Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis


Abstract
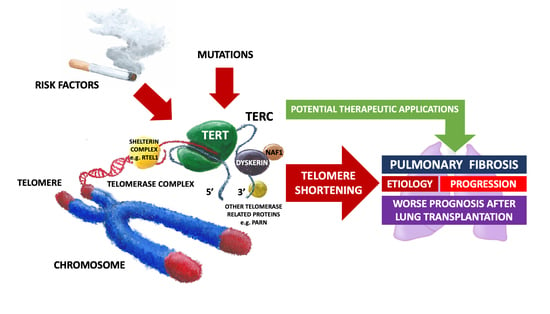
1. Introduction
2. Contribution of Telomere Abnormalities to the Etiology of IPF
2.1. Smoking, Pulmonary Fibrosis and Telomere Shortening
2.2. Oxidative Stress and Mitochondria—Other Players in the Game
2.3. Does Sex Make a Difference
2.4. Familial Interstitial Pneumonia
3. Telomere Abnormalities and the Natural History of IPF
4. Telomere Length and Lung Transplantation in IPF
5. Telomere Length and Antifibrotic Treatment
6. Targeting Telomerase in Therapy of IPF
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Karakatsani, A.; Papakosta, D.; Rapti, A.; Antoniou, K.M.; Dimadi, M.; Markopoulou, A.; Latsi, P.; Polychronopoulos, V.; Birba, G.; Ch, L.; et al. Epidemiology of interstitial lung diseases in Greece. Respir. Med. 2009, 103, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Von Plessen, C.; Grinde, O.; Gulsvik, A. Incidence and prevalence of cryptogenic fibrosing alveolitis in a Norwegian community. Respir. Med. 2003, 97, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Coultas, D.B.; Zumwalt, R.E.; Black, W.C.; Sobonya, R.E. The epidemiology of interstitial lung diseases. Am. J. Respir. Crit. Care Med. 1994, 150, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Mori, T.; Yamada, N.; Yamaguchi, M.; Hosoda, Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am. J. Respir. Crit. Care Med. 1994, 150, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Johnston, I.; Britton, J. What causes cryptogenic fibrosing alveolitis? A case-control study of environmental exposure to dust. BMJ 1990, 301, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Ley, B.; Collard, H.R.; King, T.E. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Rudd, R.M.; Prescott, R.J.; Chalmers, J.C.; Johnston, I.D.A.; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society Study on cryptogenic fibrosing alveolitis: Response to treatment and survival. Thorax 2007, 62, 62–66. [Google Scholar] [CrossRef]
- Lawson, W.E.; Loyd, J.E. The genetic approach in pulmonary fibrosis: Can it provide clues to this complex disease? Proc. Am. Thorac. Soc. 2006, 3, 345–349. [Google Scholar] [CrossRef][Green Version]
- Loyd, J.E. Pulmonary fibrosis in families. Am. J. Respir. Cell Mol. Biol. 2003, 29, S47–S50. [Google Scholar] [PubMed]
- Marshall, R.P.; Puddicombe, A.; Cookson, W.O.; Laurent, G.J. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax 2000, 55, 143–146. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Steele, M.P.; Speer, M.C.; Loyd, J.E.; Brown, K.K.; Herron, A.; Slifer, S.H.; Burch, L.H.; Wahidi, M.M.; Phillips, J.A.; Sporn, T.A.; et al. Clinical and pathologic features of familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2005, 172, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar]
- Kanematsu, T.; Kitaichi, M.; Nishimura, K.; Nagai, S.; Izumi, T. Clubbing of the fingers and smooth-muscle proliferation in fibrotic changes in the lung in patients with idiopathic pulmonary fibrosis. Chest 1994, 105, 339–342. [Google Scholar] [CrossRef]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Murthy, M.; Balch, W.E.; Chandel, N.S.; Meiners, S.; Eickelberg, O.; Selman, M.; Pardo, A.; White, E.S.; Levy, B.D.; et al. Blue journal conference. Aging and susceptibility to lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 261–269. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 3, 807–827. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Armanios, M. Telomeres and age-related disease: How telomere biology informs clinical paradigms. J. Clin. Invest. 2013, 123, 996–1002. [Google Scholar] [CrossRef]
- Li, J.S.; Miralles Fusté, J.; Simavorian, T.; Bartocci, C.; Tsai, J.; Karlseder, J.; Lazzerini Denchi, E. TZAP: A telomere-associated protein involved in telomere length control. Science 2017, 355, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomere length regulation. Annu. Rev. Biochem. 1996, 65, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Hug, N.; Lingner, J. Telomere length homeostasis. Chromosoma 2006, 115, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, D.; Songyang, Z. The telosome/shelterin complex and its functions. Genome Biol. 2008, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Grolimund, L.; Aeby, E.; Hamelin, R.; Armand, F.; Chiappe, D.; Moniatte, M.; Lingner, J. A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat. Commun. 2013, 4, 2848. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Wain, H.M.; Bruford, E.A.; Lovering, R.C.; Lush, M.J.; Wright, M.W.; Povey, S. Guidelines for human gene nomenclature. Genomics 2002, 79, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Kipling, D. Telomerase: Immortality enzyme or oncogene? Nat. Genet. 1995, 9, 104–106. [Google Scholar] [CrossRef]
