Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies

 , , , ,
, , , , 
Abstract
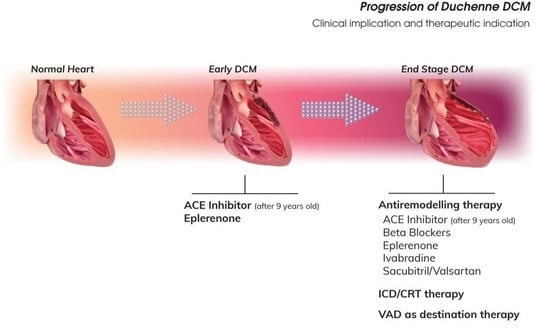
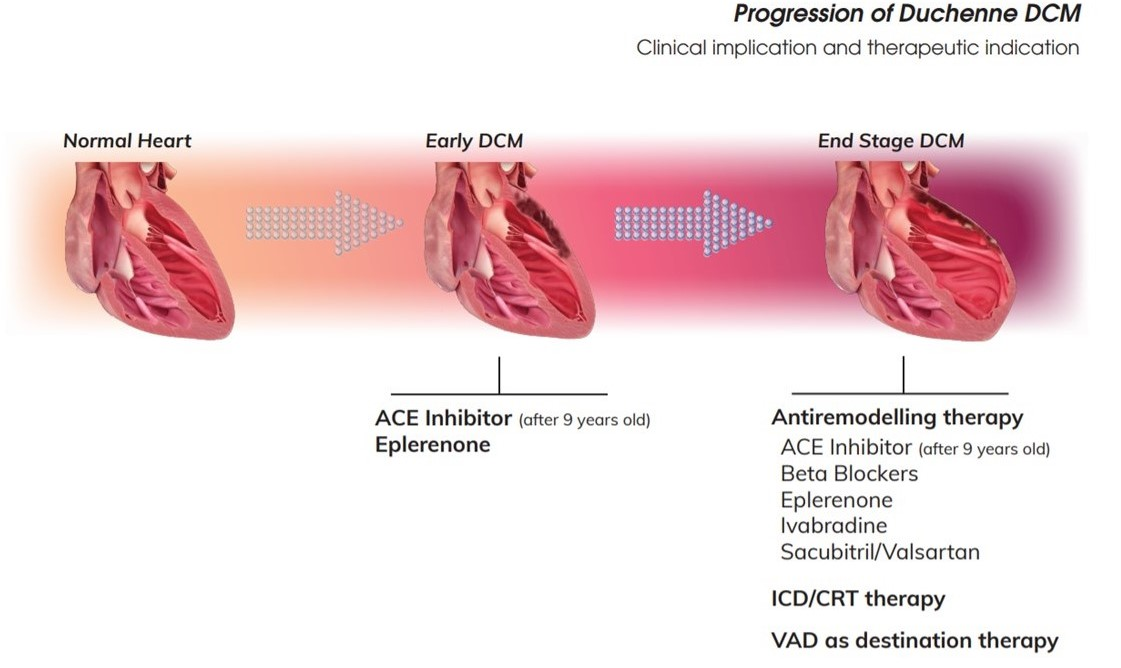{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pathophysiology of DMD-DCM
Clinical Course
3. Cardiovascular Management
4. Cardiovascular Biomarkers
5. Imaging
5.1. Transthoracic Echocardiography
5.2. Cardiac Magnetic Resonance
6. Therapeutic Strategy for DCM
6.1. Early DCM
6.2. DCM with Mid-Range Reduction of LVEF
6.3. Patients with Severe Ventricular Dysfunction
6.4. End Stage of DCM DMD
6.5. Symptomatic Drugs
7. Advanced Cardiac Therapies
7.1. Heart Transplant and Mechanical Assist Device
7.2. Ethical Aspects
8. Arrhythmias in DMD
8.1. Electrophysiologic Characteristics
8.2. CRT and Implantable Cardioverter Defibrillator
9. DMD Target Therapy
10. Female DMD Carriers
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE: | angiotensin Converting Enzyme |
| ACE inhibitors: | ACEi |
| ARNI: | Angiotensin Receptor Neprilysin Inhibitor |
| ARB: | Angiotensin Receptor Blocker |
| BB: | Beta Blockers |
| DCM: | Dilated Cardiomyopathy |
| DMD: | Duchenne Muscular Dystrophy |
| DTI: | Doppler Tissue Imaging |
| EF: | Ejection Fraction |
| FS: | Fractional Shortening |
| GLS: | Global Longitudinal Strain |
| HF: | Heart Failure |
| ICC: | Intra Class Coefficient |
| LV: | Left Ventricle |
| MR: | Magnetic Resonance |
| MPI: | Mechanical Performance Index |
| PWD: | Pulse Wave Doppler |
| RV: | Right Ventricle |
| TDI: | Tissue Doppler Index |
References
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- McNally, E.M.; Wyatt, E.J. Mutation-Based Therapy for Duchenne Muscular Dystrophy: Antisense Treatment Arrives in the Clinic. Circulation 2017, 136, 979–981. [Google Scholar] [CrossRef]
- Buddhe, S.; Cripe, L.H.; Friedland-Little, J.; Kertesz, N.; Eghtesady, P.; Finder, J.; Hor, K.N.; Judge, D.P.; Kinnett, K.; McNally, E.M.; et al. Cardiac Management of the Patient With Duchenne Muscular Dystrophy. Pediatrics 2018, 142, S72–S81. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.; Apkon, S.D.; Blackwell, A.; Case, L.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Connuck, D.M.; Sleeper, L.A.; Colan, S.D.; Cox, G.F.; Towbin, J.A.; Lowe, A.M.; Wilkinson, J.D.; Orav, E.J.; Cuniberti, L.; Salbert, B.A.; et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: A comparative study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 2008, 155, 998–1005. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.I.; Palladino, A.; Petretta, V.R.; Politano, L. Cardiomyopathies: Diagnosis of types and stages. Acta Myol. 2004, 23, 97–102. [Google Scholar] [PubMed]
- Politano, L.; Nigro, V.; Petretta, V.R.; Passamano, L.; Papparella, S.; Di Somma, S.; Comi, L.I. Development of Cardiomyopathy in Female Carriers of Duchenne and Becker Muscular Dystrophies. JAMA 1996, 275, 1335. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Campbell, K.P. Three muscular dystrophies: Loss of cytoskeleton-extracellular matrix linkage. Cell 1995, 80, 675–679. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell. Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef]
- Wallace, G.Q.; McNally, E.M. Mechanisms of Muscle Degeneration, Regeneration, and Repair in the Muscular Dystrophies. Annu. Rev. Physiol. 2009, 71, 37–57. [Google Scholar] [CrossRef]
- Yilmaz, A.; Gdynia, H.-J.; Baccouche, H.; Mahrholdt, H.; Meinhardt, G.; Basso, C.; Thiene, G.; Sperfeld, A.-D.; Ludolph, A.C.; Sechtem, U. Cardiac involvement in patients with Becker muscular dystrophy: New diagnostic and pathophysiological insights by a CMR approach. J. Cardiovasc. Magn. Reson. 2008, 10, 50. [Google Scholar] [CrossRef]
