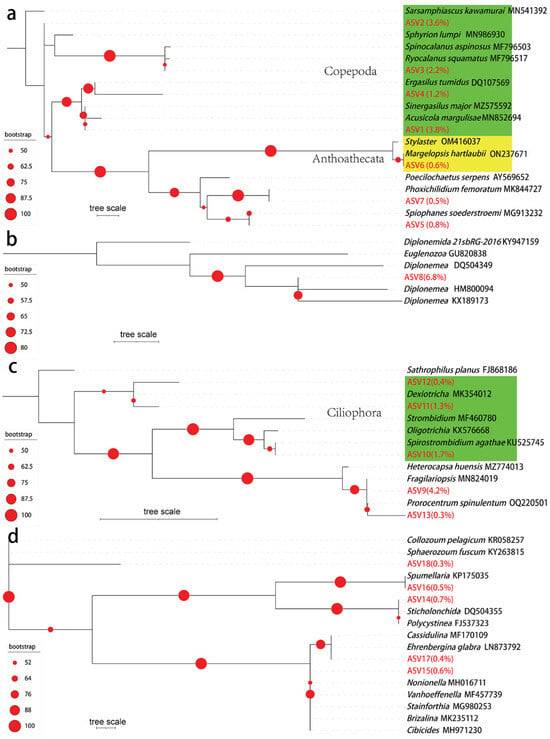
Figure 4.
Phylogenetic tree and average abundance of major Metazoa, Discoba, Alveolata, and Retaria ASVs. The maximum likelihood phylogenetic trees were reconstructed using the representative joint 18S miTags for the major ASVs from Metazoa (a), Discoba (b), Alveolata (c), and Retaria (d), along with the references from the NCBI. Average relative abundances of the ASVs in all the samples are shown in parentheses following the ASV IDs. The green color in (a) represents Copepoda, while the yellow color represents Anthoathecata. The green color in (c) represents Ciliophora. The red dots shown on the tree denote the bootstrap values.
4. Discussion
In this study, we compared the eukaryotic microbial communities in samples collected by different methods and revealed biases likely induced by the methods, in addition to the previously known biases introduced by primer selection and PCR conditions [28,29]. The high relative abundance of some protists in the MISNAC column samples likely account for the easy lysis of these naked microorganisms by the buffer injected into the filtration chamber of MISNAC, considering their soft, single-layered cell membrane. Therefore, the DNA of these organisms was probably released earlier and was absorbed in higher amounts than that of the other multicellular organisms, such as Retaria. However, our finding of low abundances of Copepoda, Discoba, and Ciliophora in the NISKIN and ISMIFF samples does not support this hypothesis. Although all the microorganisms in these NISKIN and ISMIFF samples were filtered for DNA extraction, they were all minor groups in the corresponding communities. It seems that the DNA molecules of Copepoda, Discoba, and Ciliophora would be missing if they were not collected by the columns in situ. Unlike the report of their high abundance in a sunlit marine zone [30], there are few reports regarding the prevalence of these protists in communities of deep sea water, except for pelagic and hadal diplonemid [31]. A drastic decrease in Dinoflagellata and Ciliophora with sampling depth was also recently shown [8]. However, this might be a result of drastic environmental changes imposed on the microorganisms, particularly exposure to low hydrostatic pressure and high temperature on board. It is known that marine water holds a huge amount of gases [32], and the deep ocean is a reservoir of CO2 and methane [33,34]. During the recovery of the lander on board, the rapid upward movement of the samples would probably make the microbes suffer from an increase in cellular inner pressure, as dissolved gases accumulate in cytoplasm of the microbes due to quickly descending pressure [13]. If the gases could not be expelled in a timely manner, the cells would burst as a result of the enlarging gas bubbles. In the deep ocean, organisms also maintain high levels of osmolytes to protect their cells against pressure [35]. With lowering pressure, the originally high level of osmolytes is degraded to prevent explosion of the cells as a result of an extra amount of water in the cytoplasm. However, the microbial cells would possibly be broken if the osmolytes were not degraded in a timely manner during sample collection from the deep sea. These two possible determining factors may be responsible for the declining abundance of these protists, which results in underestimation of the previously ignored dominant eukaryotic microbial groups in terms of their contribution to the deep-sea ecosystem. Therefore, compared with traditional sampling methods, MISNAC can minimize the impact of the sampling process to better represent the in situ eukaryotic microbial composition.
