Multiplex Bead Array Assay of a Panel of Circulating Cytokines and Growth Factors in Patients with Albuminuric and Non-Albuminuric Diabetic Kidney Disease


 ,
,
Abstract
:1. Introduction
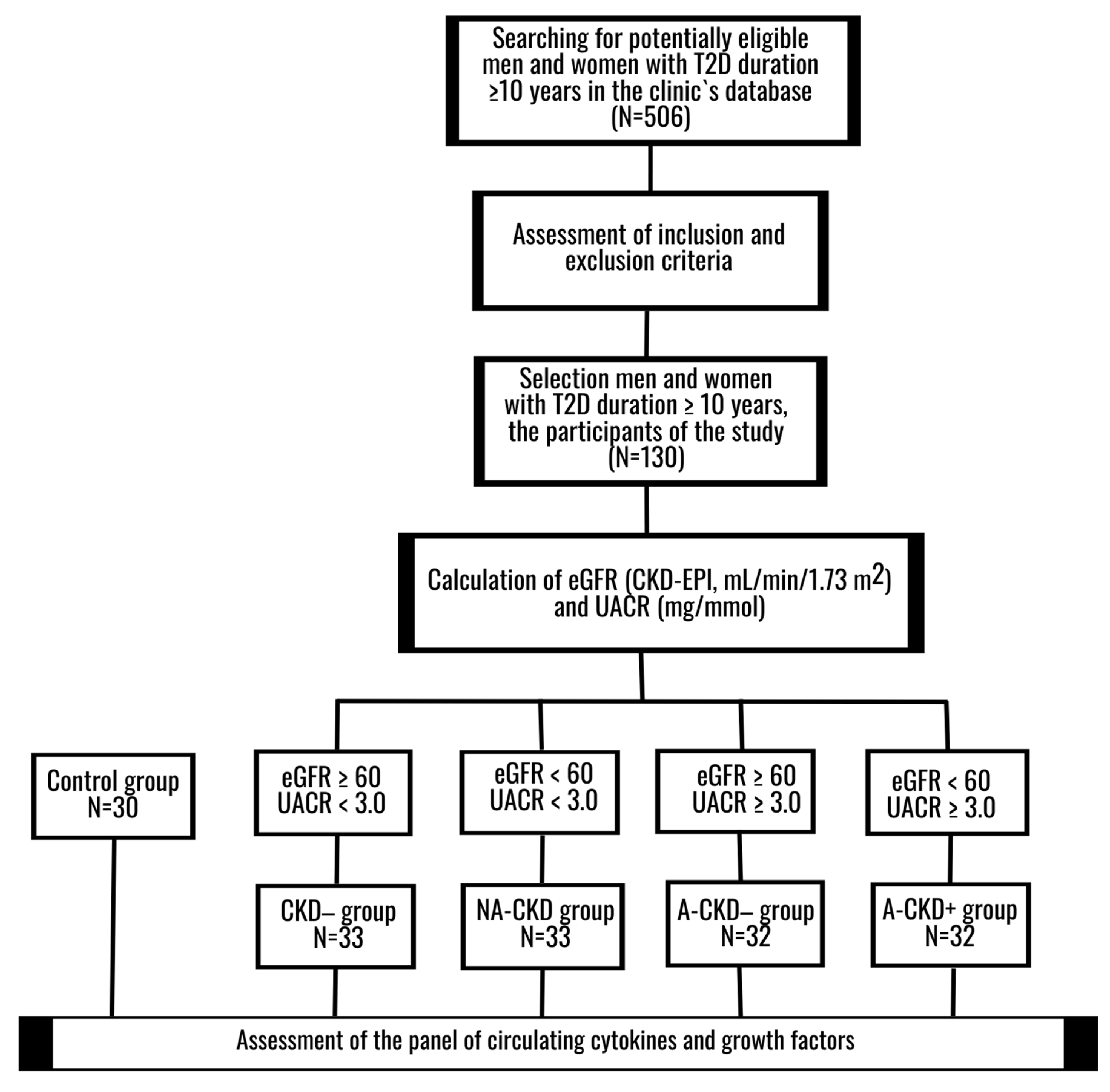
2. Materials and Methods
2.1. Design
2.2. Ethical Principals
2.3. Participants
2.4. Methods
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics of T2D Patients
3.2. IL-2, IL-4, IL-7, IL-9, IL-13, IL-15
3.3. IL-5, GM-CSF, G-CSF, IL-6, and IL-12
3.4. IL-10 and IFN-γ
3.5. IL-1β, IL-1Ra, TNF-α, and IL-17A
3.6. MCP-1, MIP-1α, MIP-1β, RANTES, Eotaxin, IP-10, and IL-8
3.7. bFGF, VEGF, and PGDF-BB
3.8. Correlation Analysis and Logistic Regression Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Discovery Cohort | Validation Cohort |
|---|---|---|
| N | 86 | 44 |
| Sex, M/F, n (%) | 29/57 (33.7%/66.3%) | 16/28 (36.4%/63.4%) |
| Age, years | 65 (58; 69) | 65.5 (58.5; 70) |
| Smokers, n (%) | 10 (11.6%) | 5 (11.4%) |
| BMI, kg/m2 | 33.4 (29.4; 37.4) | 33.2 (29.8; 38.9) |
| Diabetes duration, years | 13.5 (11; 17) | 14 (11; 18) |
| CKD−/NA-CKD/A-CKD−/A-CKD+, n | 22/22/21/21 | 11/11/11/11 |
Appendix B
| Family | Protein |
|---|---|
| Archetypical cytokines signaling through classical-cytokine receptors Type I helical Cytokine families signaling through Class I cytokine receptors (CRF1 family or Hematopoietin family) | |
| IL-2 Family, or Common Gamma Chain Receptor Family | IL-2, IL-4 subfamily(IL-4, IL-13), IL-9, IL-15, and IL-7 subfamily(IL-7) |
| Common Beta Chain Receptor Cytokine Family | IL-5, Colony Stimulating Factor 2/Granulocyte Monocyte-Stimulating Factor (CSF2/GM-CSF) |
| Prolactin family | Colony Stimulating Factor 3/Granulocyte-Stimulating Factor (CSF3/G-CSF) |
| IL-6 Family | IL-6 |
| IL-12 Family | IL-12 (p70) |
| Type II Cytokine families signaling through Class II cytokine receptors (CRF2 family or IL-10/IFN superfamily) | |
| IL-10 Family | IL-10 |
| Type II IFN | IFNγ |
| Cytokine families signaling through immunoglobulin (Ig) superfamily cytokine receptors non-Receptor tyrosine-kinase (RTK) | |
| IL-1 Family | IL-1β, IL-1Ra |
| Cytokine TNF family signaling through TNF receptor family | |
| Family A | TNFα (TNFSF2) |
| Chemokine superfamily signaling through chemokine receptors (seven-transmembrane heptahelical(serpentine) receptors associated with G-protein trimeric system) | |
| Chemokine CC Motif Ligand Family (CCL) | MCP1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5) and eotaxin(CCL11) |
| Chemokine CXC Motif Ligand Family (CXCL) | IP-10 (CXCL10), IL-8 (CXCL8) |
| Orphan and other cytokine family members | |
| IL-17 Family | IL-17A |
| Growth factors and signaling proteins | |
| Platelet-Derived Growth Factor Family | PDGF-BB (Platelet-Derived Growth Factor subunit B) |
| Vascular Endothelial Growth Factor Family | VEGF (Vascular Endothelial Growth Factor)/Vascular Permeability Factor (VPF) |
| Fibroblast Growth Factor Family | bFGF(FGF2/FGF-β) |
References
- Bethesda: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases. 2018 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States. 2018. Available online: https://www.usrds.org/2018/view/Default.aspx (accessed on 16 July 2019).
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: https://www.diabetesatlas.org (accessed on 27 April 2020).