- Frankel, K.A.; Rosser, R.J. The pathology of the heart in progressive muscular dystrophy: Epimyocardial fibrosis. Hum. Pathol. 1976, 7, 375–386. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.M.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010, 9, 177–189. [Google Scholar] [CrossRef]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.; Politano, L.; Bain, R. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Hammerer-Lercher, A.; Erlacher, P.; Bittner, R.; Korinthenberg, R.; Skladal, D.; Sorichter, S.; Sperl, W.; Puschendorf, B.; Mair, J. Clinical and Experimental Results on Cardiac Troponin Expression in Duchenne Muscular Dystrophy. Clin. Chem. 2001, 47, 451–458. [Google Scholar] [CrossRef]
- Buyse, G.M.; Goemans, N.; Hauwe, M.V.D.; Thijs, D.; De Groot, I.J.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. [Google Scholar] [CrossRef]
- Castro-Gago, M.; Gómez-Lado, C.; Puñal, J.E. Cardiac troponin I for accurate evaluation of cardiac status in myopathic patients. Brain Dev. 2009, 31, 184. [Google Scholar] [CrossRef]
- Voleti, S.; Olivieri, L.; Hamann, K.; Gordish-Dressman, H.; Spurney, C. Troponin I Levels Correlate with Cardiac MR LGE and Native T1 Values in Duchenne Muscular Dystrophy Cardiomyopathy and Identify Early Disease Progression. Pediatr. Cardiol. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, A.; Yokota, N.; Miyagawa, M.; Kawamura, J.; Ishihara, T.; Aoyagi, T.; Ishikawa, K. Plasma levels of atrial natriuretic peptide in patients with Duchenne’s progressive muscular dystrophy. Am. Heart J. 1990, 120, 1154–1158. [Google Scholar] [CrossRef]
- Villa, C.R.; Kaddourah, A.; Mathew, J.; Ryan, T.D.; Wong, B.L.Y.; Goldstein, S.L.; Jefferies, J.L. Identifying evidence of cardio-renal syndrome in patients with Duchenne muscular dystrophy using cystatin C. Neuromuscul. Disord. 2016, 26, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Power, L.C.; O’Grady, G.L.; Hornung, T.S.; Jefferies, C.; Gusso, S.; Hofman, P.L. Imaging the heart to detect cardiomyopathy in Duchenne muscular dystrophy: A review. Neuromuscul. Disord. 2018, 28, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Soslow, J.H.; Xu, M.; Slaughter, J.C.; Stanley, M.; Crum, K.; Markham, L.W.; Parra, D.A. Evaluation of Echocardiographic Measures of Left Ventricular Function in Patients with Duchenne Muscular Dystrophy: Assessment of Reproducibility and Comparison to Cardiac Magnetic Resonance Imaging. J. Am. Soc. Echocardiogr. 2016, 29, 983–991. [Google Scholar] [CrossRef]
- Buddhe, S.; Lewin, M.; Olson, A.; Ferguson, M.; Soriano, B.D. Comparison of left ventricular function assessment between echocardiography and MRI in Duchenne muscular dystrophy. Pediatr. Radiol. 2016, 46, 1399–1408. [Google Scholar] [CrossRef]
- Cirino, R.H.D.; Scola, R.H.; Ducci, R.D.-P.; Camarozano, A.C.; Kay, C.S.K.; Lorenzoni, P.J.; Werneck, L.C.; Carmes, E.R.; Da Cunha, C.L.P. Evaluation of Left-Sided Heart Chambers with Novel Echocardiographic Techniques in Men With Duchenne or Becker Muscular Dystrophy. Am. J. Cardiol. 2019, 123, 972–978. [Google Scholar] [CrossRef]
- Spurney, C.F.; McCaffrey, F.M.; Cnaan, A.; Morgenroth, L.P.; Ghelani, S.J.; Gordish-Dressman, H.; Arrieta, A.; Connolly, A.M.; Lotze, T.E.; McDonald, C.M.; et al. Feasibility and Reproducibility of Echocardiographic Measures in Children with Muscular Dystrophies. J. Am. Soc. Echocardiogr. 2015, 28, 999–1008. [Google Scholar] [CrossRef]
- Levy, P.T.; Machefsky, A.; Sanchez, A.A.; Patel, M.D.; Rogal, S.; Fowler, S.; Yaeger, L.; Hardi, A.; Holland, M.R.; Hamvas, A.; et al. Reference Ranges of Left Ventricular Strain Measures by Two-Dimensional Speckle-Tracking Echocardiography in Children: A Systematic Review and Meta-Analysis. J. Am. Soc. Echocardiogr. 2016, 29, 209–225.e6. [Google Scholar] [CrossRef]