- Blasco, M.A. Mice with bad ends: Mouse models for the study of telomeres and telomerase in cancer and aging. EMBO J. 2005, 24, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Reddy, R.; Barsky, L.; Scholes, J.; Chen, H.; Shi, W.; Driscoll, B. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L57–L70. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A.; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Shi, Y.; Liu, Y.; Pan, X.H.; Zhang, K.X. Telomere shortening activates TGF-β/Smads signaling in lungs and enhances both lipopolysaccharide and bleomycin-induced pulmonary fibrosis. Acta Pharmacol. Sin. 2018, 39, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, Á.; Gómez-López, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife 2018, 7, e31299. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Crestani, B.; Dieude, P.; Nunes, H.; Allanore, Y.; Kannengiesser, C.; Airo, P.; Matucci-Cerinic, M.; Wallaert, B.; Israel-Biet, D.; et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS ONE 2013, 8, e70621. [Google Scholar] [CrossRef]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef]
- Snetselaar, R.; van Moorsel, C.H.M.; Kazemier, K.M.; van der Vis, J.J.; Zanen, P.; van Oosterhout, M.F.M.; Grutters, J.C. Telomere length in interstitial lung diseases. Chest 2015, 148, 1011–1018. [Google Scholar] [CrossRef]
- Borie, R.; Tabèze, L.; Thabut, G.; Nunes, H.; Cottin, V.; Marchand-Adam, S.; Prevot, G.; Tazi, A.; Cadranel, J.; Mal, H.; et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur. Respir. J. 2016, 48, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Sui, B.; Hu, C.; Jin, Y. Mitochondrial metabolic failure in telomere attrition-provoked aging of bone marrow mesenchymal stem cells. Biogerontology 2016, 17, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Mendez, M.; Choi, K.; Subbotina, N.; Courey, A.; Cunningham, A.; Dave, A.; Engelhardt, J.F.; Liu, X.; White, E.S.; et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care. Med. 2010, 181, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Snetselaar, R.; van Batenburg, A.A.; van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; van der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE. 2017, 12, e0189467. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.A.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Hanumanthu, V.S.; Strong, M.A.; DeZern, A.E.; Stanley, S.E.; Takemoto, C.M.; Danilova, L.; Applegate, C.D.; Bolton, S.G.; Mohr, D.W.; et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proc. Natl. Acad. Sci. USA 2018, 115, E2358–E2365. [Google Scholar] [CrossRef] [PubMed]
- Juge, P.A.; Borie, R.; Kannengiesser, C.; Gazal, S.; Revy, P.; Wemeau-Stervinou, L.; Debray, M.P.; Ottaviani, S.; Marchand-Adam, S.; Nathan, N.; et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1602314. [Google Scholar] [CrossRef]
- Dai, J.; Cai, H.; Zhuang, Y.; Wu, Y.; Min, H.; Li, J.; Shi, Y.; Gao, Q.; Yi, L. Telomerase gene mutations and telomere length shortening in patients with idiopathic pulmonary fibrosis in a Chinese population. Respirology 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Collopy, L.C.; Walne, A.J.; Cardoso, S.; de la Fuente, J.; Mohamed, M.; Toriello, H.; Tamary, H.; Ling, A.J.; Lloyd, T.; Kassam, R.; et al. Triallelic and epigenetic-like inheritance in human disorders of telomerase. Blood 2015, 126, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.A.; Fox, G.; Bhatia, R.; Sala, E.; Noble, B.; Denic, N.; Fernandez, D.; Duguid, N.; Dohey, A.; Kamel, F.; et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: Clinical and genetic features. Respir. Res. 2012, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Garcia, C.K. Epigenetic inheritance of telomere length obscures identification of causative PARN locus. J. Med. Genet. 2016, 53, 356–358. [Google Scholar] [CrossRef]
- Stanley, S.E.; Gable, D.L.; Wagner, C.L.; Carlile, T.M.; Hanumanthu, V.S.; Podlevsky, J.D.; Khalil, S.E.; DeZern, A.E.; Rojas-Duran, M.F.; Applegate, C.D.; et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Dressen, A.; Abbas, A.R.; Cabanski, C.; Reeder, J.; Ramalingam, T.R.; Neighbors, M.; Bhangale, T.R.; Brauer, M.J.; Hunkapiller, J.; Reeder, J.; et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: A candidate gene sequencing study. Lancet Respir. Med. 2018, 6, 603–614. [Google Scholar] [CrossRef]
- Alder, J.K.; Guo, N.; Kembou, F.; Parry, E.M.; Anderson, C.J.; Gorgy, A.I.; Walsh, M.F.; Sussan, T.; Biswal, S.; Mitzner, W.; et al. Telomere length is a determinant of emphysema susceptibility. Am. J. Respir. Crit. Care Med. 2011, 184, 904–912. [Google Scholar] [CrossRef]
- Telomerase Database. Available online: http://telomerase.asu.edu/diseases.html (accessed on 7 July 2019).
