Modifications of Titin Contribute to the Progression of Cardiomyopathy and Represent a Therapeutic Target for Treatment of Heart Failure


Abstract
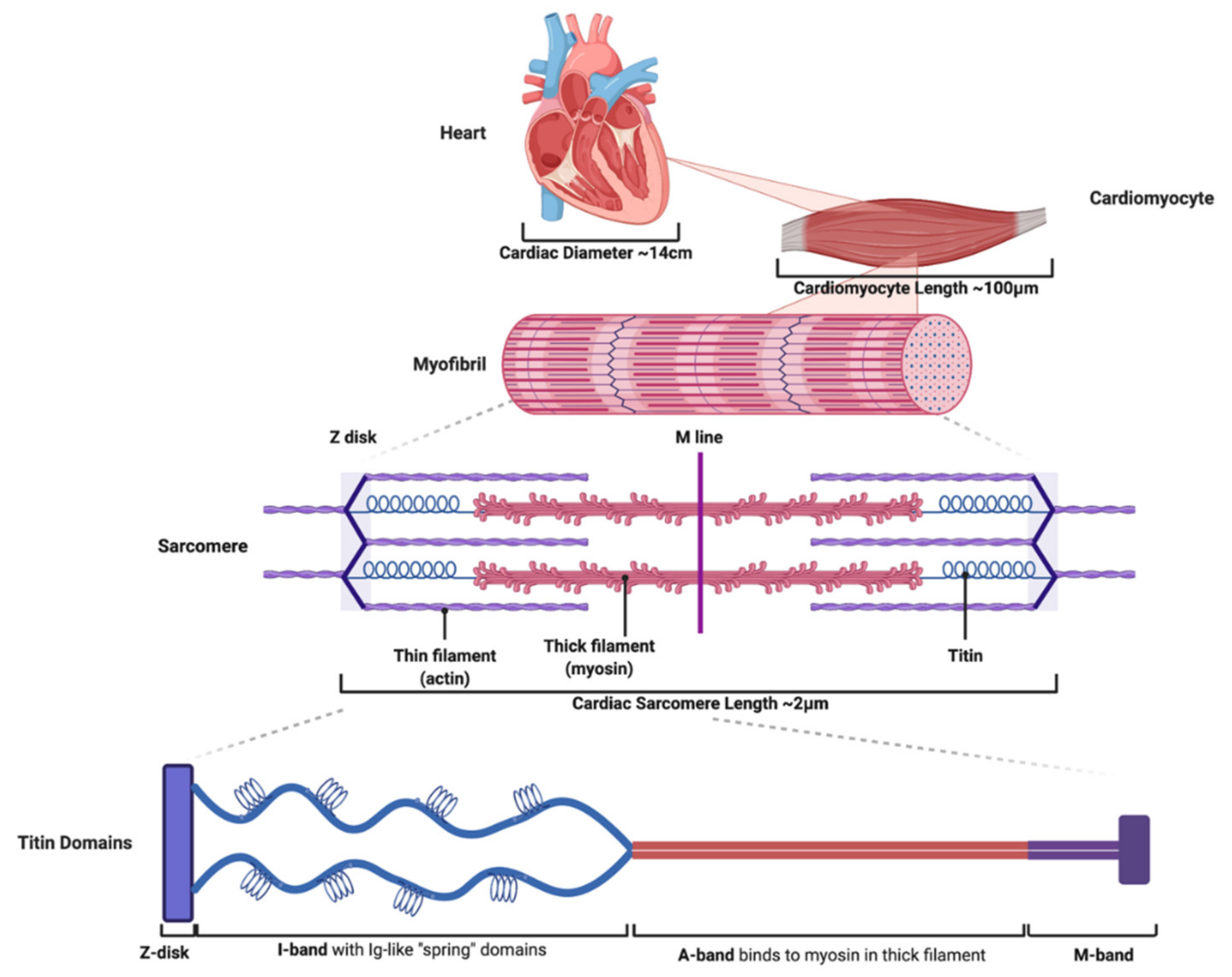
:1. Titin Is an Essential Molecule within the Sarcomere Where It Contributes to Cardiac Function
1.1. Titin Contains Four Structural Domains That Impart Important Molecular Properties to the Sarcomere during Cardiac Function
1.2. Titin’s Structure Is Essential to Normal Cardiac Function, Passive Tension, and Length Dependent Activation
1.3. Abnormalities in Titin Contribute to Systolic and Diastolic Heart Failure
2. Truncation Mutations in TTN Cause Dilated Cardiomyopathy (DCM)
2.1. TTN Truncation Variations in Highly Constitutively Expressed Exons Cause DCM
2.2. TTNtv Cause DCM through Haploinsufficiency, Increased Metabolic Stress, and Activation of the mTOR Signalling Cascade
2.3. TTNtv Are Associated with Late Presentation Of DCM, Are More Pathogenic in European Ancestry, and Are Associated with Early Atrial Arrhythmias
2.4. TTNtv Are Associated with Worse Outcomes When Combined with Additional Cardiac Risk Factors
2.5. Inhibition of the mTOR Pathway and Antisense Oligo Nucleotide Mediated Exon Skipping Are Potential Therapies for TTNtv Related DCM
3. Titin Transcriptional Modifications Are Associated with Development of Cardiomyopathy and Represent a Therapeutic Target for Inherited and Acquired Heart Failure
3.1. Transcriptional Changes of Titin Isosforms Alter Passive Tension and Myocardial Stiffness in Heart Failure
3.2. RBM20 Is a Key Regulator of Titin Isoform Preference
3.3. Modifications of Titin Isoforms Represent a Therapeutic Target for Treatment of Heart Failure
4. Post-Translational Modifications of Titin Affect Myocardial Physiology and Can Be Modified to Treat Heart Failure
4.1. Phosphorylation of the PEVK Element of Titin Increases Cardiomyocyte Passive Tension
4.2. Phosphorylation of the N2B Unique Sequence (N2Bus) Element of Titin Decreases Cardiomyocyte Passive Tension
4.3. Diabetes Contributes to Diastolic Dysfunction via Abnormal Phosphorylation of Titin, Which Can Be Reversed by Treatment with Metformin, and Neuregulin-1 (NRG-1)
4.4. Activation of Cyclic Guanosine Monophosphate (cGMP) and PKG Decreases Titin Passive Tension and Represents a Viable Therapy for Treatment of Heart Failure
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 2013, 127, e6–e245. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Petrie, M.C.; Murdoch, D.R.; Davie, A.P. Clinical epidemiology of heart failure: Public and private health burden. Eur. Heart J. 1998, 19 (Suppl. P), P9–P16. [Google Scholar]
- Zipes, D.P.; Braunwald, E. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 7th ed.; W.B. Saunders: Philadelphia, PA, USA, 2005; Volume xxi, 2183p, p. 75. [Google Scholar]
- Furst, D.O.; Osborn, M.; Nave, R.; Weber, K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: A map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell Biol. 1988, 106, 1563–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labeit, S.; Kolmerer, B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 1995, 270, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Whiting, A.; Wardale, J.; Trinick, J. Does titin regulate the length of muscle thick filaments? J. Mol. Biol. 1989, 205, 263–268. [Google Scholar] [CrossRef]
