
Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. 16S rRNA Sequencing
2.3. Sequence Processing
2.4. Biodiversity Analysis
3. Results
3.1. Composition of Amphipod Bacterial Communities
3.2. Alpha Indexes in Amphipod Gut Bacterial Communities
3.3. Relationships between Amphipod Gut Bacterial Communities
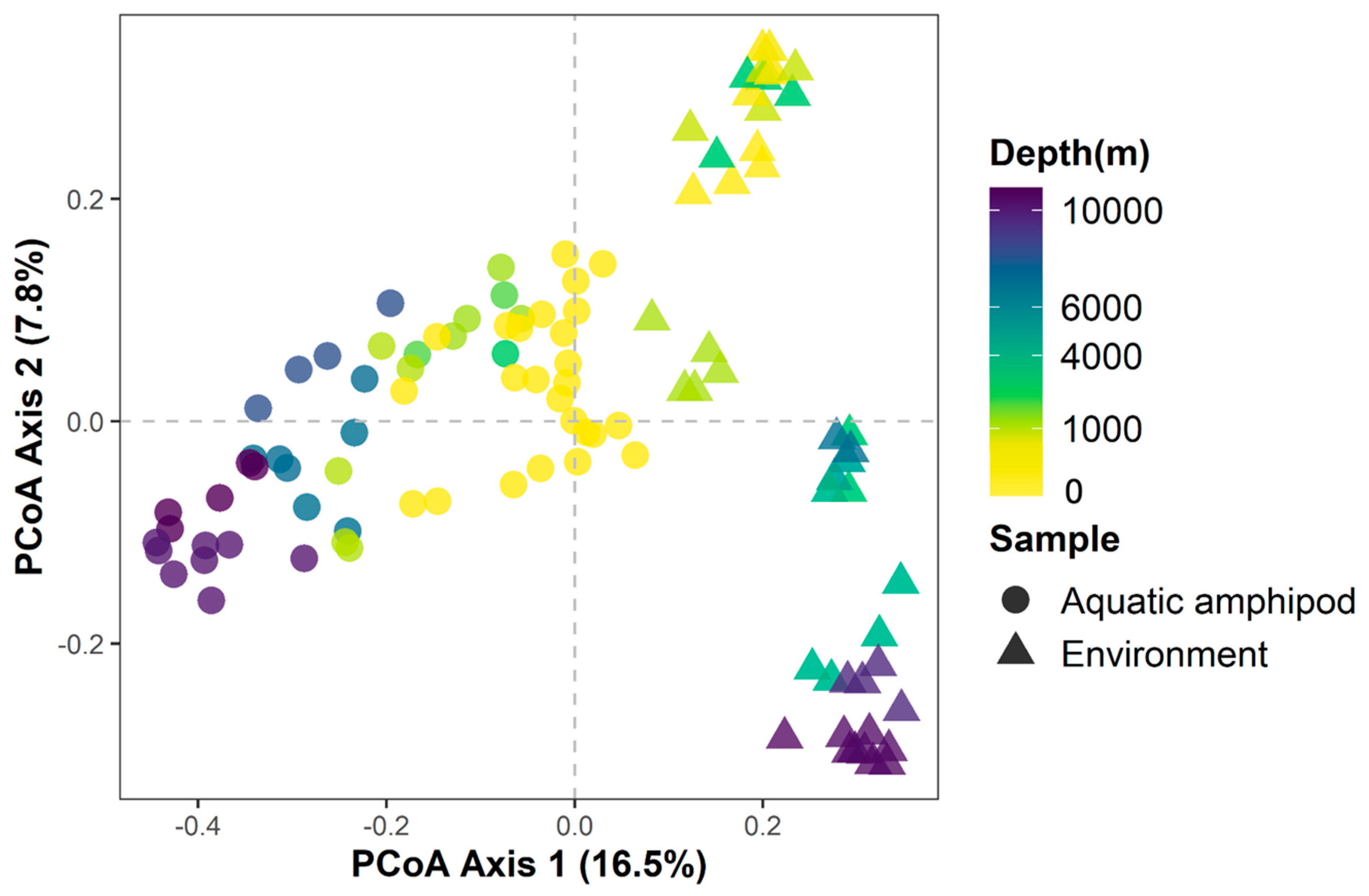
3.4. Relationships between Amphipod Gut and Environmental Bacterial Communities
3.5. Discriminant Taxa for Gut Microbial Communities
4. Discussion
4.1. Taxonomic Features of Amphipod Gut Microbiota
4.2. Alpha Diversities of Marine Amphipod Gut Microbiota
4.3. Factors Associated with Intestinal Microbiota
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Väinölä, R.; Witt, J.D.S.; Grabowski, M.; Bradbury, J.H.; Jazdzewski, K.; Sket, B. Global diversity of amphipods (Amphipoda; Crustacea) in freshwater. In Freshwater Animal Diversity Assessment; Balian, E.V., Lévêque, C., Segers, H., Martens, K.V., Väinölä, R., Witt, J.D.S., Grabowski, M., Bradbury, J.H., Jazdzewski, K., Sket, B., Eds.; Developments in Hydrobiology; Springer: Dordrecht, The Netherlands, 2008; pp. 241–255. [Google Scholar]
- Jamieson, A.J.; Fujii, T.; Mayor, D.J.; Solan, M.; Priede, I.G. Hadal trenches: The ecology of the deepest places on Earth. Trends Ecol. Evol. 2010, 25, 190–197. [Google Scholar] [CrossRef]
- Fujii, T.; Kilgallen, N.M.; Rowden, A.A.; Jamieson, A.J. Deep-sea amphipod community structure across abyssal to hadal depths in the Peru-Chile and Kermadec trenches. Mar. Ecol. Prog. Ser. 2013, 492, 125–138. [Google Scholar] [CrossRef]
- Lindeman, D. Natural history of the terrestrial amphipod Cerrorchestia hyloraina Lindeman (Crustacea: Amphipoda; Talitridae) in a Costa Rican cloud forest. J. Nat. Hist. 1991, 25, 623–638. [Google Scholar] [CrossRef]
- Glazier, D.S. Amphipoda. In Encyclopedia of Inland Waters; Likens, G.E.G., White, D.S., Eds.; Academic Press: Oxford, UK, 2009; pp. 89–115. [Google Scholar]
- Horton, T.; Gofas, S.; Kroh, A.; Poore, G.C.B.; Read, G.; Rosenberg, G.; Stöhr, S.; Bailly, N.; Boury-Esnault, N.; Brandão, S.N.; et al. Improving nomenclatural consistency: A decade of experience in the World Register of Marine Species. Eur. J. Taxon. 2017, 389, 1–24. [Google Scholar] [CrossRef]