- Kärkkäinen, M.; Kettunen, H.P.; Nurmi, H.; Selander, T.; Purokivi, M.; Kaarteenaho, R. Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 160. [Google Scholar] [CrossRef]
- Morlá, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agustí, A.G.N. Telomere shortening in smokers with and without COPD. Eur. Respir. J. 2006, 27, 525–528. [Google Scholar] [CrossRef]
- Valdes, A.M.; Andrew, T.; Gardner, J.P.; Kimura, M.; Oelsner, E.; Cherkas, L.F.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet Lond. Engl. 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Białas, A.J.; Sitarek, P.; Miłkowska-Dymanowska, J.; Piotrowski, W.J.; Górski, P. The Role of Mitochondria and Oxidative/Antioxidative Imbalance in Pathobiology of Chronic Obstructive Pulmonary Disease. Oxid. Med. Cell. Longev. 2016, 2016, 7808576. [Google Scholar] [CrossRef] [PubMed]
- Faner, R.; Rojas, M.; MacNee, W.; Agusti, A. Abnormal Lung Aging in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Am. J. Crit. Care. Med. 2012, 186, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Carleo, A.; Bergantini, L.; Landi, C.; Prasse, A.; Bargagli, E. Oxidant/Antioxidant Disequilibrium in Idiopathic Pulmonary Fibrosis Pathogenesis. Inflammation 2019. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Dalavanga, Y.; Poulakis, N.; Sixt, S.U.; Guzman, J.; Costabel, U. Decreased expression of haem oxygenase-1 by alveolar macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2008, 31, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Mazur, W.; Lindholm, P.; Vuorinen, K.; Myllärniemi, M.; Salmenkivi, K.; Kinnula, V.L. Cell-specific elevation of NRF2 and sulfiredoxin-1 as markers of oxidative stress in the lungs of idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. APMIS. 2010, 118, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Homma, T.; Kobayashi, S.; Seo, H.G. Mutual interaction between oxidative stress and endoplasmic reticulum stress in the pathogenesis of diseases specifically focusing on non-alcoholic fatty liver disease. World. J. Biol. Chem. 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R. Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Murray, S.; Fell, C.D.; Flaherty, K.R.; Toews, G.B.; Myers, J.; Colby, T.V.; Travis, W.D.; Kazerooni, E.A.; Gross, B.H.; et al. Sex differences in physiological progression of idiopathic pulmonary fibrosis. Eur. Respir. J. 2008, 31, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Harari, S.; Caminati, A.; Confalonieri, M.; Poletti, V.; Vancheri, C.; Pesci, A.; Rogliani, P.; Luppi, F.; Agostini, C.; Rottoli, P.; et al. The prognostic role of Gender-Age-Physiology system in idiopathic pulmonary fibrosis patients treated with pirfenidone. Clin. Respir. J. 2019, 13, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Armbruster, B.N.; Price, D.T.; Counter, C.M. In vivo regulation of hTERT expression and telomerase activity by androgen. J. Urol. 2003, 170, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Bayne, S.; Liu, J.P. Hormones and growth factors regulate telomerase activity in ageing and cancer. Mol. Cell. Endocrinol. 2005, 240, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Townsley, D.M.; Dumitriu, B.; Liu, D.; Biancotto, A.; Weinstein, B.; Chen, C.; Hardy, N.; Mihalek, A.D.; Lingala, S.; Kim, Y.J.; et al. Danazol Treatment for Telomere Diseases. N. Engl. J. Med. 2016, 374, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Borie, R. Clinical implications of telomere dysfunction in lung fibrosis. Curr. Opin. Pulm. Med. 2018, 24, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68. [Google Scholar] [CrossRef]