- Maruyama, K.; Kimura, S.; Yoshidomi, H.; Sawada, H.; Kikuchi, M. Molecular size and shape of beta-connectin, an elastic protein of striated muscle. J. Biochem. 1984, 95, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- LeWinter, M.M.; Wu, Y.; Labeit, S.; Granzier, H. Cardiac titin: Structure, functions and role in disease. Clin. Chim. Acta 2007, 375, 1–9. [Google Scholar] [CrossRef]
- Gotthardt, M.; Hammer, R.E.; Hubner, N.; Monti, J.; Witt, C.C.; McNabb, M.; Richardson, J.A.; Granzier, H.; Labeit, S.; Herz, J. Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J. Biol. Chem. 2003, 278, 6059–6065. [Google Scholar] [CrossRef] [Green Version]
- LeWinter, M.M.; Granzier, H. Cardiac titin: A multifunctional giant. Circulation 2010, 121, 2137–2145. [Google Scholar] [CrossRef] [Green Version]
- Linke, W.A.; Granzier, H. A spring tale: New facts on titin elasticity. Biophys. J. 1998, 75, 2613–2614. [Google Scholar] [CrossRef] [Green Version]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Barlow, D.P.; Gautel, M.; Gibson, T.; Holt, J.; Hsieh, C.L.; Francke, U.; Leonard, K.; Wardale, J.; Whiting, A.; et al. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature 1990, 345, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Bonow, R.O. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 9th ed.; Saunders: Philadelphia, PA, USA, 2012; Volume xxiv, 1961p. [Google Scholar]
- Fukuda, N.; Granzier, H.L. Titin/connectin-based modulation of the Frank-Starling mechanism of the heart. J. Muscle Res. Cell Motil. 2005, 26, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The Giant Protein Titin’s Role in Cardiomyopathy: Genetic, Transcriptional, and Post-translational Modifications of TTN and Their Contribution to Cardiac Disease. Front. Physiol. 2019, 10, 1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagueh, S.F.; Shah, G.; Wu, Y.; Torre-Amione, G.; King, N.M.; Lahmers, S.; Witt, C.C.; Becker, K.; Labeit, S.; Granzier, H.L. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004, 110, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin mutation in familial restrictive cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef]
- Golbus, J.R.; Puckelwartz, M.J.; Fahrenbach, J.P.; Dellefave-Castillo, L.M.; Wolfgeher, D.; McNally, E.M. Population-based variation in cardiomyopathy genes. Circ. Cardiovasc. Genet. 2012, 5, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Jansweijer, J.A.; Nieuwhof, K.; Russo, F.; Hoorntje, E.T.; Jongbloed, J.D.; Lekanne Deprez, R.H.; Postma, A.V.; Bronk, M.; van Rijsingen, I.A.; de Haij, S.; et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Zaunbrecher, R.J.; Abel, A.N.; Beussman, K.; Leonard, A.; von Frieling-Salewsky, M.; Fields, P.A.; Pabon, L.; Reinecke, H.; Yang, X.; Macadangdang, J.; et al. Cronos Titin Is Expressed in Human Cardiomyocytes and Necessary for Normal Sarcomere Function. Circulation 2019, 140, 1647–1660. [Google Scholar] [CrossRef]
- Van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260.e29. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [Green Version]
- Chopra, A.; Kutys, M.L.; Zhang, K.; Polacheck, W.J.; Sheng, C.C.; Luu, R.J.; Eyckmans, J.; Hinson, J.T.; Seidman, J.G.; Seidman, C.E.; et al. Force Generation via beta-Cardiac Myosin, Titin, and alpha-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Dev. Cell 2018, 44, 87–96.e5. [Google Scholar] [CrossRef] [Green Version]