- Hughes, L.E.; Ahyong, S.T. Collecting and processing amphipods. J. Crustac. Biol. 2016, 36, 584–588. [Google Scholar] [CrossRef]
- Wade, S.; Corbin, T.; McDowell, L.M.; Bradbury, J.; Authority, S.A.E.P. Critter Catalogue: A Guide to the Aquatic Invertebrates of South Australian Inland Waters; Environment Protection Authority: Washington, DC, USA, 2004. [Google Scholar]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.; Carey, H.V.; Domazet-Loso, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Annu. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Abdelrhman, K.F.; Bacci, G.; Marras, B.; Nistri, A.; Schintu, M.; Ugolini, A.; Mengoni, A. Exploring the bacterial gut microbiota of supralittoral talitrid amphipods. Res. Microbiol. 2017, 168, 74–84. [Google Scholar] [CrossRef]
- Abdelrhman, K.F.; Bacci, G.; Nistri, A.; Mengoni, A.; Ugolini, A. Diet and gut microbiota of two supralittoral amphipods Orchestia montagui and Talitrus saltator living in different microhabitats. Estuar. Coast. Shelf S 2017, 197, 119–125. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Wang, Y.; Li, J.Y.; Yan, G.Y.; He, L.S. Comparative analysis of the gut microbial communities between two dominant amphipods from the Challenger Deep, Mariana Trench. Deep Sea Res. Part I 2019, 151, 103081. [Google Scholar] [CrossRef]
- Chan, J.; Geng, D.; Pan, B.; Zhang, Q.; Xu, Q. Gut microbial divergence between three hadal amphipod species from the isolated hadal trenches. Microb. Ecol. 2022, 84, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Hatada, Y.; Tsubouchi, T.; Nagahama, T.; Takami, H. The Hadal Amphipod Hirondellea gigas possessing a unique cellulase for digesting wooden debris buried in the deepest seafloor. PLoS ONE 2012, 7, e42727. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Herlemann, D.P.; Labrenz, M.; Jurgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Jari, O.; Blanchet, F.G.; Friendly, M.; Roeland Kindt, P.L.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; et al. vegan: Community Ecology Package. R. Package Version 2018; version 2.5–7. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 11 December 2020).