- Ley, B.; Swigris, J.; Day, B.M.; Stauffer, J.L.; Raimundo, K.; Chou, W.; Collard, H.R. Pirfenidone Reduces Respiratory-related Hospitalizations in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 756–761. [Google Scholar] [CrossRef]
- Bando, M.; Yamauchi, H.; Ogura, T.; Taniguchi, H.; Watanabe, K.; Azuma, A.; Homma, S.; Sugiyama, Y.; Japan Pirfenidone Clinical Study Group. Clinical Experience of the Long-term Use of Pirfenidone for Idiopathic Pulmonary Fibrosis. Intern. Med. 2016, 55, 443–448. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet Lond. Engl. 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Raghu, G.; Johnson, W.C.; Lockhart, D.; Mageto, Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: Results of a prospective, open-label Phase II study. Am. J. Respir. Crit. Care Med. 1999, 159, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; du Bois, R.M.; Jenkins, G.; Kolb, M.; Maher, T.M.; Raghu, G.; Vancheri, C.; Laurent, G.J. Mapping the future for pulmonary fibrosis: Report from the 17th International Colloquium on Lung and Airway Fibrosis. Eur. Respir. J. 2013, 42, 230–238. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet Lond. Engl. 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef]
- Silhan, L.L.; Shah, P.D.; Chambers, D.C.; Snyder, L.D.; Riise, G.C.; Wagner, C.L.; Hellström-Lindberg, E.; Orens, J.B.; Mewton, J.F.; Danoff, S.K.; et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur. Respir. J. 2014, 44, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Kannengiesser, C.; Hirschi, S.; Le Pavec, J.; Mal, H.; Bergot, E.; Jouneau, S.; Naccache, J.M.; Revy, P.; Boutboul, D.; et al. Severe hematologic complications after lung transplantation in patients with telomerase complex mutations. J. Heart Lung Transplant. 2015, 34, 538–546. [Google Scholar] [CrossRef]
- Zhang, Y.; Jones, K.D.; Achtar-Zadeh, N.; Green, G.; Kukreja, J.; Xu, B.; Wolters, P.J. Histopathological and molecular analysis of idiopathic pulmonary fibrosis lungs from patients treated with pirfenidone or nintedanib. Histopathology 2019, 74, 341–349. [Google Scholar] [CrossRef]
- Le Saux, C.J.; Davy, P.; Brampton, C.; Ahuja, S.S.; Fauce, S.; Shivshankar, P.; Nguyen, H.; Ramaseshan, M.; Tressler, R.; Pirot, Z.; et al. A novel telomerase activator suppresses lung damage in a murine model of idiopathic pulmonary fibrosis. PLoS ONE 2013, 8, e58423. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Function | OMIM Number | Mutation | Reference Sequence | Amino Acid Substitution | Comment | Ref. |
|---|---|---|---|---|---|---|---|
| TERT | Enzyme in telomerase complex | 187270 | 97C>T | coding DNA | Pro33Ser | [38] | |
| 164T>A | coding DNA | Leu55Gln | [37] | ||||
| 277+1G>A | coding DNA | [37] | |||||
| (334_336)delC | coding DNA | Pro112ProfsX16 | [37] | ||||
| 430G>A | coding DNA | Val144Met | [38,46] | ||||
| coding DNA | Val170Leu | uncertain description | [47,48] | ||||
| 1234C>T | coding DNA | His412Tyr | [49] | ||||
| 1456C>T | coding DNA | Arg486Cys | [38] | ||||
| coding DNA | Arg622His | uncertain description | [50] | ||||
| 1885G>C | coding DNA | Gly629Arg | [51] | ||||
| 1989G>C | coding DNA | Ser663Arg | [51] | ||||
| coding DNA | Val664Leu | uncertain description | [48] | ||||
| 2069G>C | coding DNA | Trp690Ser | [51] | ||||
| 2081C>T | coding DNA | Val694Glu | [51] | ||||
| 2110C>T | coding DNA | Pro704Ser | [51] | ||||
| 2225C>T | coding DNA | Arg742His | [51] | ||||
| 2240delT | coding DNA | Val747AlafsX20 | [38] | ||||
| 2329G>A | coding DNA | Val777Leu | [50,52] | ||||