- Vikhorev, P.G.; Smoktunowicz, N.; Munster, A.B.; Copeland, O.; Kostin, S.; Montgiraud, C.; Messer, A.E.; Toliat, M.R.; Li, A.; Dos Remedios, C.G.; et al. Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Sci. Rep. 2017, 7, 14829. [Google Scholar] [CrossRef]
- Zhou, Q.; Kesteven, S.; Wu, J.; Aidery, P.; Gawaz, M.; Gramlich, M.; Feneley, M.P.; Harvey, R.P. Pressure Overload by Transverse Aortic Constriction Induces Maladaptive Hypertrophy in a Titin-Truncated Mouse Model. Biomed Res. Int. 2015, 2015, 163564. [Google Scholar] [CrossRef]
- Ware, J.S.; Cook, S.A. Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nat. Rev. Cardiol. 2018, 15, 241–252. [Google Scholar] [CrossRef]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knoll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibayama, J.; Yuzyuk, T.N.; Cox, J.; Makaju, A.; Miller, M.; Lichter, J.; Li, H.; Leavy, J.D.; Franklin, S.; Zaitsev, A.V. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure: Integrated metabolomics and proteomics study. PLoS ONE 2015, 10, e0118974. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; de Marvao, A.; Adami, E.; Fiedler, L.R.; Ng, B.; Khin, E.; Rackham, O.J.; van Heesch, S.; Pua, C.J.; Kui, M.; et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet. 2017, 49, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neishabouri, S.H.; Hutson, S.M.; Davoodi, J. Chronic activation of mTOR complex 1 by branched chain amino acids and organ hypertrophy. Amino Acids 2015, 47, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Shimoshige, S.; Miki, T.; Tanno, M.; Mochizuki, A.; Fujito, T.; Yuda, S.; Muranaka, A.; Ogasawara, M.; Hashimoto, A.; et al. Clinical impact of myocardial mTORC1 activation in nonischemic dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2016, 91, 6–9. [Google Scholar] [CrossRef]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genom. 2018, 19, 812. [Google Scholar] [CrossRef] [Green Version]
- Gramlich, M.; Michely, B.; Krohne, C.; Heuser, A.; Erdmann, B.; Klaassen, S.; Hudson, B.; Magarin, M.; Kirchner, F.; Todiras, M.; et al. Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J. Mol. Cell. Cardiol. 2009, 47, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Tran, D.; Baalbaki, M.; Tang, L.F.; Poon, A.; Pelonero, A.; Titus, E.W.; Yuan, C.; Shi, C.; Patchava, S.; et al. An internal promoter underlies the difference in disease severity between N- and C-terminal truncation mutations of Titin in zebrafish. Elife 2015, 4, e09406. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.H.; Dvornikov, A.V.; Zhu, P.; Ma, X.; Kim, M.; Ding, Y.; Xu, X. Exon- and contraction-dependent functions of titin in sarcomere assembly. Development 2016, 143, 4713–4722. [Google Scholar] [CrossRef] [Green Version]
- Huttner, I.G.; Wang, L.W.; Santiago, C.F.; Horvat, C.; Johnson, R.; Cheng, D.; von Frieling-Salewsky, M.; Hillcoat, K.; Bemand, T.J.; Trivedi, G.; et al. A-Band Titin Truncation in Zebrafish Causes Dilated Cardiomyopathy and Hemodynamic Stress Intolerance. Circ. Genom. Precis. Med. 2018, 11, e002135. [Google Scholar] [CrossRef] [Green Version]
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.J.; Liu, B.P. Prevalence of TTN mutations in patients with dilated cardiomyopathy: A meta-analysis. Herz 2019. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, C.M.; Damrauer, S.M.; Levin, M.G.; Birtwell, D.; Carey, D.J.; Golden, A.M.; Hartzel, D.N.; Hu, Y.; Judy, R.; Kelly, M.A.; et al. Genomics-First Evaluation of Heart Disease Associated With Titin-Truncating Variants. Circulation 2019, 140, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Weng, L.C.; Roselli, C.; Lin, H.; Haggerty, C.M.; Shoemaker, M.B.; Barnard, J.; Arking, D.E.; Chasman, D.I.; Albert, C.M.; et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA 2018, 320, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef] [Green Version]