- Patrianakos, A.P.; Zacharaki, A.; Kalogerakis, A.; Solidakis, G.; Parthenakis, F.; Vardas, P. Two-dimensional global and segmental longitudinal strain: Are the results from software in different high-end ultrasound systems comparable? Echo Res. Pr. 2015, 2, 29–39. [Google Scholar] [CrossRef][Green Version]
- Amedro, P.; Vincenti, M.; De La Villeon, G.; Lavastre, K.; Barrea, C.; Guillaumont, S.; Bredy, C.; Gamon, L.; Meli, A.C.; Cazorla, O.; et al. Speckle-Tracking Echocardiography in Children with Duchenne Muscular Dystrophy: A Prospective Multicenter Controlled Cross-Sectional Study. J. Am. Soc. Echocardiogr. 2019, 32, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Power, A.; Poonja, S.; Disler, D.; Myers, K.; Patton, D.J.; Mah, J.K.; Fine, N.M.; Greenway, S.C. Echocardiographic Image Quality Deteriorates with Age in Children and Young Adults with Duchenne Muscular Dystrophy. Front. Cardiovasc. Med. 2017, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, M.; Hor, K.N.; Al-Khalidi, H.R.; Benson, D.W.; Jefferies, J.L.; Taylor, M.D.; Egnaczyk, G.F.; Raman, S.V.; Basu, S.K.; Cripe, L.H.; et al. Comparison of right and left ventricular function and size in Duchenne muscular dystrophy. Eur. J. Radiol. 2015, 84, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.C.; Magalhães, T.A.; Meira, Z.M.A.; Rassi, C.H.R.E.; Andrade, A.C.D.S.; Gutierrez, P.S.; Azevedo, C.F.; Gurgel-Giannetti, J.; Vainzof, M.; Zatz, M.; et al. Myocardial Fibrosis Progression in Duchenne and Becker Muscular Dystrophy. JAMA Cardiol. 2017, 2, 190–199. [Google Scholar] [CrossRef]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Halnon, N.J.; Kissel, J.T.; He, X.; Tran, T.; Smart, S.; McCarthy, B.; Taylor, M.D.; et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 14, 153–161. [Google Scholar] [CrossRef]
- Florian, A.-R.; Rösch, S.; Bietenbeck, M.; Engelen, M.; Sechtem, U.; Stypmann, J.; Waltenberger, J.; Yilmaz, A. Cardiac involvement in female Duchenne and Becker muscular dystrophy carriers in comparison to their first-degree male relatives: A comparative cardiovascular magnetic resonance study. Eur. Heart J. Cardiovasc. Imaging 2015, 17, 326–333. [Google Scholar] [CrossRef]
- Lang, S.M.; Shugh, S.; Mazur, W.; Sticka, J.J.; Rattan, M.S.; Jefferies, J.L.; Taylor, M.D. Myocardial Fibrosis and Left Ventricular Dysfunction in Duchenne Muscular Dystrophy Carriers Using Cardiac Magnetic Resonance Imaging. Pediatr. Cardiol. 2015, 36, 1495–1501. [Google Scholar] [CrossRef]
- Florian, A.-R.; Ludwig, A.; Engelen, M.; Waltenberger, J.; Rösch, S.; Sechtem, U.; Yilmaz, A. Left ventricular systolic function and the pattern of late-gadolinium-enhancement independently and additively predict adverse cardiac events in muscular dystrophy patients. J. Cardiovasc. Magn. Reson. 2014, 16, 81. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Markousis-Mavrogenis, G.; Papavasiliou, A.; Kolovou, G. Cardiac involvement in Duchenne and Becker muscular dystrophy. World J. Cardiol. 2015, 7, 410–414. [Google Scholar] [CrossRef]
- Giglio, V.; Puddu, P.E.; Camastra, G.; Sbarbati, S.; Della Sala, S.W.; Ferlini, A.; Gualandi, F.; Ricci, E.; Sciarra, F.; Ansalone, G.; et al. Patterns of late gadolinium enhancement in Duchenne muscular dystrophy carriers. J. Cardiovasc. Magn. Reson. 2014, 16, 45. [Google Scholar] [CrossRef]
- Wexberg, P.; Avanzini, M.; Mascherbauer, J.; Pfaffenberger, S.; Freudenthaler, B.; Bittner, R.E.; Bernert, G.; Weidinger, F. Myocardial late gadolinium enhancement is associated with clinical presentation in Duchenne muscular dystrophy carriers. J. Cardiovasc. Magn. Reson. 2016, 18, 61. [Google Scholar] [CrossRef]