In deep-sea surficial sediments, heterotrophic protists such as Discoba, Ciliophora, and Rhizaria have been identified as the dominant groups [2]. Rhizaria was reported as the dominant group across the SCS water column, in addition to a large proportion of novel eukaryotic phylotypes [36]. In this study, the discovery of prevalent Discoba and Ciliophora in the sediments supports our hypothesis that the low relative abundances of naked protists in some of our water samples can probably be ascribed to the sampling method. When DNA was extracted from the sediment samples, released DNA from broken cells would have been included in the final total DNA [5,37]. As a control, the DNA of some possibly broken protists in Niskin and ISMIFF samples was filtered out through the membrane in this study. Retaria DNA was retained on the filtration membrane, as shown in our community structures, which probably resulted from the hard carbonate shell of Retaria species that are less affected by the change in external temperature and pressure. Moreover, the shell of the Retaria species protected the tissues from being lysed by the buffer flooded into the MISNAC chamber. In contrast, prokaryotic cells are all single cells and are equally vulnerable to the lysis buffer. Our previous study showed consistency in prokaryotic community structure of MISNAC column and membrane samples [15], indicating that the DNA of deep-sea prokaryotes in waters can be effectively collected by the columns. Our analysis showed that the diversity of the eukaryotic microbial community structure obtained by the MISNAC column collection method was higher than that of communities collected by other methods, which indicates that a wider spectrum of microorganisms was sampled by the MISNAC columns for in situ DNA extraction compared with other methods. Our result with respect to the community structures also suggests that the 18S miTags from the MISNAC membranes and columns should be combined for a complete understanding of the eukaryotic microbial communities in the deep ocean, considering the low digestion efficiency of Retaria by the buffer for DNA extraction in this study.
All the ASVs in our phylogenetic trees were single-cellular protists with respect to the taxonomy of their closest relatives. Some of the relatives were only identified in fresh water and coastal water, such as parasitic Sinergasilus [38], while Diplonemea was widely distributed from the coastal to abyssal benthic floor [37,39]. Spirostrombidium consists of ciliates affiliated with Ciliophora that have been identified in global coastal areas [40]; however, their distribution in the deep ocean has been rarely reported. Although recent studies have identified abundant Ciliophora in deep-sea sediments, the taxa at lower taxonomic levels were not presented [2]. In contrast to previous studies [36], we found dominant Ciliophora in deep-sea waters of the SCS. The Prorocentrum species are known as dinoflagellates that can produce toxins in coastal zones [41]. Prorocentrum spinulentum protists are tiny (9–12 um) and oval in shape [42]. As far as we know, they have not been reported in the deep ocean yet. Fragilariopsis includes some cold-adapted diatom species inhabiting Antarctic waters [43]. As a relative of Retaria ASV15, Nonionella has been found to be able to perform complete denitrification on the ocean floor [44]. The foraminiferal genus Vanhoeffenella is common in the deep sea and has been morphologically characterized as an eye-like cell [45]. Therefore, this study identified some protist species that dominate the deep-sea water column, which expands the known distribution of these protists in global oceans. They are the ‘dark matter’ deep-sea eukaryotes that cannot be highlighted with traditional sampling methods, despite their importance in the ecosystem.