- Linschoten, M.; Teske, A.J.; Baas, A.F.; Vink, A.; Dooijes, D.; Baars, H.F.; Asselbergs, F.W. Truncating Titin (TTN) Variants in Chemotherapy-Induced Cardiomyopathy. J. Card. Fail. 2017, 23, 476–479. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchia, J.; Garcia-Pinilla, J.M.; Pascual-Figal, D.A.; Nunez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A.; Mazzarotto, F.; et al. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef]
- Zhou, J.; Ng, B.; Ko, N.S.J.; Fiedler, L.R.; Khin, E.; Lim, A.; Sahib, N.E.; Wu, Y.; Chothani, S.P.; Schafer, S.; et al. Titin truncations lead to impaired cardiomyocyte autophagy and mitochondrial function in vivo. Hum. Mol. Genet. 2019, 28, 1971–1981. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, M.; Pane, L.S.; Zhou, Q.; Chen, Z.; Murgia, M.; Schotterl, S.; Goedel, A.; Metzger, K.; Brade, T.; Parrotta, E.; et al. Antisense-mediated exon skipping: A therapeutic strategy for titin-based dilated cardiomyopathy. Embo Mol. Med. 2015, 7, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.K.; Neupane, B.; Pradhan, K.; Zhou, Q.; Testa, L.; Pelzl, L.; Maleck, C.; Gawaz, M.; Gramlich, M. The assembly and evaluation of antisense oligonucleotides applied in exon skipping for titin-based mutations in dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 131, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin diversity--alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Sun, M. RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophys. Rev. 2018, 10, 15–25. [Google Scholar] [CrossRef]
- Makarenko, I.; Opitz, C.A.; Leake, M.C.; Neagoe, C.; Kulke, M.; Gwathmey, J.K.; del Monte, F.; Hajjar, R.J.; Linke, W.A. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ. Res. 2004, 95, 708–716. [Google Scholar] [CrossRef]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksaite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef]
- Li, D.; Morales, A.; Gonzalez-Quintana, J.; Norton, N.; Siegfried, J.D.; Hofmeyer, M.; Hershberger, R.E. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2010, 3, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012, 9, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Molgaard, H.; Moller, J.E.; Eiskjaer, H.; Mogensen, J. Pathogenic RBM20-Variants Are Associated With a Severe Disease Expression in Male Patients With Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef]
- Streckfuss-Bomeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Ozcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Yin, Z.; Ren, J.; McCormick, R.J.; Ford, S.P.; Guo, W. RBM20 is an essential factor for thyroid hormone-regulated titin isoform transition. J. Mol. Cell. Biol. 2015, 7, 88–90. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Yin, Z.; Tan, B.; Guo, W. Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2363–2371. [Google Scholar] [CrossRef]
- Hinze, F.; Dieterich, C.; Radke, M.H.; Granzier, H.; Gotthardt, M. Reducing RBM20 activity improves diastolic dysfunction and cardiac atrophy. J. Mol. Med. (Berl.) 2016, 94, 1349–1358. [Google Scholar] [CrossRef]