- Viazzi, F.; Russo, G.T.; Ceriello, A.; Fioretto, P.; Giorda, C.; De Cosmo, S.; Pontremoli, R. Natural history and risk factors for diabetic kidney disease in patients with T2D: Lessons from the AMD-annals. J. Nephrol. 2019, 32, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Korbut, A.I.; Klimontov, V.V.; Vinogradov, I.V.; Romanov, V.V. Risk factors and urinary biomarkers of non-albuminuric and albuminuric chronic kidney disease in patients with type 2 diabetes. World J. Diabetes 2019, 10, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Solini, A.; Bonora, E.; Fondelli, C.; Orsi, E.; Nicolucci, A.; Penno, G.; RIACE Study Group. Chronic kidney disease in type 2 diabetes: Lessons from the Renal Insufficiency and Cardiovascular Events (RIACE) Italian Multicentre Study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Di Vincenzo, A.; Bettini, S.; Russo, L.; Mazzocut, S.; Mauer, M.; Fioretto, P. Renal structure in type 2 diabetes: Facts and misconceptions [published online ahead of print, 2020 Jul 12]. J. Nephrol. 2020. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Martín-Núñez, E.; Muros-de-Fuentes, M.; Mora-Fernández, C.; Navarro-González, J.F. Inflammatory cytokines in diabetic nephropathy. J. Diabetes Res. 2015, 2015, 948417. [Google Scholar] [CrossRef]
- Pérez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Inflammation in diabetic kidney disease. Nephron 2019, 143, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Opazo-Ríos, L.; Guerrero-Hue, M.; García-Caballero, C.; Vázquez-Carballo, C.; Mas, S.; Sanz, A.B.; Herencia, C.; Mezzano, S.; et al. Pathogenic pathways and therapeutic approaches targeting inflammation in diabetic nephropathy. Int. J. Mol. Sci. 2020, 21, 3798. [Google Scholar] [CrossRef]
- Kelly, K.J.; Dominguez, J.H. Rapid progression of diabetic nephropathy is linked to inflammation and episodes of acute renal failure. Am. J. Nephrol. 2010, 32, 469–475. [Google Scholar] [CrossRef]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; IJpelaar, D.H.T. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Zhao, Y. Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS ONE 2019, 14, e0221991. [Google Scholar] [CrossRef]
- Wu, C.C.; Chen, J.S.; Lu, K.C.; Chen, C.C.; Lin, S.H.; Chu, P.; Sytwu, H.K.; Lin, Y.F. Aberrant cytokines/chemokines production correlate with proteinuria in patients with overt diabetic nephropathy. Clin. Chim. Acta 2010, 411, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Araújo, L.S.; Torquato, B.G.S.; da Silva, C.A.; Dos Reis Monteiro, M.L.G.; Dos Santos Martins, A.L.M.; da Silva, M.V.; Dos Reis, M.A.; Machado, J.R. Renal expression of cytokines and chemokines in diabetic nephropathy. BMC Nephrol. 2020, 21, 308. [Google Scholar] [CrossRef] [PubMed]
- Salti, T.; Khazim, K.; Haddad, R.; Campisi-Pinto, S.; Bar-Sela, G.; Cohen, I. Glucose induces IL-1α-dependent inflammation and extracellular matrix proteins expression and deposition in renal tubular epithelial cells in diabetic kidney disease. Front. Immunol. 2020, 11, 1270. [Google Scholar] [CrossRef] [PubMed]
- Stefan, G.; Stancu, S.; Zugravu, A.; Petre, N.; Mandache, E.; Mircescu, G. Histologic predictors of renal outcome in diabetic nephropathy: Beyond renal pathology society classification. Medicine 2019, 98, e16333. [Google Scholar] [CrossRef] [PubMed]