In summary, we proposed an efficient method to collect deep-sea eukaryotic microbial DNA for studies of microbial ecosystems. This study is the first to use MISNAC to analyze the community structure of in situ eukaryotic microorganisms in the deep sea, demonstrating that in situ nucleic acid collection can help to restore the in situ habitat of eukaryotic microorganisms in extreme deep-sea environments. This device can provide nucleic acids for subsequent research on deep-sea eukaryotic microorganisms to explain the geobiochemical effects of more unknown eukaryotic microorganisms in the deep sea. As far as we know, no live deep-sea video platform is available for the in situ imaging of deep-sea pico- and nanoplankton [46]. Direct observation with an in situ microscope may provide direct evidence for the dominance of the single-cellular protists revealed by this study. We also conclude that the deep-sea in situ nucleic acid collection carried out by the MISNAC device meets the needs of metagenomics and metatranscriptomics analyses to explore the role of microbially mediated nutrient and element cycles in the deep sea in the future [16]. Metatranscriptomic data are also needed for further analysis of the dominant species reported in this study to determine their in situ transcriptional expressions. Further research should focus on refining and expanding the application of MISNAC and other in situ sampling approaches to improve our understanding of deep-sea ecosystems, their response to environmental changes, and the crucial roles played by eukaryotic microorganisms in organic matter degradation and elemental cycling in these extreme habitats.
5. Conclusions
In this study, we revealed the influence of sampling methods on the detection of deep-sea eukaryotic microorganisms under high hydrostatic pressure. The utilization of MISNAC revealed a greater diversity of protists in the deep-sea water column, particularly naked protists that were previously overlooked. These findings highlight the importance of employing advanced sampling methods such as MISNAC to expand our understanding of the deep-sea water column’s protist community. Overall, our study emphasizes the significance of considering sampling methodologies for accurate assessment and exploration of deep-sea eukaryotic microorganisms and their ecological roles in organic matter degradation and elemental cycling in the deep ocean.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jmse12010061/s1, Table S1: Basic information of samples; Table S2: Statistics of metagenomic sequencing data and 18S miTags.
Author Contributions
Y.W. conceived the study. M.Y. performed the experiments. P.X. analyzed data, summarized the results, and drafted the manuscript. H.Z. provided assistance with phylogenetic analysis. Z.G., L.H., Y.J. and Y.W. critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hainan Provincial Natural Science Foundation of China grant number [No. 322CXTD531].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
No ethical clearance was required, as no animals were collected in this study.
Data Availability Statement
The original data presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank the Supercomputer Center of Sanya University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hutchins, D.A.; Fu, F. Microorganisms and ocean global change. Nat. Microbiol. 2017, 2, 17058. [Google Scholar] [CrossRef] [PubMed]
- Schoenle, A.; Hohlfeld, M.; Hermanns, K.; Mahé, F.; de Vargas, C.; Nitsche, F.; Arndt, H. High and specific diversity of protists in the deep-sea basins dominated by diplonemids, kinetoplastids, ciliates and foraminiferans. Comm. Biol. 2021, 4, 501. [Google Scholar] [CrossRef] [PubMed]