| 2383-2A>G | coding DNA | [49] | |||||
| 2583-2A>C | coding DNA | Leu862_Leu884del | [37] | ||||
| 2593C>T | coding DNA | Arg865Cys | [38] | ||||
| 2594G>A | coding DNA | Arg865His | [38] | ||||
| coding DNA | Arg865Ala | uncertain description | [46] | ||||
| coding DNA | Thr874Arg | uncertain description | [46] | ||||
| 2620A>G | coding DNA | Thr874Ala | [51] | ||||
| 2648T>G | coding DNA | Phe883Cys | [53] | ||||
| 2812C>T | coding DNA | Arg938Trp | [51] | ||||
| coding DNA | Phe1032Ile | uncertain description | [50] | ||||
| 3323C>T | coding DNA | Pro1108Leu | [49] | ||||
| 3329C>T | coding DNA | Thr1110Met | [37] | ||||
| 3346_3522del177 | coding DNA | Glu1116fsX | [38] | ||||
| PARN | mRNA stability | 604212 | coding DNA | Phe8LeufsX | uncertain description | [48] | |
| 246-2A>G | coding DNA | [46,54] | |||||
| 529C>T | coding DNA | Gln177X | [54,55] | ||||
| 563_564insT | coding DNA | Ile188IlefsX7 | [54] | ||||
| 14676110C>A | coding DNA; genomic | Glu374X | uncertain description | [51] | |||
| 1262A>G | coding DNA | Lys421Arg | [54] | ||||
| 14647946G>C | coding DNA; genomic | Leu461Val | uncertain description | [51] | |||
| 14540858CCT>C | coding DNA; genomic | Glu585AspfsX5 | uncertain description | [51] | |||
| RTEL1 | DNA helicase | 608833 | 62320919A>AC | coding DNA; genomic | Arg675ThrfsX15 | uncertain description | [51] |
| 623321112G>A | coding DNA; genomic | Gly703Arg | uncertain description | [51] | |||
| 2214-2A>C | coding DNA | [51] | |||||
| 62321477T>C | coding DNA; genomic | Trp751Arg | uncertain description | [51] | |||
| 62321483C>T | coding DNA; genomic | Arg753Cys | uncertain description | [51] | |||
| 2992C>T | coding DNA | Arg998X | [51] | ||||
| 62324600C>T | coding DNA; genomic | Arg1010X | uncertain description | [51] | |||
| 602delG | coding DNA | Gly201GlufsX15 | [54] | ||||
| 1451C>T | coding DNA | Pro484Leu | [54] | ||||
| 1940C>T | coding DNA | Pro647Leu | [54] | ||||
| 2005C>T | coding DNA | Gln693X | [54] | ||||
| 2695T>C | coding DNA | Phe899Leu | [49] | ||||
| 2824G>A | coding DNA | Asp942Asn | [49] | ||||
| 2875C>T | coding DNA | His959Tyr | [49] | ||||
| NAF1 | catalyzes the addition of repetitive DNA sequences to telomeres | 617868 | 956_957delAA | coding DNA | Lys319Argfs*21 | [56] | |
| 984insA | coding DNA | Ser329Ilefs*12 | PF combined with emphysema | [56] | |||
| DKC1 | telomerase stabilization and maintenance | 300126 | 1213A>G | coding DNA | Thr405Ala | [57] | |
| Region | |||||||
| TERC | Template in telomerase complex | 602322 | 37a>g | RNA | P1b | [38] | |
| 98g>a | RNA | P2b | [37] | ||||
| 108c>u | RNA | P3 | [50] | ||||
| 325g>u | RNA | P5 | [47] | ||||
| 375_377delgga | RNA | Box H | PF combined with emphysema | [58] | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilgili, H.; Białas, A.J.; Górski, P.; Piotrowski, W.J. Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2019, 8, 1232. https://doi.org/10.3390/jcm8081232
Bilgili H, Białas AJ, Górski P, Piotrowski WJ. Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis. Journal of Clinical Medicine. 2019; 8(8):1232. https://doi.org/10.3390/jcm8081232
Chicago/Turabian StyleBilgili, Hasancan, Adam J. Białas, Paweł Górski, and Wojciech J. Piotrowski. 2019. "Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis" Journal of Clinical Medicine 8, no. 8: 1232. https://doi.org/10.3390/jcm8081232
APA StyleBilgili, H., Białas, A. J., Górski, P., & Piotrowski, W. J. (2019). Telomere Abnormalities in the Pathobiology of Idiopathic Pulmonary Fibrosis. Journal of Clinical Medicine, 8(8), 1232. https://doi.org/10.3390/jcm8081232