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Nishida, A.H.; Ochman, H. Rates of gut microbiome divergence in mammals. Mol. Ecol. 2018, 27, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- Eustace, R.M.; Kilgallen, N.M.; Lacey, N.C.; Jamieson, A.J. Population Structure of the Hadal Amphipod Hirondellea Gigas (Amphipoda: Lysianassoidea) from the Izu-Bonin Trench. J. Crustac. Biol. 2013, 33, 793–801. [Google Scholar] [CrossRef]
- Zhang, W.; Watanabe, H.K.; Ding, W.; Lan, Y.; Tian, R.-M.; Sun, J.; Chen, C.; Cai, L.; Li, Y.; Oguri, K. Gut microbial divergence between two populations of the hadal amphipod Hirondellea gigas. Appl. Environ. Microbiol. 2019, 85, e02032-18. [Google Scholar] [CrossRef]
- Lian, C.-A.; Yan, G.-Y.; Huang, J.-M.; Danchin, A.; Wang, Y.; He, L.-S. Genomic characterization of a novel gut symbiont from the hadal snailfish. Front. Microbiol. 2020, 10, 2978. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Dittmer, J.; Bouchon, D.; Cordaux, R. Phylogenomics of “Candidatus Hepatoplasma crinochetorum,” a lineage of mollicutes associated with noninsect arthropods. Genome Biol. Evol. 2014, 6, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.M.; Wang, S.L.; Gao, Z.M.; Zhang, A.Q.; Danchin, A.; He, L.S. Genomic characterization of symbiotic mycoplasmas from the stomach of deep-sea isopod bathynomus sp. Environ. Microbiol. 2016, 18, 2646–2659. [Google Scholar] [CrossRef]
- Brune, A.; Resh, V.; Cardé, R. Encyclopedia of Insects; Academic Press: Cambridge, MA, USA, 2003. [Google Scholar]
- Fraune, S.; Zimmer, M. Host-specificity of environmentally transmitted Mycoplasma-like isopod symbionts. Environ. Microbiol. 2008, 10, 2497–2504. [Google Scholar] [CrossRef]
- Wang, Y.; Stingl, U.; Anton-Erxleben, F.; Geisler, S.; Brune, A.; Zimmer, M. “Candidatus Hepatoplasma crinochetorum,” a new, stalk-forming lineage of Mollicutes colonizing the midgut glands of a terrestrial isopod. Appl. Environ. Microbiol. 2004, 70, 6166–6172. [Google Scholar] [CrossRef]
- Breznak, J.A.; Brune, A. Role of microorganisms in the digestion of lignocellulose by termites. Ann. Rev. Entomol. 1994, 39, 453–487. [Google Scholar] [CrossRef]
- Lauro, F.M.; Stratton, T.K.; Chastain, R.A.; Ferriera, S.; Johnson, J.; Goldberg, S.M.; Yayanos, A.A.; Bartlett, D.H. Complete Genome Sequence of the Deep-Sea Bacterium Psychromonas Strain CNPT3. Genome Announc. 2013, 1, 10–1128. [Google Scholar] [CrossRef]
- Denner, E.B.; Mark, B.; Busse, H.J.; Turkiewicz, M.; Lubitz, W. Psychrobacter proteolyticus sp. nov., a psychrotrophic, halotolerant bacterium isolated from the Antarctic krill Euphausia superba Dana, excreting a cold-adapted metalloprotease. Syst. Appl. Microbiol. 2001, 24, 44–53. [Google Scholar] [CrossRef]
- Maruyama, A.; Honda, D.; Yamamoto, H.; Kitamura, K.; Higashihara, T. Phylogenetic analysis of psychrophilic bacteria isolated from the Japan Trench, including a description of the deep-sea species Psychrobacter pacificensis sp. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Romanenko, L.A.; Tanaka, N.; Frolova, G.M.; Mikhailov, V.V. Psychrobacter fulvigenes sp. nov., isolated from a marine crustacean from the Sea of Japan. Int. J. Syst. Evol. Microbiol. 2009, 59, 1480–1486. [Google Scholar] [CrossRef]
- Kämpfer, P.; Albrecht, A.; Buczolits, S.; Busse, H.-J. Psychrobacter faecalis sp. nov., a new species from a bioaerosol originating from pigeon faeces. Syst. Appl. Microbiol. 2002, 25, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Gennari, M.; Parini, M.; Volpon, D.; Serio, M. Isolation and characterization by conventional methods and genetic transformation of Psychrobacter and Acinetobacter from fresh and spoiled meat, milk and cheese. Int. J. Food Microbiol. 1992, 15, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Juni, E.; Heym, G.A. Psychrobacter immobilis gen. nov., sp. nov.: Genospecies Composed of Gram-Negative, Aerobic, Oxidase-Positive Coccobacilli. Int. J. Syst. Bacteriol. 1986, 36, 388–391. [Google Scholar] [CrossRef]