- Tandon, A.; Villa, C.R.; Hor, K.N.; Jefferies, J.L.; Gao, Z.; Towbin, J.A.; Wong, B.L.; Mazur, W.; Fleck, R.J.; Sticka, J.J.; et al. Myocardial Fibrosis Burden Predicts Left Ventricular Ejection Fraction and Is Associated with Age and Steroid Treatment Duration in Duchenne Muscular Dystrophy. J. Am. Heart Assoc. 2015, 4, e001338. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, L.J.; Kellman, P.; McCarter, R.J.; Cross, R.R.; Hansen, M.S.; Spurney, C.F. Native T1 values identify myocardial changes and stratify disease severity in patients with Duchenne muscular dystrophy. J. Cardiovasc. Magn. Reson. 2016, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Soslow, J.H.; Damon, S.M.; Crum, K.; Lawson, M.A.; Slaughter, J.C.; Xu, M.; Arai, A.E.; Sawyer, D.B.; Parra, D.A.; Damon, B.M.; et al. Increased myocardial native T1 and extracellular volume in patients with Duchenne muscular dystrophy. J. Cardiovasc. Magn. Reson. 2015, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Roujol, S.; Weingärtner, S.; Foppa, M.; Chow, K.; Kawaji, K.; Ngo, L.H.; Kellman, P.; Manning, W.J.; Thompson, R.; Nezafat, R. Accuracy, precision, and reproducibility of four T1 mapping sequences: A head-to-head comparison of MOLLI, ShMOLLI, SASHA, and SAPPHIRE. Radiology 2014, 272, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Kellman, P.; Bandettini, W.P.; Mancini, C.; Hammer-Hansen, S.; Hansen, M.S.; Arai, A.E. Characterization of myocardial T1-mapping bias caused by intramyocardial fat in inversion recovery and saturation recovery techniques. J. Cardiovasc. Magn. Reson. 2015, 17, 1–11. [Google Scholar] [CrossRef]
- Siegel, B.; Olivieri, L.; Gordish-Dressman, H.; Spurney, C. Myocardial Strain Using Cardiac MR Feature Tracking and Speckle Tracking Echocardiography in Duchenne Muscular Dystrophy Patients. Pediatr. Cardiol. 2017, 39, 478–483. [Google Scholar] [CrossRef]
- Aikawa, T.; Takeda, A.; Oyama-Manabe, N.; Naya, M.; Yamazawa, H.; Koyanagawa, K.; Ito, Y.M.; Anzai, T. Progressive left ventricular dysfunction and myocardial fibrosis in Duchenne and Becker muscular dystrophy: A longitudinal cardiovascular magnetic resonance study. Pediatr. Cardiol. 2018, 40, 384–392. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Duboc, D.; Meune, C.; Lerebours, G.; Devaux, J.-Y.; Vaksmann, G.; Bécane, H.-M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2005, 45, 855–857. [Google Scholar] [CrossRef]
- Allen, H.D.; Flanigan, K.M.; Thrush, P.T.; Dvorchik, I.; Yin, H.; Canter, C.; Connolly, A.M.; Parrish, M.; McDonald, C.M.; Braunlin, E.; et al. A Randomized, Double-Blind Trial of Lisinopril and Losartan for the Treatment of Cardiomyopathy in Duchenne Muscular Dystrophy. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Cardona, A.; He, X.; Halnon, N.; Markham, L.; Soslow, J.H.; Puchalski, M.D.; Auerbach, S.R.; et al. Stabilization of Early Duchenne Cardiomyopathy With Aldosterone Inhibition: Results of the Multicenter AIDMD Trial. J. Am. Heart Assoc. 2019, 8, e013501. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.; Margossian, R.; Darras, B.T.; Colan, S.D.; Jenkins, K.J.; Geva, T.; Powell, A.J. Safety and Efficacy of Carvedilol Therapy for Patients with Dilated Cardiomyopathy Secondary to Muscular Dystrophy. Pediatr. Cardiol. 2007, 29, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Fayssoil, A.; Ben Yaou, R.; Ogna, A.; Leturcq, F.; Nardi, O.; Clair, B.; Wahbi, K.; Lofaso, F.; Laforet, P.; Duboc, D.; et al. Clinical profiles and prognosis of acute heart failure in adult patients with dystrophinopathies on home mechanical ventilation. ESC Heart Fail. 2017, 4, 527–534. [Google Scholar] [CrossRef]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J. Genetic Predictors and Remodeling of Dilated Cardiomyopathy in Muscular Dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef]
- Kajimoto, H.; Ishigaki, K.; Okumura, K.; Tomimatsu, H.; Nakazawa, M.; Saito, K.; Osawa, M.; Nakanishi, T. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ. J. 2006, 70, 991–994. [Google Scholar] [CrossRef]