- Konenkov, V.I.; Klimontov, V.V.; Myakina, N.E.; Tyan, N.V.; Fazullina, O.N.; Romanov, V.V. Increased serum concentrations of inflammatory cytokines in type 2 diabetic patients with chronic kidney disease. Ther. Arch. 2015, 87, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Araújo, L.S.; da Silva, M.V.; da Silva, C.A.; Borges, M.F.; Palhares, H.M.D.C.; Rocha, L.P.; Corrêa, R.R.M.; Rodrigues Júnior, V.; Dos Reis, M.A.; Machado, J.R. Analysis of serum inflammatory mediators in type 2 diabetic patients and their influence on renal function. PLoS ONE 2020, 15, e0229765. [Google Scholar] [CrossRef] [Green Version]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Dom, Z.I.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Sierra-Mondragon, E.; Molina-Jijon, E.; Namorado-Tonix, C.; Rodríguez-Muñoz, R.; Pedraza-Chaverri, J.; Reyes, J.L. All-trans retinoic acid ameliorates inflammatory response mediated by TLR4/NF-κB during initiation of diabetic nephropathy. J. Nutr. Biochem. 2018, 60, 47–60. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Di, Y.P. Fast and efficient measurement of clinical and biological samples using immunoassay-based multiplexing systems. Methods Mol. Biol. 2020, 2102, 129–147. [Google Scholar] [CrossRef]
- Lioudaki, E.; Stylianou, K.G.; Petrakis, I.; Kokologiannakis, G.; Passam, A.; Mikhailidis, D.P.; Daphnis, E.K.; Ganotakis, E.S. Increased urinary excretion of podocyte markers in normoalbuminuric patients with diabetes. Nephron 2015, 131, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Abe, M.; Moritani, H.; Mitori, H.; Kondo, M.; Tanaka-Amino, K.; Eguchi, M.; Imasato, A.; Inoki, Y.; Kajiyama, H.; et al. Potential of urinary nephrin as a biomarker reflecting podocyte dysfunction in various kidney disease models. Exp. Biol. Med. 2016, 241, 1865–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Shazly, A.A.A.; Sallam, A.M.; El-Hefnawy, M.H.; El-Mesallamy, H.O. Epidermal growth factor receptor and podocin predict nephropathy progression in type 2 diabetic patients through interaction with the autophagy influencer ULK-1. J. Diabetes Complicat. 2019, 33, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Wang, Y.; Cai, G.; Liang, J.; Yue, C.; Wang, F.; Song, J.; Wang, J.; Liu, M.; Luo, J.; et al. Elevated serum concentrations of HE4 as a novel biomarker of disease severity and renal fibrosis in kidney disease. Oncotarget 2016, 7, 67748–67759. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Yang, Q.; Li, X.; Qin, Y. Potential association between elevated serum human epididymis protein 4 and renal fibrosis: A systemic review and meta-analysis. Medicine 2017, 96, e7824. [Google Scholar] [CrossRef]
- García Morán, G.A.; Parra-Medina, R.; Cardona, A.G.; Cardona, A.G.; Quintero-Ronderos, P.; Rodríguez, É.G. Cytokines, chemokines and growth factors. In Autoimmunity: From Bench to Bedside; Anaya, J.M., Shoenfeld, Y., Rojas-Villarraga, A., Eds.; El Rosario University Press: Bogota, Colombia, 2013; ISBN 9789587383768. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459450/ (accessed on 24 April 2020).
- Bafico, A.; Aaronson, S.A. Classification of growth factors and their receptors. In Holland-Frei Cancer Medicine, 6th ed.; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Eds.; BC Decker: Hamilton, ON, Canada, 2003; ISBN 101550092138. Available online: https://www.ncbi.nlm.nih.gov/books/NBK12423/ (accessed on 24 April 2020).
- Sherbet, G.V.; Gajanan, V. Growth Factors and Their Receptors in Cell Differentiation, Cancer and Cancer Therapy; Elsevier: Amsterdam, The Netherlands; Boston, MA, USA, 2011; 347p. [Google Scholar]
- Pfister, I.B.; Zandi, S.; Gerhardt, C.; Spindler, J.; Reichen, N.; Garweg, J.G. Risks and Challenges in Interpreting Simultaneous Analyses of Multiple Cytokines. Transl. Vis. Sci. Technol. 2020, 9, 27. [Google Scholar] [CrossRef]
- EUTOX Uremic Toxin Database. Available online: https://www.uremic-toxins.org/eutox-database/ (accessed on 27 April 2020).