- Methawasin, M.; Strom, J.G.; Slater, R.E.; Fernandez, V.; Saripalli, C.; Granzier, H. Experimentally Increasing the Compliance of Titin Through RNA Binding Motif-20 (RBM20) Inhibition Improves Diastolic Function In a Mouse Model of Heart Failure With Preserved Ejection Fraction. Circulation 2016, 134, 1085–1099. [Google Scholar] [CrossRef]
- Liss, M.; Radke, M.H.; Eckhard, J.; Neuenschwander, M.; Dauksaite, V.; von Kries, J.P.; Gotthardt, M. Drug discovery with an RBM20 dependent titin splice reporter identifies cardenolides as lead structures to improve cardiac filling. PLoS ONE 2018, 13, e0198492. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kong, X.; Zhang, M.; Yang, X.; Xu, X. RNA binding protein 24 deletion disrupts global alternative splicing and causes dilated cardiomyopathy. Protein. Cell 2019, 10, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Schotterl, S.; Backes, D.; Brunner, E.; Hahn, J.K.; Ionesi, E.; Aidery, P.; Sticht, C.; Labeit, S.; Kandolf, R.; et al. Inhibition of miR-208b improves cardiac function in titin-based dilated cardiomyopathy. Int. J. Cardiol. 2017, 230, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Herwig, M.; Linke, W.A. Tampering with springs: Phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys. Rev. 2017, 9, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [Green Version]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Rain, S.; Bos Dda, S.; Handoko, M.L.; Westerhof, N.; Stienen, G.; Ottenheijm, C.; Goebel, M.; Dorfmuller, P.; Guignabert, C.; Humbert, M.; et al. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J. Am. Heart Assoc. 2014, 3, e000716. [Google Scholar] [CrossRef] [Green Version]
- Kotter, S.; Gout, L.; Von Frieling-Salewsky, M.; Muller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Kruger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef]
- Belin, R.J.; Sumandea, M.P.; Allen, E.J.; Schoenfelt, K.; Wang, H.; Solaro, R.J.; de Tombe, P.P. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ. Res. 2007, 101, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Solaro, R.J. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J. Biol. Chem. 2008, 283, 26829–26833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, C.; Granzier, H. Tuning the molecular giant titin through phosphorylation: Role in health and disease. Trends Cardiovasc. Med. 2013, 23, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysiak, J.; Unger, A.; Beckendorf, L.; Hamdani, N.; von Frieling-Salewsky, M.; Redfield, M.M.; Dos Remedios, C.G.; Sheikh, F.; Gergs, U.; Boknik, P.; et al. Protein phosphatase 5 regulates titin phosphorylation and function at a sarcomere-associated mechanosensor complex in cardiomyocytes. Nat. Commun. 2018, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Slater, R.E.; Strom, J.G.; Methawasin, M.; Liss, M.; Gotthardt, M.; Sweitzer, N.; Granzier, H.L. Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J. Gen. Physiol. 2019, 151, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Hopf, A.E.; Andresen, C.; Kotter, S.; Isic, M.; Ulrich, K.; Sahin, S.; Bongardt, S.; Roll, W.; Drove, F.; Scheerer, N.; et al. Diabetes-Induced Cardiomyocyte Passive Stiffening Is Caused by Impaired Insulin-Dependent Titin Modification and Can Be Modulated by Neuregulin-1. Circ. Res. 2018, 123, 342–355. [Google Scholar] [CrossRef]