- Matsumura, T.; Tamura, T.; Kuru, S.; Kikuchi, Y.; Kawai, M. Carvedilol can prevent cardiac events in Duchenne muscular dystrophy. Intern. Med. 2010, 49, 1357–1363. [Google Scholar] [CrossRef]
- Ogata, H.; Ishikawa, Y.; Ishikawa, Y.; Minami, R. Beneficial effects of beta-blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J. Cardiol. 2009, 53, 72–78. [Google Scholar] [CrossRef]
- Saito, T.; Matsumura, T.; Miyai, I.; Nozaki, S.; Shinno, S. Carvedilol effectiveness for left ventricular-insufficient patients with Duchenne muscular dystrophy. Rinsho Shinkeigaku 2001, 41, 691–694. [Google Scholar]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of Angiotensin-Converting Enzyme Inhibitors and/or Beta Blockers on the Cardiomyopathy in Duchenne Muscular Dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Adorisio, R.; Calvieri, C.; Cantarutti, N.; D’Amico, A.; Catteruccia, M.; Bertini, E.; Baban, A.; Filippelli, S.; Perri, G.; Amodeo, A.; et al. Heart rate reduction strategy using ivabradine in end-stage Duchenne cardiomyopathy. Int. J. Cardiol. 2019, 280, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Berger, F.; Jokinen, E.; Kantor, P.F.; Daubeney, P.E. Ivabradine in Children With Dilated Cardiomyopathy and Symptomatic Chronic Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Riddle, V.; Siegler, M.A.; Dickerson, D.; Backonja, M.; Kramer, W.G.; Nagaraju, K.; Gordish-Dressman, H.; Damsker, J.M.; McCall, J.M. Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids 2018, 134, 43–52. [Google Scholar] [CrossRef]
- Shaddy, R.E.; Canter, C.; Halnon, N.; Kochilas, L.; Rossano, J.; Bonnet, D.; Bush, C.; Zhao, Z.; Kantor, P.F.; Burch, M.; et al. Design for the sacubitril/valsartan (LCZ696) compared with enalapril study of pediatric patients with heart failure due to systemic left ventricle systolic dysfunction (PANORAMA-HF study). Am. Heart J. 2017, 193, 23–34. [Google Scholar] [CrossRef]
- Das, B.B. Current State of Pediatric Heart Failure. Children 2018, 5, 88. [Google Scholar] [CrossRef]
- Mullens, W.; Damman, K.; Harjola, V.-P.; Mebazaa, A.; Rocca, H.-P.B.-L.; Martens, P.; Testani, J.M.; Tang, W.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef]
- Lewis, A.B.; Chabot, M. The effect of treatment with angiotensin-converting enzyme inhibitors on survival of pediatric patients with dilated cardiomyopathy. Pediatr. Cardiol. 1993, 14, 9–12. [Google Scholar]
- Kirk, R.; Dipchand, A.I.; Rosenthal, D.N.; Addonizio, L.; Burch, M.; Chrisant, M.; Dubin, A.; Everitt, M.; Gajarski, R.; Mertens, L.; et al. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary. J. Heart Lung Transpl. 2014, 33, 888–909. [Google Scholar] [CrossRef]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Kaltman, J.R.; Benson, D.W.; Canter, C.E.; Cripe, L.H.; Duan, D.; Finder, J.D.; Groh, W.J.; Hoffman, E.P.; Judge, D.P.; et al. Contemporary Cardiac Issues in Duchenne Muscular Dystrophy. Circulation 2015, 131, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Piperata, A.; Bottio, T.; Toscano, G.; Avesani, M.; Vianello, A.; Gerosa, G. Is heart transplantation a real option in patients with Duchenne syndrome? Inferences from a case report. ESC Heart Fail. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rose, E.A.; Gelijns, A.C.; Moskowitz, A.J.; Heitjan, D.F.; Stevenson, L.W.; Dembitsky, W.; Long, J.W.; Ascheim, D.D.; Tierney, A.R.; Levitan, R.G.; et al. Long-Term Use of a Left Ventricular Assist Device for End-Stage Heart Failure. N. Engl. J. Med. 2001, 345, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Starling, R.C.; Naka, Y.; Boyle, A.J.; Gonzalez-Stawinski, G.; John, R.; Jorde, U.; Russell, S.D.; Conte, J.V.; Aaronson, K.D.; McGee, E.C.; et al. Results of the Post-U.S. Food and Drug Administration-Approval Study with a Continuous Flow Left Ventricular Assist Device as a Bridge to Heart Transplantation. J. Am. Coll. Cardiol. 2011, 57, 1890–1898. [Google Scholar] [CrossRef]