- Acinas, S.G.; Sánchez, P.; Salazar, G.; Cornejo-Castillo, F.M.; Sebastián, M.; Logares, R.; Royo-Llonch, M.; Paoli, L.; Sunagawa, S.; Hingamp, P.; et al. Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Comm. Biol. 2021, 4, 604. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-Y.; Liu, J.; Xue, C.-X.; Tian, J.; Zhang, X.-H. Shift and metabolic potentials of microbial eukaryotic communities across the full depths of the Mariana Trench. Front. Microbiol. 2021, 11, 603296. [Google Scholar] [CrossRef] [PubMed]
- Cordier, T.; Angeles, I.B.; Henry, N.; Lejzerowicz, F.; Berney, C.; Morard, R.; Brandt, A.; Cambon-Bonavita, M.A.; Guidi, L.; Lombard, F.; et al. Patterns of eukaryotic diversity from the surface to the deep-ocean sediment. Sci. Adv. 2022, 8, eabj9309. [Google Scholar] [CrossRef] [PubMed]
- Shu, W.-S.; Huang, L.-N. Microbial diversity in extreme environments. Nat. Rev. Microbiol. 2022, 20, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C. New perspectives in benthic deep-sea microbial ecology. Front. Mar. Sci. 2015, 2, 17. [Google Scholar] [CrossRef]
- Giner, C.R.; Pernice, M.C.; Balagué, V.; Duarte, C.M.; Gasol, J.M.; Logares, R.; Massana, R. Marked changes in diversity and relative activity of picoeukaryotes with depth in the world ocean. ISME J. 2020, 14, 437–449. [Google Scholar] [CrossRef]
- Wang, Y.; Tian, R.M.; Gao, Z.M.; Bougouffa, S.; Qian, P.-Y. Optimal eukaryotic 18S and universal 16S/18S ribosomal RNA primers and their application in a study of symbiosis. PLoS ONE 2014, 9, e90053. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.S. Comparative analyses of the V4 and V9 regions of 18S rDNA for the extant eukaryotic community using the Illumina platform. Sci. Rep. 2020, 10, 6519. [Google Scholar] [CrossRef]
- Zhou, Y.-L.; Mara, P.; Cui, G.-J.; Edgcomb, V.P.; Wang, Y. Microbiomes in the Challenger Deep slope and bottom-axis sediments. Nat. Commun. 2022, 13, 1515. [Google Scholar] [CrossRef] [PubMed]
- Logares, R.; Sunagawa, S.; Salazar, G.; Cornejo-Castillo, F.M.; Ferrera, I.; Sarmento, H.; Hingamp, P.; Ogata, H.; de Vargas, C.; Lima-Mendez, G.; et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 2014, 16, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Gooday, A.J.; Schoenle, A.; Dolan, J.R.; Arndt, H. Protist diversity and function in the dark ocean—Challenging the paradigms of deep-sea ecology with special emphasis on foraminiferans and naked protists. Eur. J. Protistol. 2020, 75, 125721. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gilna, P.; Li, W.Z. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 2009, 25, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-F.; Li, W.-L.; Li, J.; Chen, J.; Xin, Y.-Z.; He, L.-S.; Wang, Y. Multiple in situ nucleic acid collections (MISNAC) from deep-sea waters. Front. Mar. Sci. 2020, 7, 00081. [Google Scholar] [CrossRef]
- Wei, Z.; Li, Q.; Lu, R.; Zheng, P.; Wang, Y. In situ genomics and transcriptomics of SAR202 subclusters revealed subtle distinct activities in deep-sea water. Microorganisms 2022, 10, 1629. [Google Scholar] [CrossRef]
- Pachiadaki, M.G.; Taylor, C.; Oikonomou, A.; Yakimov, M.M.; Stoeck, T.; Edgcomb, V. In situ grazing experiments apply new technology to gain insights into deep-sea microbial food webs. Deep. Sea Res. II 2016, 129, 223–231. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Xu, H.; Luo, X.; Qian, J.; Pang, X.; Song, J.; Qian, G.; Chen, J.; Chen, S. FastUniq: A fast de novo duplicates removal tool for paired short reads. PLoS ONE 2012, 7, e52249. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; Desantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.; von Haeseler, A.; Minh, B. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Kounosu, A.; Murase, K.; Yoshida, A.; Maruyama, H.; Kikuchi, T. Improved 18S and 28S rDNA primer sets for NGS-based parasite detection. Sci. Rep. 2019, 9, 15789. [Google Scholar] [CrossRef]
- Frau, A.; Kenny, J.G.; Lenzi, L.; Campbell, B.J.; Ijaz, U.Z.; Duckworth, C.A.; Burkitt, M.D.; Hall, N.; Anson, J.; Darby, A.C.; et al. DNA extraction and amplicon production strategies deeply inf luence the outcome of gut mycobiome studies. Sci. Rep. 2019, 9, 9328. [Google Scholar] [CrossRef]