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni EstanTodos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef]
- Norlander, A.E.; Madhur, M.S. Inflammatory cytokines regulate renal sodium transporters: How, where, and why? Am. J. Physiol.-Ren. Physiol. 2017, 313, F141–F144. [Google Scholar] [CrossRef] [Green Version]
- Feigerlová, E.; Battaglia-Hsu, S.F. IL-6 signaling in diabetic nephropathy: From pathophysiology to therapeutic perspectives. Cytokine Growth Factor Rev. 2017, 37, 57–65. [Google Scholar] [CrossRef]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 signaling pathway and its role in kidney disease: An update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Magno, A.L.; Heart, L.Y.; Carnagarin, R.; Schlaich, M.P.; Matthews, V.B. Current knowledge of IL-6 cytokine family members in acute and chronic kidney disease. Biomedicines 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, R.; Jayakumar, C.; Chen, F.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Low-dose IL-17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J. Am. Soc. Nephrol. 2016, 27, 745–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Liu, X.; Yin, J.; Wu, H.; Cai, X.; Wang, N.; Qian, Y.; Wang, F. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism 2018, 83, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sangoi, M.B.; Carvalho, J.A.M.; Guarda, N.S.; Duarte, T.; Duarte, M.M.M.F.; Premaor, M.O.; Comim, F.V.; Moretto, M.B.; Moresco, R.N. Association between urinary levels of interleukin-6, interleukin-10 and tumor necrosis factor-alpha with glomerular and tubular damage indicators in patients with type 2 diabetes. Clin. Lab. 2019, 65. [Google Scholar] [CrossRef]
- Cortvrindt, C.; Speeckaert, R.; Moerman, A.; Delanghe, J.R.; Speeckaert, M.M. The role of interleukin-17A in the pathogenesis of kidney diseases. Pathology 2017, 49, 247–258. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.J.; Chen, X.; Kwan, T.; Chadban, S.J.; Wu, H. Interleukin 17A promotes diabetic kidney injury. Sci. Rep. 2019, 9, 2264. [Google Scholar] [CrossRef] [Green Version]
- Perlman, A.S.; Chevalier, J.M.; Wilkinson, P.; Liu, H.; Parker, T.; Levine, D.M.; Sloan, B.J.; Gong, A.; Sherman, R.; Farrell, F.X. Serum inflammatory and immune mediators are elevated in early stage diabetic nephropathy. Ann. Clin. Lab. Sci. 2015, 45, 256–263. [Google Scholar]
- Coto, E.; Gómez, J.; Suárez, B.; Tranche, S.; Díaz-Corte, C.; Ortiz, A.; Ruiz-Ortega, M.; Coto-Segura, P.; Batalla, A.; López-Larrea, C. Association between the IL17RA rs4819554 polymorphism and reduced renal filtration rate in the Spanish RENASTUR cohort. Hum. Immunol. 2015, 76, 75–78. [Google Scholar] [CrossRef]
- Kuo, H.L.; Huang, C.C.; Lin, T.Y.; Lin, C.Y. IL-17 and CD40 ligand synergistically stimulate the chronicity of diabetic nephropathy. Nephrol. Dial. Transplant. 2018, 33, 248–256. [Google Scholar] [CrossRef]
- Norlander, A.E.; Saleh, M.A.; Kamat, N.V.; Ko, B.; Gnecco, J.; Zhu, L.; Dale, B.L.; Iwakura, Y.; Hoover, R.S.; McDonough, A.A.; et al. Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II-induced hypertension. Hypertension 2016, 68, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Nishida, M.; Hamaoka, K. How does G-CSF act on the kidney during acute tubular injury? Nephron Exp. Nephrol. 2006, 104, e123–e128. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.J.; Ryu, J.H.; Piao, H.; Hwang, J.H.; Han, D.; Lee, S.K.; Jang, J.Y.; Lee, J.; Koo, T.Y.; Yang, J. Granulocyte colony-stimulating factor attenuates renal ischemia-reperfusion injury by inducing myeloid-derived suppressor cells. J. Am. Soc. Nephrol. 2020, 31, 731–746. [Google Scholar] [CrossRef] [PubMed]