- Slaughter, M.S.; Rogers, J.G.; Milano, C.A.; Russell, S.D.; Conte, J.V.; Feldman, D.; Sun, B.; Tatooles, A.J.; Delgado, R.M.; Long, J.W.; et al. Advanced Heart Failure Treated with Continuous-Flow Left Ventricular Assist Device. N. Engl. J. Med. 2009, 361, 2241–2251. [Google Scholar] [CrossRef]
- Enciso, J.S. Mechanical Circulatory Support: Current Status and Future Directions. Prog. Cardiovasc. Dis. 2016, 58, 444–454. [Google Scholar] [CrossRef]
- Delgado, R.; Radovancevic, B.; Massin, E.K.; Frazier, O.H.; Benedict, C. Neurohormonal changes after implantation of a left ventricular assist system. ASAIO J. 1998, 44, 299–302. [Google Scholar]
- Barbone, A.; Holmes, J.W.; Heerdt, P.M.; Andrew, H.S.; Naka, Y.; Joshi, N.; Daines, M.; Marks, A.R.; Oz, M.C.; Burkhoff, D. Comparison of Right and Left Ventricular Responses to Left Ventricular Assist Device Support in Patients with Severe Heart Failure. Circulation 2001, 104, 670–675. [Google Scholar] [CrossRef]
- Madigan, J.D.; Barbone, A.; Choudhri, A.F.; Morales, D.L.; Cai, B.; Oz, M.C.; Burkhoff, D. Time course of reverse remodeling of the left ventricle during support with a left ventricular assist device. J. Thorac. Cardiovasc. Surg. 2001, 121, 902–908. [Google Scholar] [CrossRef]
- Klotz, S.; Foronjy, R.F.; Dickstein, M.L.; Gu, A.; Garrelds, I.M.; Danser, A.J.; Oz, M.C.; D’Armiento, J.; Burkhoff, D. Mechanical Unloading During Left Ventricular Assist Device Support Increases Left Ventricular Collagen Cross-Linking and Myocardial Stiffness. Circulation 2005, 112, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.R.; Oz, M.C.; Chen, J.M.; Packer, M.; Rose, E.A.; Burkhoff, D. Reversal of Chronic Ventricular Dilation in Patients With End-Stage Cardiomyopathy by Prolonged Mechanical Unloading. Circulation 1995, 91, 2717–2720. [Google Scholar] [CrossRef] [PubMed]
- Zafeiridis, A.; Jeevanandam, V.; Houser, S.R.; Margulies, K.B. Regression of cellular hypertrophy after left ventricular assist device support. Circulation 1998, 98, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Altemose, G.T.; Gritsus, V.; Jeevanandam, V.; Goldman, B.; Margulies, K.B. Altered myocardial phenotype after mechanical support in human beings with advanced cardiomyopathy. J. Heart Lung Transpl. 1997, 16, 765–773. [Google Scholar]
- Dipla, K.; Mattiello, J.A.; Jeevanandam, V.; Houser, S.R.; Margulies, K.B. Myocyte recovery after mechanical circulatory support in humans with end-stage heart failure. Circulation 1998, 97, 2316–2322. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, A.; Adorisio, R. Left ventricular assist device in Duchenne Cardiomyopathy: Can we change the natural history of cardiac disease? Int. J. Cardiol. 2012, 161, e43. [Google Scholar] [CrossRef]
- Brancaccio, G.; Filippelli, S.; Michielon, G.; Iacobelli, R.; Alfieri, S.; Gandolfo, F.; Pongiglione, G.; Albanese, S.; Perri, G.; Parisi, F.; et al. Ventricular Assist Devices as a Bridge to Heart Transplantation or as Destination Therapy in Pediatric Patients. Transpl. Proc. 2012, 44, 2007–2012. [Google Scholar] [CrossRef]
- Kilic, A.; Acker, M.A.; Atluri, P. Dealing with surgical left ventricular assist device complications. J. Thorac. Dis. 2015, 7, 2158–2164. [Google Scholar]
- Ryan, T.D.; Jefferies, J.L.; Sawnani, H.; Wong, B.L.Y.; Gardner, A.; Del Corral, M.; Lorts, A.; Morales, D.L.S. Implantation of the HeartMate II and HeartWare Left Ventricular Assist Devices in Patients With Duchenne Muscular Dystrophy. ASAIO J. 2014, 60, 246–248. [Google Scholar] [CrossRef]