- Wright, A.C.; Hill, R.T.; Johnson, J.A.; Roghman, M.C.; Colwell, R.R.; Morris, J.G., Jr. Distribution of Vibrio vulnificus in the Chesapeake Bay. Appl. Environ. Microbiol. 1996, 62, 717–724. [Google Scholar] [CrossRef]
- Kim, Y.O.; Park, I.S.; Park, S.; Nam, B.H.; Park, J.M.; Kim, D.G.; Yoon, J.H. Tenacibaculum haliotis sp. nov., isolated from the gut of an abalone Haliotis discus hannai. Int. J. Syst. Evol. Microbiol. 2017, 67, 3268–3273. [Google Scholar] [CrossRef]
- Småge, S.B.; Frisch, K.; Vold, V.; Duesund, H.; Brevik, Ø.J.; Olsen, R.H.; Sjaatil, S.T.; Klevan, A.; Brudeseth, B.; Watanabe, K.; et al. Induction of tenacibaculosis in Atlantic salmon smolts using Tenacibaculum finnmarkense and the evaluation of a whole cell inactivated vaccine. Aquaculture 2018, 495, 858–864. [Google Scholar] [CrossRef]
- Hyun, D.W.; Kim, M.S.; Shin, N.R.; Kim, J.Y.; Kim, P.S.; Whon, T.W.; Yun, J.H.; Bae, J.W. Shimia haliotis sp. nov., a bacterium isolated from the gut of an abalone, Haliotis discus hannai. Int. J. Syst. Evol. Microbiol. 2013, 63, 4248–4253. [Google Scholar] [CrossRef]
- Pagan-Jimenez, M.; Ruiz-Calderon, J.F.; Dominguez-Bello, M.G.; Garcia-Arraras, J.E. Characterization of the intestinal microbiota of the sea cucumber Holothuria glaberrima. PLoS ONE 2019, 14, e0208011. [Google Scholar] [CrossRef] [PubMed]
- El-Bouhy, Z.; El-Nobi, G.; El-Murr, A.; Abd El-Hakim, S. Study on Vibriosis in Mugil Capito in El-Dakahlia and Damitta Governorates, Egypt. Abbassa Int. J. Aquat 2016, 9, 2016. [Google Scholar]
- Haenen, O.L.; van Zanten, E.; Jansen, R.; Roozenburg, I.; Engelsma, M.Y.; Dijkstra, A.; Boers, S.A.; Voorbergen-Laarman, M.; Moller, A.V. Vibrio vulnificus outbreaks in Dutch eel farms since 1996: Strain diversity and impact. Dis. Aquat. Organ 2014, 108, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Gillan, D.C.; Dubilier, N. Novel epibiotic thiothrix bacterium on a marine amphipod. Appl. Environ. Microbiol. 2004, 70, 3772–3775. [Google Scholar] [CrossRef]
- Bauermeister, J.; Ramette, A.; Dattagupta, S. Repeatedly evolved host-specific ectosymbioses between sulfur-oxidizing bacteria and amphipods living in a cave ecosystem. PLoS ONE 2012, 7, e50254. [Google Scholar] [CrossRef]
- Dattagupta, S.; Schaperdoth, I.; Montanari, A.; Mariani, S.; Kita, N.; Valley, J.W.; Macalady, J.L. A novel symbiosis between chemoautotrophic bacteria and a freshwater cave amphipod. ISME J. 2009, 3, 935–943. [Google Scholar] [CrossRef]
- Senghor, B.; Sokhna, C.; Ruimy, R.; Lagier, J.-C. Gut microbiota diversity according to dietary habits and geographical provenance. Human. Microb. J. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Gerringer, M.E.; Popp, B.N.; Linley, T.D.; Jamieson, A.J.; Drazen, J.C. Comparative feeding ecology of abyssal and hadal fishes through stomach content and amino acid isotope analysis. Deep Sea Res. Part I Oceanogr. Res. Pap. 2017, 121, 110–120. [Google Scholar] [CrossRef]
- Eardly, D.F.; Carton, M.W.; Gallagher, J.M.; Patching, J.W. Bacterial abundance and activity in deep-sea sediments from the eastern North Atlantic. Prog. Oceanogr. 2001, 50, 245–259. [Google Scholar] [CrossRef]
- Guerra-García, J.M.; Tierno de Figueroa, J.M.; Navarro-Barranco, C.; Ros, M.; Sánchez-Moyano, J.E.; Moreira, J. Dietary analysis of the marine Amphipoda (Crustacea: Peracarida) from the Iberian Peninsula. J. Sea Res. 2014, 85, 508–517. [Google Scholar] [CrossRef]