- So, B.I.; Song, Y.S.; Fang, C.H.; Park, J.Y.; Lee, Y.; Shin, J.H.; Kim, H.; Kim, K.S. G-CSF prevents progression of diabetic nephropathy in rat. PLoS ONE 2013, 8, e77048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbas, O.; Yapislar, H.; Oltulu, F.; Yavasoğlu, A.; Aktug, H.; Taskiran, D. Nephro-protective effect of granulocyte colony-stimulating factor in streptozotocin induced diabetic rats. Biotech. Histochem. 2014, 89, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ruster, C.; Wolf, G. The role of chemokines and chemokine receptors in diabetic nephropathy. Front. Biosci. 2008, 13, 944–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhavsar, I.; Miller, C.S.; Al-Sabbagh, M. Macrophage inflammatory protein-1 alpha (MIP-1 alpha)/CCL3: As a biomarker. In General Methods in Biomarker Research and Their Applications; Preedy, V., Patel, V., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 243–249. Print ISBN 978-94-007-7695-1, Online ISBN 978-94-007-7696-8. [Google Scholar] [CrossRef]
- Zheng, G.; Wang, Y.; Mahajan, D.; Qin, X.; Wang, Y.; Wang, Y.; Alexander, S.I.; Harris, D.C. The role of tubulointerstitial inflammation. Kidney Int. 2005, 67 (Suppl. S94), S96–S100. [Google Scholar] [CrossRef] [Green Version]
- Correa-Costa, M.; Braga, T.T.; Felizardo, R.J.; Andrade-Oliveira, V.; Perez, K.R.; Cuccovia, I.M.; Hiyane, M.I.; da Silva, J.S.; Câmara, N.O. Macrophage trafficking as key mediator of adenine-induced kidney injury. Mediat. Inflamm. 2014, 2014, 291024. [Google Scholar] [CrossRef]
- Strutz, F. The role of FGF-2 in renal fibrogenesis. Front. Biosci. 2009, 1, 125–131. [Google Scholar] [CrossRef]
- Xu, Z.; Dai, C. Ablation of FGFR2 in fibroblasts ameliorates kidney fibrosis after ischemia/reperfusion injury in mice. Kidney Dis. 2017, 3, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Golembiewska, E.; Lindholm, B.; Stenvinkel, P. End-stage renal disease, inflammation and cardiovascular outcomes. In Expanded Hemodialysis—Innovative Clinical Approach in Dialysis. Contrib Nephrol.; Karger Publishers: Basel, Switzerland, 2017; Volume 191, pp. 32–43. [Google Scholar] [CrossRef]
- Wei, T.; Shu, Q.; Ning, J.; Wang, S.; Li, C.; Zhao, L.; Zheng, H.; Gao, H. The protective effect of basic fibroblast growth factor on diabetic nephropathy through remodeling metabolic phenotype and suppressing oxidative stress in mice. Front. Pharmacol. 2020, 11, 66. [Google Scholar] [CrossRef]
- Sheng, W.S.; Xu, H.L.; Zheng, L.; Zhuang, Y.D.; Jiao, L.Z.; Zhou, J.F.; ZhuGe, D.L.; Chi, T.T.; Zhao, Y.Z.; Lan, L. Intrarenal delivery of bFGF-loaded liposome under guiding of ultrasound-targeted microbubble destruction prevent diabetic nephropathy through inhibition of inflammation. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. S2), 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, R.C.; Quimby, K.R.; Greenidge, A.R. M1/M2 macrophages in diabetic nephropathy: Nrf2/HO-1 as therapeutic targets. Curr. Pharm. Des. 2018, 24, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, R.; Xie, J.; Xiong, H.; He, J.C.; Chen, N. Interleukin-10 deficiency aggravates kidney inflammation and fibrosis in the unilateral ureteral obstruction mouse model. Lab. Investig. 2013, 93, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Soranno, D.E.; Lu, H.D.; Weber, H.M.; Rai, R.; Burdick, J.A. Immunotherapy with injectable hydrogels to treat obstructive nephropathy. J. Biomed. Mater. Res. Part A 2014, 102, 2173–2180. [Google Scholar] [CrossRef] [Green Version]
- Rodell, C.B.; Rai, R.; Faubel, S.; Burdick, J.A.; Soranno, D.E. Local immunotherapy via delivery of interleukin-10 and transforming growth factor β antagonist for treatment of chronic kidney disease. J. Control Release 2015, 206, 131–139. [Google Scholar] [CrossRef]
- Gnudi, L.; Benedetti, S.; Woolf, A.S.; Long, D.A. Vascular growth factors play critical roles in kidney glomeruli. Clin. Sci. 2015, 129, 1225–1236. [Google Scholar] [CrossRef]
- Lin, S.; Teng, J.; Li, J.; Sun, F.; Yuan, D.; Chang, J. Association of chemerin and vascular endothelial growth factor (VEGF) with diabetic nephropathy. Med. Sci. Monit. 2016, 22, 3209–3214. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Antoniadi, G.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. The renal endothelium in diabetic nephropathy. Ren. Fail. 2013, 35, 592–599. [Google Scholar] [CrossRef]