- Iodice, F.; Testa, G.; Averardi, M.; Brancaccio, G.; Amodeo, A.; Cogo, P. Implantation of a left ventricular assist device as a destination therapy in Duchenne muscular dystrophy patients with end stage cardiac failure: Management and lessons learned. Neuromuscul. Disord. 2015, 25, 19–23. [Google Scholar] [CrossRef]
- Stoller, U.; Araj, F.; Amin, A.; Fitzsimmons, C.; Morlend, R.; Thibodeau, J.T.; Ramaciotti, C.; Drazner, M.H.; Meyer, D.M.; Mammen, P.P.A. Implantation of a left ventricular assist device to provide long-term support for end-stage Duchenne muscular dystrophy-associated cardiomyopathy. ESC Heart Fail. 2017, 4, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Perri, G.; Filippelli, S.; Adorisio, R.; Iacobelli, R.; Iodice, F.; Testa, G.; Paglietti, M.G.; D’Amario, M.; Massetti, M.; Amodeo, A. Left ventricular assist device as destination therapy in cardiac end-stage dystrophinopathies: Midterm results. J. Thorac. Cardiovasc. Surg. 2017, 153, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Wittlieb-Weber, C.A.; Villa, C.; Conway, J.; Bock, M.J.; Gambetta, K.E.; Johnson, J.N.; Lal, A.K.; Schumacher, K.R.; Law, S.P.; Deshpande, S.R.; et al. Use of advanced heart failure therapies in Duchenne muscular dystrophy. Prog. Pediatr. Cardiol. 2019, 53, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Magnetta, D.A.; Kang, J.; Wearden, P.D.; Smith, K.J.; Feingold, B. Cost-Effectiveness of Ventricular Assist Device Destination Therapy for Advanced Heart Failure in Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2018, 39, 1242–1248. [Google Scholar] [CrossRef]
- Adorisio, R.; D’Amario, D.; Perri, G.; Amodeo, A. Comment on: ’Implantation of a left ventricular assist device to provide long term support for end-stage Duchenne muscular dystrophy-associated cardiomyopathy. ESC Heart Fail. 2018, 5, 651–652. [Google Scholar] [CrossRef]
- Feudtner, C. Collaborative Communication in Pediatric Palliative Care: A Foundation for Problem-Solving and Decision-Making. Pediatr. Clin. N. Am. 2007, 54, 583–607. [Google Scholar] [CrossRef]
- Arbustini, E.; Di Toro, A.; Giuliani, L.; Favalli, V.; Narula, N.; Grasso, M. Cardiac Phenotypes in Hereditary Muscle Disorders. J. Am. Coll. Cardiol. 2018, 72, 2485–2506. [Google Scholar] [CrossRef]
- Fayssoil, A.; Abasse, S.; Silverston, K. Cardiac Involvement Classification and Therapeutic Management in Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2017, 4, 17–23. [Google Scholar] [CrossRef]
- Fayssoil, A.; Ben Yaou, R.; Ogna, A.; Chaffaut, C.; Leturcq, F.; Nardi, O.; Wahbi, K.; Duboc, D.; Lofaso, F.; Prigent, H.; et al. Left bundle branch block in Duchenne muscular dystrophy: Prevalence, genetic relationship and prognosis. PLoS ONE 2018, 13, e0190518. [Google Scholar] [CrossRef]
- Perloff, J.K. Cardiac rhythm and conduction in Duchenne’s muscular dystrophy: A prospective study of 20 patients. J. Am. Coll. Cardiol. 1984, 3, 1263–1268. [Google Scholar] [CrossRef]
- Corrado, G.; Lissoni, A.; Beretta, S.; Terenghi, L.; Tadeo, G.; Foglia-Manzillo, G.; Tagliagambe, L.M.; Spata, M.; Santarone, M. Prognostic value of electrocardiograms, ventricular late potentials, ventricular arrhythmias, and left ventricular systolic dysfunction in patients with Duchenne muscular dystrophy. Am. J. Cardiol. 2002, 89, 838–841. [Google Scholar] [CrossRef]
- Segawa, K.; Komaki, H.; Mori-Yoshimura, M.; Oya, Y.; Kimura, K.; Tachimori, H.; Kato, N.; Sasaki, M.; Takahashi, Y. Cardiac conduction disturbances and aging in patients with Duchenne muscular dystrophy. Medicine (Baltimore) 2017, 96, e8335. [Google Scholar] [CrossRef] [PubMed]
- Fragakis, N.; Sotiriadou, M.; Krexi, L.; Vassilikos, V. Electrical storm in a patient with Duchenne muscular dystrophy cardiomyopathy triggered by abrupt β-blocker interruption. Ann. Noninvasive Electrocardiol. 2017, 22, e12477. [Google Scholar] [CrossRef] [PubMed]