- Kruger, M.; Kotter, S.; Grutzner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Bishu, K.; Hamdani, N.; Mohammed, S.F.; Kruger, M.; Ohtani, T.; Ogut, O.; Brozovich, F.V.; Burnett, J.C., Jr.; Linke, W.A.; Redfield, M.M. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation 2011, 124, 2882–2891. [Google Scholar] [CrossRef] [Green Version]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Butler, J.; Lam, C.S.P.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; O’Connor, C.M.; Pieske, B.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; et al. Rationale and Design of the VITALITY-HFpEF Trial. Circ. Heart Fail. 2019, 12, e005998. [Google Scholar] [CrossRef]
- Filippatos, G.; Maggioni, A.P.; Lam, C.S.P.; Pieske-Kraigher, E.; Butler, J.; Spertus, J.; Ponikowski, P.; Shah, S.J.; Solomon, S.D.; Scalise, A.V.; et al. Patient-reported outcomes in the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED ejection fraction (SOCRATES-PRESERVED) study. Eur. J. Heart Fail. 2017, 19, 782–791. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proposed Titin Modifying Therapy | Mechanism of Action | Effect on Cardiac Function |
|---|---|---|
| mTOR inhibitor: rapamycin | Decrease mTOR complex signaling that is activated Titin truncation variations (TTNtv) mediated mRNA decay | Improve DCM phenotype for patients with TTNtv |
| Antisense oligonucleotide mediated exon skipping | Bind mRNA during transcription to skip exon containing missense mutation and prevent early termination | Improve DCM phenotype for patients with TTNtv |
| T3 hormone, insulin | Increase RBM20 expression to transcriptionally select shorter, stiffer N2B TTN isoform | Increase passive tension to treat DCM |
| Cardenolides: digoxin and digitoxin | Decrease RBM20 expression to transcriptionally select longer, softer N2BA TTN isoform | Decrease passive tension to treat diastolic dysfunction |
| Metformin, insulin | Increase ERK2 mediated phosphorylation of N2Bus element | Decrease passive tension to treat diastolic dysfunction |
| Neuregulin-1 (NRG-1) | Increase ERK2 mediated phosphorylation of N2Bus element, inhibit PKC⍺ phosphorylation of PEVK element | Decrease passive tension to treat diastolic dysfunction |
| cGMP agonists: sildenafil, vericiguat | Increases cGMP activity to increase PKG mediated phosphorylation of N2Bus element | Decrease passive tension to treat diastolic dysfunction |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tharp, C.; Mestroni, L.; Taylor, M. Modifications of Titin Contribute to the Progression of Cardiomyopathy and Represent a Therapeutic Target for Treatment of Heart Failure. J. Clin. Med. 2020, 9, 2770. https://doi.org/10.3390/jcm9092770
Tharp C, Mestroni L, Taylor M. Modifications of Titin Contribute to the Progression of Cardiomyopathy and Represent a Therapeutic Target for Treatment of Heart Failure. Journal of Clinical Medicine. 2020; 9(9):2770. https://doi.org/10.3390/jcm9092770
Chicago/Turabian StyleTharp, Charles, Luisa Mestroni, and Matthew Taylor. 2020. "Modifications of Titin Contribute to the Progression of Cardiomyopathy and Represent a Therapeutic Target for Treatment of Heart Failure" Journal of Clinical Medicine 9, no. 9: 2770. https://doi.org/10.3390/jcm9092770
APA StyleTharp, C., Mestroni, L., & Taylor, M. (2020). Modifications of Titin Contribute to the Progression of Cardiomyopathy and Represent a Therapeutic Target for Treatment of Heart Failure. Journal of Clinical Medicine, 9(9), 2770. https://doi.org/10.3390/jcm9092770