- Majumder, S.; Advani, A. VEGF and the diabetic kidney: More than too much of a good thing. J. Diabetes Complicat. 2017, 31, 273–279. [Google Scholar] [CrossRef]
- Schrijvers, B.F.; Flyvbjerg, A.; De Vriese, A.S. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int. 2004, 65, 2003–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.J. Eotaxin-1 (CCL11). Front. Immunol. 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, T.; Rothenberg, M.E. The regulatory function of eosinophils. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Gauckler, P.; Shin, J.I.; Mayer, G.; Kronbichler, A. Eosinophilia and kidney disease: More than just an incidental finding? J. Clin. Med. 2018, 7, 529. [Google Scholar] [CrossRef] [Green Version]
- Laurentius, T.; Raffetseder, U.; Fellner, C.; Kob, R.; Nourbakhsh, M.; Floege, J.; Bertsch, T.; Bollheimer, L.C.; Ostendorf, T. High-fat diet-induced obesity causes an inflammatory microenvironment in the kidneys of aging Long-Evans rats. J. Inflamm. 2019, 16, 14. [Google Scholar] [CrossRef]
- Mansouri, L.; Paulsson, J.M.; Moshfegh, A.; Jacobson, S.H.; Lundahl, J. Leukocyte proliferation and immune modulator production in patients with chronic kidney disease. PLoS ONE 2013, 8, e73141. [Google Scholar] [CrossRef] [Green Version]
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Klimontov, V.V.; Tyan, N.V.; Fazullina, O.N.; Myakina, N.E.; Lykov, A.P.; Konenkov, V.I. Clinical and metabolic factors associated with chronic low-grade inflammation in type 2 diabetic patients. Diabetes Mellit. 2016, 19, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Klimontov, V.V.; Korbut, A.I. Albuminuric and non-albuminuric patterns of chronic kidney disease in type 2 diabetes. Diabetes Metab. Syndr. 2019, 13, 474–479. [Google Scholar] [CrossRef]
- Fioretto, P.; Caramori, M.L.; Mauer, M. The kidney in diabetes: Dynamic pathways of injury and repair. The Camillo Golgi Lecture 2007. Diabetologia 2008, 51, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Ekinci, E.I.; Jerums, G.; Skene, A.; Crammer, P.; Power, D.; Cheong, K.Y.; Panagiotopoulos, S.; McNeil, K.; Baker, S.T.; Fioretto, P.; et al. Renal structure in normoalbuminuric and albuminuric patients with type 2 diabetes and impaired renal function. Diabetes Care 2013, 36, 3620–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles-Osorio, M.L.; Sabath, E. Tubular dysfunction and non-albuminuric renal disease in subjects with type 2 diabetes mellitus. Rev. Investig. Clin. 2014, 66, 234–239. [Google Scholar]
- Kosmas, C.E.; Silverio, D.; Tsomidou, C.; Salcedo, M.D.; Montan, P.D.; Guzman, E. The impact of insulin resistance and chronic kidney disease on inflammation and cardiovascular disease. Clin. Med. Insights Endocrinol. Diabetes 2018, 11, 1179551418792257. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J. Immunol. Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, J.B.; Li, S.; Ton, T.G.N.; Peng, Y.; Hansen, M.K.; Neslusan, C.; Riley, R.; Liu, J.; Gilbertson, D.T. Association of diabetes-related kidney disease with cardiovascular and non-cardiovascular outcomes: A retrospective cohort study. BMC Endocr. Disord. 2019, 19, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytvyn, Y.; Bjornstad, P.; van Raalte, D.H.; Heerspink, H.L.; Cherney, D.Z.I. The new biology of diabetic kidney disease-mechanisms and therapeutic implications. Endocr. Rev. 2020, 41, 202–231. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Wang, S.; Wang, M.; Lu, W. Renoprotection of dapagliflozin in human renal proximal tubular cells via the inhibition of the high mobility group box 1-receptor for advanced glycation end products-nuclear factor-κB signaling pathway. Mol. Med. Rep. 2018, 18, 3625–3630. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Butler, A.E.; Atkin, S.L.; Katsiki, N.; Sahebkar, A. Sodium-glucose cotransporter 2 inhibitors and inflammation in chronic kidney disease: Possible molecular pathways. J. Cell. Physiol. 2018, 234, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Xu, S.; Wang, Z.; Liu, H.; Peng, L.; Fang, Q.; Deng, T.; Zhang, W.; Lou, J. Recombinant human GLP-1 (rhGLP-1) alleviating renal tubulointestitial injury in diabetic STZ-induced rats. Biochem. Biophys. Res. Commun. 2018, 495, 793–800. [Google Scholar] [CrossRef]