- de Vargas, C.; Audic, S.; Henry, N.; Decelle, J.; Mahé, F.; Logares, R.; Lara, E.; Berney, C.; Le Bescot, N.; Probert, I.; et al. Eukaryotic plankton diversity in the sunlit ocean. Science 2015, 348, 1261605. [Google Scholar] [CrossRef]
- Eloe, E.A.; Shulse, C.N.; Fadrosh, D.W.; Williamson, S.J.; Allen, E.E.; Bartlett, D.H. Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 2011, 3, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.G.; Robinson, R.J. Notes on the determination of dissolved oxygen in sea wate. J. Mar. Res. 1939, 2, 1–8. [Google Scholar] [CrossRef]
- Luo, J.; Xie, Y.; Hou, M.Z.; Xiong, Y.; Wu, X.; Lüddeke, C.T.; Huang, L. Advances in subsea carbon dioxide utilization and storage. Energy Rev. 2023, 2, 100016. [Google Scholar] [CrossRef]
- Kholmogorov, A.; Ponomarev, V.; Syrbu, N.S.; Shkorba, S. Dissolved methane transport in the Tatar Strait and the deepest basin of the Japan (East) Sea from its possible sources. Water 2023, 15, 821. [Google Scholar] [CrossRef]
- Yancey, P.H.; Blake, W.R.; Conley, J. Unusual organic osmolytes in deep-sea animals: Adaptations to hydrostatic pressure and other perturbants. Comp. Biochem. Physiol. A 2002, 133, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiao, N.; Ren, R.; Warren, A. Distribution and Diversity of Microbial Eukaryotes in Bathypelagic Waters of the South China Sea. J. Eukaryot. Microbiol. 2017, 64, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Scheckenbach, F.; Hausmann, K.; Wylezich, C.; Weitere, M.; Arndt, H. Large-scale patterns in biodiversity of microbial eukaryotes from the abyssal sea floor. Proc. Natl. Acad. Sci. USA 2010, 107, 115–120. [Google Scholar] [CrossRef]
- Kvach, Y.; Tkachenko, M.Y.; Seifertová, M.; Ondračková, M. Insights into the diversity, distribution and phylogeny of three ergasilid copepods (Hexanauplia: Ergasilidae) in lentic water bodies of the Morava river basin, Czech Republic. Limnologica 2021, 91, 125922. [Google Scholar] [CrossRef]
- Yabuki, A.; Kawato, M.; Nagano, Y.; Tsuchida, S.; Yoshida, T.; Fujiwara, Y. Structural Comparison of Diplonemid Communities around the Izu Peninsula, Japan. Microbes Environ. 2021, 36, ME21012. [Google Scholar] [CrossRef]
- Song, W.; Xu, D.; Chen, X.; Warren, A.; Shin, M.K.; Song, W.; Li, L. Overview of the diversity, phylogeny and biogeography of Strombidiid Oligotrich ciliates (Protista, Ciliophora), With a brief revision and a key to the known genera. Front. Microbiol. 2021, 12, 700940. [Google Scholar] [CrossRef]
- Durán-Riveroll, L.M.; Cembella, A.D.; Okolodkov, Y.B. A review on the biodiversity and biogeography of toxigenic benthic marine dinoflagellates of the coasts of Latin America. Front. Mar. Sci. 2019, 6, 00148. [Google Scholar] [CrossRef]
- Tillmann, U.; Gottschling, M.; Wietkamp, S.; Hoppenrath, M. Morphological and Phylogenetic Characterisation of Prorocentrum spinulentum, sp. nov. (Prorocentrales, Dinophyceae), a Small Spiny Species from the North Atlantic. Microorganisms 2023, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Mock, T.; Otillar, R.P.; Strauss, J.; McMullan, M.; Paajanen, P.; Schmutz, J.; Salamov, A.; Sanges, R.; Toseland, A.; Ward, B.J.; et al. Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 2017, 541, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Risgaard-Petersen, N.; Langezaal, A.M.; Ingvardsen, S.; Schmid, M.C.; Jetten, M.S.M.; Op den Camp, H.J.M.; Derksen, J.W.M.; Piña-Ochoa, E.; Eriksson, S.P.; Peter Nielsen, L.; et al. Evidence for complete denitrification in a benthic foraminifer. Nature 2006, 443, 93–96. [Google Scholar] [CrossRef]
- Voltski, I.; Gooday, A.J.; Pawlowski, J. Eyes of the deep-sea floor: The integrative taxonomy of the foraminiferal genus Vanhoeffenella. Protist 2018, 169, 235–267. [Google Scholar] [CrossRef]
- Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Acinas, S.G.; Babin, M.; Bork, P.; Boss, E.; Bowler, C.; Cochrane, G.; et al. Tara Oceans: Towards global ocean ecosystems biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).