- Navarro-Barranco, C.; Tierno-de-Figueroa, J.M.; Guerra-García, J.M.; Sánchez-Tocino, L.; García-Gómez, J.C. Feeding habits of amphipods (Crustacea: Malacostraca) from shallow soft bottom communities: Comparison between marine caves and open habitats. J. Sea Res. 2013, 78, 1–7. [Google Scholar] [CrossRef]
- Luo, M.; Gieskes, J.; Chen, L.; Shi, X.; Chen, D. Provenances, distribution, and accumulation of organic matter in the southern Mariana Trench rim and slope: Implication for carbon cycle and burial in hadal trenches. Mar. Geol. 2017, 386, 98–106. [Google Scholar] [CrossRef]
- Gunter, G. Notes on sea beach ecology. Food sources on sandy beaches and localized diatom blooms bordering gulf beaches. Gulf Caribb. Res. 1979, 6, 305–307. [Google Scholar] [CrossRef]
- Tap, J.; Furet, J.P.; Bensaada, M.; Philippe, C.; Roth, H.; Rabot, S.; Lakhdari, O.; Lombard, V.; Henrissat, B.; Corthier, G.; et al. Gut microbiota richness promotes its stability upon increased dietary fibre intake in healthy adults. Environ. Microbiol. 2015, 17, 4954–4964. [Google Scholar] [CrossRef] [PubMed]
- Leser, T.D.; Mølbak, L. Better living through microbial action: The benefits of the mammalian gastrointestinal microbiota on the host. Environ. Microbiol. 2009, 11, 2194–2206. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef]
- Hessler, R.R.; Ingram, C.L.; Aristides Yayanos, A.; Burnett, B.R. Scavenging amphipods from the floor of the Philippine trench. Deep. Sea Res. Part I 1978, 25, 1029–1047. [Google Scholar] [CrossRef]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [PubMed]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef]
- Wu, S.; Wang, G.; Angert, E.R.; Wang, W.; Li, W.; Zou, H. Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS ONE 2012, 7, e30440. [Google Scholar] [CrossRef] [PubMed]
- Amaro, T.; Witte, H.; Herndl, G.J.; Cunha, M.R.; Billett, D.S. Deep-sea bacterial communities in sediments and guts of deposit-feeding holothurians in Portuguese canyons (NE Atlantic). Deep Sea Res. Part I 2009, 56, 1834–1843. [Google Scholar] [CrossRef]
- Huang, Q.; Sham, R.C.; Deng, Y.; Mao, Y.; Wang, C.; Zhang, T.; Leung, K.M.Y. Diversity of gut microbiomes in marine fishes is shaped by host-related factors. Mol. Ecol. 2020, 29, 5019–5034. [Google Scholar] [CrossRef] [PubMed]
- Kers, J.G.; Velkers, F.C.; Fischer, E.A.; Hermes, G.D.; Stegeman, J.A.; Smidt, H. Host and environmental factors affecting the intestinal microbiota in chickens. Front. Microbiol. 2018, 9, 235. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Sums of Sqs | Mean Sqs | F.Model | R2 | p | |
|---|---|---|---|---|---|---|
| Entire amphipod dataset | Diet | 4.620 | 2.310 | 13.568 | 0.199 | 0.0001 |
| Habitat | 2.768 | 1.384 | 8.129 | 0.119 | 0.0001 | |
| Site | 1.883 | 0.942 | 5.530 | 0.081 | 0.0001 | |
| Host family | 2.055 | 0.685 | 4.024 | 0.088 | 0.0001 | |
| Host species | 2.196 | 0.366 | 2.150 | 0.095 | 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, T.; Liao, Y.; Wang, Y.; Li, J.; He, L. Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones. J. Mar. Sci. Eng. 2023, 11, 2197. https://doi.org/10.3390/jmse11112197
Wei T, Liao Y, Wang Y, Li J, He L. Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones. Journal of Marine Science and Engineering. 2023; 11(11):2197. https://doi.org/10.3390/jmse11112197
Chicago/Turabian StyleWei, Taoshu, Yanwen Liao, Yong Wang, Junyuan Li, and Lisheng He. 2023. "Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones" Journal of Marine Science and Engineering 11, no. 11: 2197. https://doi.org/10.3390/jmse11112197
APA StyleWei, T., Liao, Y., Wang, Y., Li, J., & He, L. (2023). Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones. Journal of Marine Science and Engineering, 11(11), 2197. https://doi.org/10.3390/jmse11112197