- Chang, J.T.; Liang, Y.J.; Hsu, C.Y.; Chen, C.Y.; Chen, P.J.; Yang, Y.F.; Chen, Y.L.; Pei, D.; Chang, J.B.; Leu, J.G. Glucagon-like peptide receptor agonists attenuate advanced glycation end products-induced inflammation in rat mesangial cells. BMC Pharmacol. Toxicol. 2017, 18, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Maleki, M.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. Anti-inflammatory potentials of incretin-based therapies used in the management of diabetes. Life Sci. 2020, 241, 117152. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef] [PubMed]






| Parameter | CKD− | NA-CKD | A-CKD− | A-CKD+ |
|---|---|---|---|---|
| N | 33 | 33 | 32 | 32 |
| Sex, M/F, n | 12/21 | 12/21 | 12/20 | 12/20 |
| Age, years | 64 (58; 69) | 65 (58; 67) | 63 (57; 67) | 65 (58; 69.5) |
| Smokers, n (%) | 3 (9.1%) | 5 (15.1%) | 4 (12.5%) | 3 (9.4%) |
| BMI, kg/m2 | 34.4 (28.1; 36.9) | 34.7 (30.1; 38.9) | 33.8 (31.2; 40.0) | 32.9 (30.3; 36.7) |
| WHR | 0.97 (0.93; 1.1) | 1.02 (0.9; 1.1) | 1.00 (0.93; 1.1) | 1.00 (0.91; 1.07) |
| Diabetes duration, years | 13 (11; 16) | 14 (12; 19) | 13.5 (11; 17) | 13.5 (11; 17.5) |
| Diabetic complications and associated diseases | ||||
| Obesity, n (%) | 21 (63.6%) | 24 (72.7%) | 25 (78.2%) | 26 (81.3%) |
| Diabetic retinopathy, n (%) | 17 (51.5%) | 17 (51.5%) | 20 (62.5%) | 21 (65.6%) |
| Arterial hypertension, n (%) | 31 (93.9%) | 33 (100%) | 31 (96.9%) | 32 (100%) |
| Coronary artery disease, n (%) | 13 (39.4%) | 16 (48.5%) | 16 (50.0%) | 18 (56.2%) |
| Myocardial infarction in anamnesis, n (%) | 4 (12.1) | 7 (21.2%) | 5 (15.6%) | 10 (31.3%) |
| Cerebrovascular event in anamnesis, n (%) | 1 (3.0%) | 6 (18.2%) | 3 (9.4%) | 2 (6.3%) |
| Peripheral artery disease, n (%) | 17 (51.5%) | 22 (66.7%) | 20 (62.5%) | 23 (71.9%) |
| Treatment | ||||
| Metformin, n (%) | 26 (78.8%) | 20 (60.6%) | 23 (71.9%) | 20 (62.5%) |
| Sulfonylurea, n (%) | 8 (24.2%) | 15 (45.5%) | 12 (37.5%) | 9 (28.1%) |
| Insulin, n (%) | 21 (63.6%) | 22 (66.7%) | 21 (65.6%) | 26 (81.3%) |
| RAS blockers, n (%) | 33 (100%) | 27 (81.8%) | 26 (81.3%) | 27 (84.4%) |
| Diuretics, n (%) | 12 (36.4%) | 17 (51.5%) | 16 (50.0%) | 17 (53.1%) |
| Calcium channel blockers, n (%) | 8 (24.2%) | 16 (48.5%) | 10 (31.3%) | 14 (43.8%) |
| Antiplatelet agents, n (%) | 21 (63.6%) | 27 (81.8%) | 19 (59.4%) | 24 (75.0%) |
| Statins, n (%) | 13 (39.4%) | 23 (69.7%) | 7 (21.9%) ### | 17 (53.1%) |
| Parameter | CKD− | NA-CKD | A-CKD− | A-CKD+ | |
|---|---|---|---|---|---|
| N | 33 | 33 | 32 | 32 | |
| Renal tests | |||||
| Serum creatinine, μmol/L | 76 (68; 87) | 111 (102; 124) *** | 87 (79; 97) §§§ ### | 117 (98; 144) *** | |
| eGFR, mL/min/1.73 m2 | 84 (73; 94) | 51 (45; 55) *** | 71 (65; 77) §§§ ### | 51 (43; 55) *** | |
| UACR, mg/mmol | 0.6 (0.3; 0.9) | 0.6 (0.3; 0.9) | 13.5 (6.4; 38.4) *** ### | 14.1 (7.2; 82.5) *** ### | |
| Urinary nephrin excretion, ng/mmol | 12.1 (4.7; 21.1) | 7.8 (4.1; 16.3) | 22.6 (16.6; 36.2) *** ### | 22.2 (17.1; 32.9) *** ### | |
| Urinary podocin excretion, ng/mmol | 133 (86; 222) | 105 (65.4; 193.9) | 250 (144; 421) *** ### | 268 (134; 411) *** ### | |
| Urinary excretion of WFDC2, ng/mmol | M | 442 (310; 941) +++ | 723 (406; 811) +++ | N/D | 628 (410; 1277) +++ |
| F | 5.6 (0; 81) | 129 (0; 330) ** | N/D | 231 (110; 619) ** | |
| Other biochemical parameters | |||||
| HbA1C | % | 8.48 (7.5; 9.8) | 9.06 (7.9; 10.26) | 9.36 (8.25; 11.58) | 8.81 (7.64; 10.1) |
| mmol/mol | 69 (58; 84) | 76 (63; 89) | 79 (67; 103) | 73 (60; 87) | |