- Lillo, M.A.; Himelman, E.; Shirokova, N.; Xie, L.-H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, 130091. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Quick, A.; Cao, S.; Reynolds, J.; Chiang, D.Y.; Beavers, D.; Li, N.; Wang, G.; Rodney, G.G.; Anderson, M.E.; et al. Oxidized CaMKII (Ca2+/Calmodulin-Dependent Protein Kinase II) Is Essential for Ventricular Arrhythmia in a Mouse Model of Duchenne Muscular Dystrophy. Circ. Arrhythmia Electrophysiol. 2018, 11, e005682. [Google Scholar] [CrossRef]
- Palladino, A.; Papa, A.A.; Morra, S.; Russo, V.; Ergoli, M.; Rago, A.; Orsini, C.; Nigro, G.; Politano, L. Are there real benefits to implanting cardiac devices in patients with end-stage dilated dystrophinopathic cardiomyopathy? Review of literature and personal results. Acta Myol. 2019, 38, 1–7. [Google Scholar]
- D’Amario, D.; Gowran, A.; Canonico, F.; Castiglioni, E.; Rovina, D.; Santoro, R.; Spinelli, P.; Adorisio, R.; Amodeo, A.; Perrucci, G.L.; et al. Dystrophin Cardiomyopathies: Clinical Management, Molecular Pathogenesis and Evolution towards Precision Medicine. J. Clin. Med. 2018, 7, 291. [Google Scholar] [CrossRef]
- Bello, L.; Gordish-Dressman, H.; Morgenroth, L.P.; Henricson, E.K.; Duong, T.; Hoffman, E.P.; Cnaan, A.; McDonald, C.M.; CINRG investigators. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology 2015, 85, 1048–1055. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Sheri, N.; Nguyen, Q.; Yokota, T. Cardiac Involvement in Dystrophin-Deficient Females: Current Understanding and Implications for the Treatment of Dystrophinopathies. Genes 2020, 11, 765. [Google Scholar] [CrossRef]
- Brioschi, S.; Gualandi, F.; Scotton, C.; Armaroli, A.; Bovolenta, M.; Falzarano, M.S.; Sabatelli, P.; Selvatici, R.; D’Amico, A.; Pane, M.; et al. Genetic characterization in symptomatic female DMD carriers: Lack of relationship between X-inactivation, transcriptional DMD allele balancing and phenotype. BMC Med. Genet. 2012, 13, 73. [Google Scholar] [CrossRef]
- Finsterer, J.; Stöllberger, C. Muscle, cardiac, and cerebral manifestations in female carriers of dystrophin variants. J. Neurol. Sci. 2018, 388, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, M.; Kobayashi, M.; Adachi, K.; Matsumura, T.; Kimura, E. Female dystrophinopathy: Review of current literature. Neuromuscul. Disord. 2018, 28, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Kamakura, K. Cardiac involvement of female carrier of Duchenne muscular dystrophy. Intern. Med. 2000, 39, 2–3. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adorisio, R.; Mencarelli, E.; Cantarutti, N.; Calvieri, C.; Amato, L.; Cicenia, M.; Silvetti, M.; D’Amico, A.; Grandinetti, M.; Drago, F.; et al. Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies. J. Clin. Med. 2020, 9, 3186. https://doi.org/10.3390/jcm9103186
Adorisio R, Mencarelli E, Cantarutti N, Calvieri C, Amato L, Cicenia M, Silvetti M, D’Amico A, Grandinetti M, Drago F, et al. Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies. Journal of Clinical Medicine. 2020; 9(10):3186. https://doi.org/10.3390/jcm9103186
Chicago/Turabian StyleAdorisio, Rachele, Erica Mencarelli, Nicoletta Cantarutti, Camilla Calvieri, Liliana Amato, Marianna Cicenia, Massimo Silvetti, Adele D’Amico, Maria Grandinetti, Fabrizio Drago, and et al. 2020. "Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies" Journal of Clinical Medicine 9, no. 10: 3186. https://doi.org/10.3390/jcm9103186
APA StyleAdorisio, R., Mencarelli, E., Cantarutti, N., Calvieri, C., Amato, L., Cicenia, M., Silvetti, M., D’Amico, A., Grandinetti, M., Drago, F., & Amodeo, A. (2020). Duchenne Dilated Cardiomyopathy: Cardiac Management from Prevention to Advanced Cardiovascular Therapies. Journal of Clinical Medicine, 9(10), 3186. https://doi.org/10.3390/jcm9103186