| Total cholesterol, mmol/L | 4.92 (4.32; 5.51) | 5.14 (4.11; 6.07) | 4.89 (4.06; 5.89) | 5.5 (4.19; 6.25) | |
| LDL-cholesterol, mmol/L | 3.02 (2.7; 3.59) | 3.21 (2.51; 3.92) | 3.19 (2.6; 3.95) | 3.26 (2.54; 4.18) | |
| HDL-cholesterol, mmol/L | 1.13 (1; 1.31) | 1.22 (1.09; 1.41) | 1.07 (0.93; 1.25) # | 1.12 (1; 1.41) | |
| Triglycerides, mmol/L | 1.76 (1.15; 2.47) | 1.9 (1.5; 2.6) | 2.2 (1.64; 3.3) | 1.93 (1.54; 3.08) | |
| Uric acid, μmol/L | 298 (243; 352) | 341 (315; 392) | 322 (281; 383) | 366 (272; 418) | |
| Serum hs-CRP, mg/L | 4.1 (2.3; 8.2) | 6.2 (2.3; 8.4) | 4.4 (1.4; 7.3) | 6.7 (2.2; 13.1) | |
| Hematology and coagulation tests | |||||
| Hemoglobin, g/L | 139 (131; 148) | 141 (128; 151) | 141 (127; 152) | 141 (126; 153) | |
| RBC, × 1012/L | 4.79 (4.5; 4.9) | 4.72 (4.44; 5.06) | 4.85 (4.6; 5.13) | 4.7 (4.26; 4.87) | |
| WBC, × 109/L | 6.18 (5.93; 7.65) | 7.44 (5.61; 8.34) | 7.52 (6.03; 8.85) | 7.31 (5.53; 8.15) | |
| Platelets, × 109/L | 244 (218; 269) | 253 (207; 282) | 247 (212; 278) | 206 (181; 275) | |
| ESR, mm/h | 15 (9; 22) | 19 (12; 26.5) | 22.5 (15.5; 29) | 23.5 (18; 30) ** | |
| Fibrinogen, g/L | 4.15 (3.3; 5.3) | 3.9 (3.2; 4.45) | 4.4 (3.7; 5.5) | 4.6 (4; 5.3) | |
| SFMCs, mg/dL | 4 (3.5; 13.5) | 14 (3.5; 21) | 10.5 (6; 21.5) | 12 (8; 19) | |
| D-dimer, ng/mL | 255 (215; 309) | 251 (231; 385) | 268 (231; 297) | 277 (238; 327) | |
| Parameter | Crude OR (95% CI), p-Value | Adjusted OR (95% CI), p-Value |
|---|---|---|
| eGFR <60 mL/min × 1.73 m21 | ||
| IL-17A, pg/mL | 1.04(1.01–1.09), p = 0.004 | 1.03(1.01–1.09), p = 0.01 |
| MIP-1α, pg/mL | 1.30(1.06–1.49), p = 0.02 | 1.15(1.02–1.50), p = 0.03 |
| Serum hs-CRP, mg/L | 1.01(0.96–1.05), p = 0.10 | 1.20(1.02–1.30), p = 0.02 |
| Age, years | 1.06 (0.98–1.15), p = 0.16 | 1.04(0.99–1.14), p = 0.08 |
| UACR ≥3.0 mg/mmol2 | ||
| Eotaxin, 10 pg/mL | 0.98(0.94–1.00), p = 0.09 | 0.95(0.90–1.00), p = 0.03 |
| IL-15, 10 pg/mL | 1.03(0.98–1.06), p = 0.20 | 1.04(0.98–1.07), p = 0.09 |
| Parameter | Crude OR (95% CI), p-Value | Adjusted OR (95% CI), p-Value |
|---|---|---|
| NA-CKD1 | ||
| IL-17A, pg/mL | 1.08 (1.04–1.18), p = 0.001 | 1.06 (1.02–1.12), p = 0.004 |
| MIP-1α, pg/mL | 1.70 (1.20–2.30), p = 0.008 | 1.45 (1.02–2.06), p = 0.03 |
| A-CKD−2 | ||
| IL-13, pg/mL | 1.20 (0.96–1.50), p = 0.09 | 1.24 (1.01–1.54), p = 0.04 |
| HbA1C, % | 1.15 (0.94–1.52), p = 0.12 | 1.30 (0.98–1.62), p = 0.06 |
| A-CKD+3 | ||
| IL-6, pg/mL | 1.27 (1.02–1.64), p = 0.02 | 1.37 (1.08–1.69), p = 0.009 |
| Serum hs-CRP, mg/L | 1.06 (0.92–1.32), p = 0.21 | 1.18 (1.01–1.36), p = 0.04 |
| Age, years | 1.03 (0.90; 1.12), p = 0.26 | 1.09 (0.98–1.20), p = 0.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimontov, V.V.; Korbut, A.I.; Orlov, N.B.; Dashkin, M.V.; Konenkov, V.I. Multiplex Bead Array Assay of a Panel of Circulating Cytokines and Growth Factors in Patients with Albuminuric and Non-Albuminuric Diabetic Kidney Disease. J. Clin. Med. 2020, 9, 3006. https://doi.org/10.3390/jcm9093006
Klimontov VV, Korbut AI, Orlov NB, Dashkin MV, Konenkov VI. Multiplex Bead Array Assay of a Panel of Circulating Cytokines and Growth Factors in Patients with Albuminuric and Non-Albuminuric Diabetic Kidney Disease. Journal of Clinical Medicine. 2020; 9(9):3006. https://doi.org/10.3390/jcm9093006
Chicago/Turabian StyleKlimontov, Vadim V., Anton I. Korbut, Nikolai B. Orlov, Maksim V. Dashkin, and Vladimir I. Konenkov. 2020. "Multiplex Bead Array Assay of a Panel of Circulating Cytokines and Growth Factors in Patients with Albuminuric and Non-Albuminuric Diabetic Kidney Disease" Journal of Clinical Medicine 9, no. 9: 3006. https://doi.org/10.3390/jcm9093006
APA StyleKlimontov, V. V., Korbut, A. I., Orlov, N. B., Dashkin, M. V., & Konenkov, V. I. (2020). Multiplex Bead Array Assay of a Panel of Circulating Cytokines and Growth Factors in Patients with Albuminuric and Non-Albuminuric Diabetic Kidney Disease. Journal of Clinical Medicine, 9(9), 3006. https://doi.org/10.3390/jcm9093006