Pulmonary Fibrosis in Children

 ,
,
Abstract
1. Introduction
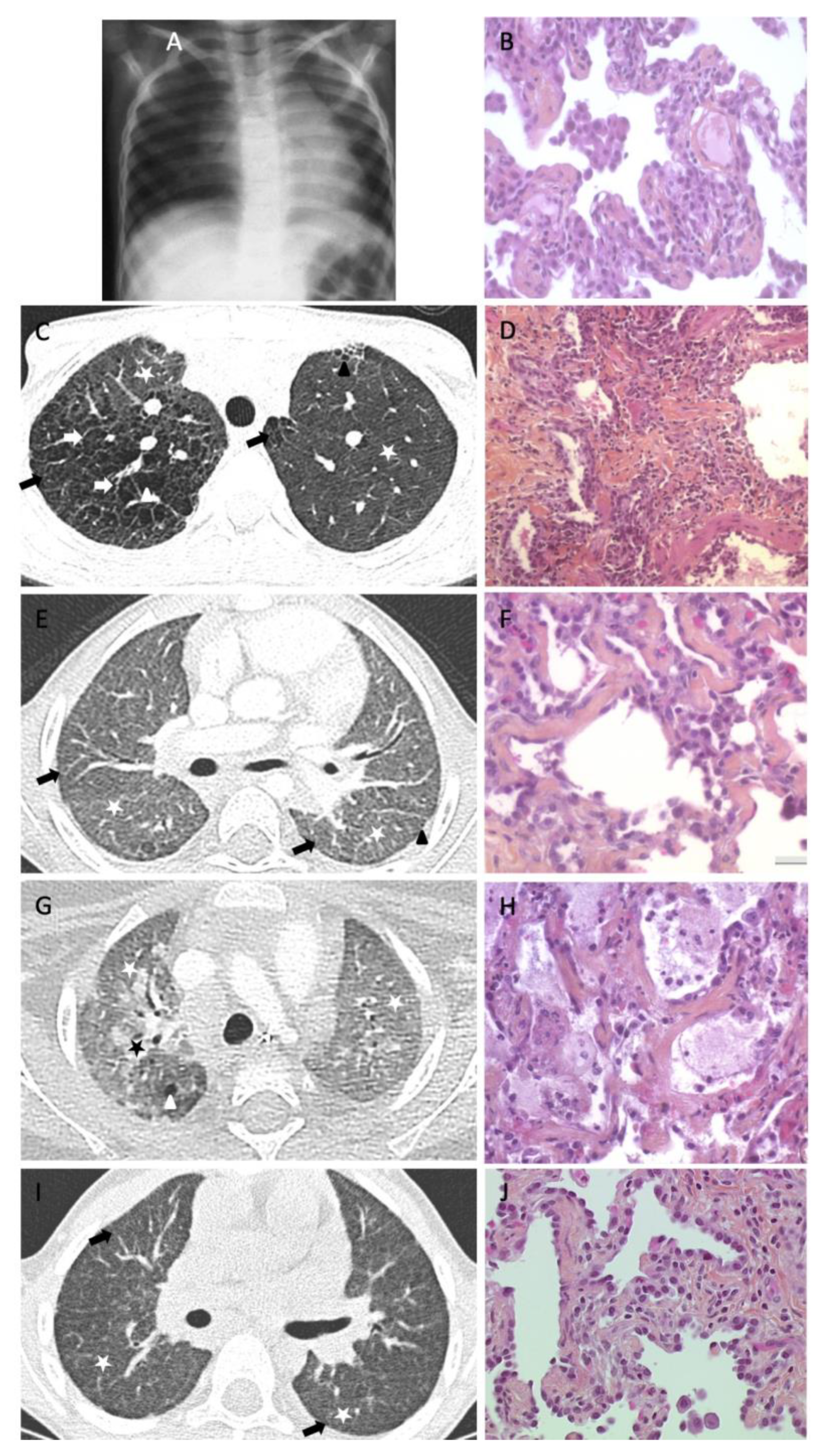
2. PF Reports in Pediatric Population
3. Lessons from Surfactant Disorders
4. Other Situations of PF Evolution through Age
5. Summary of Childhood and Adult PF Comparison
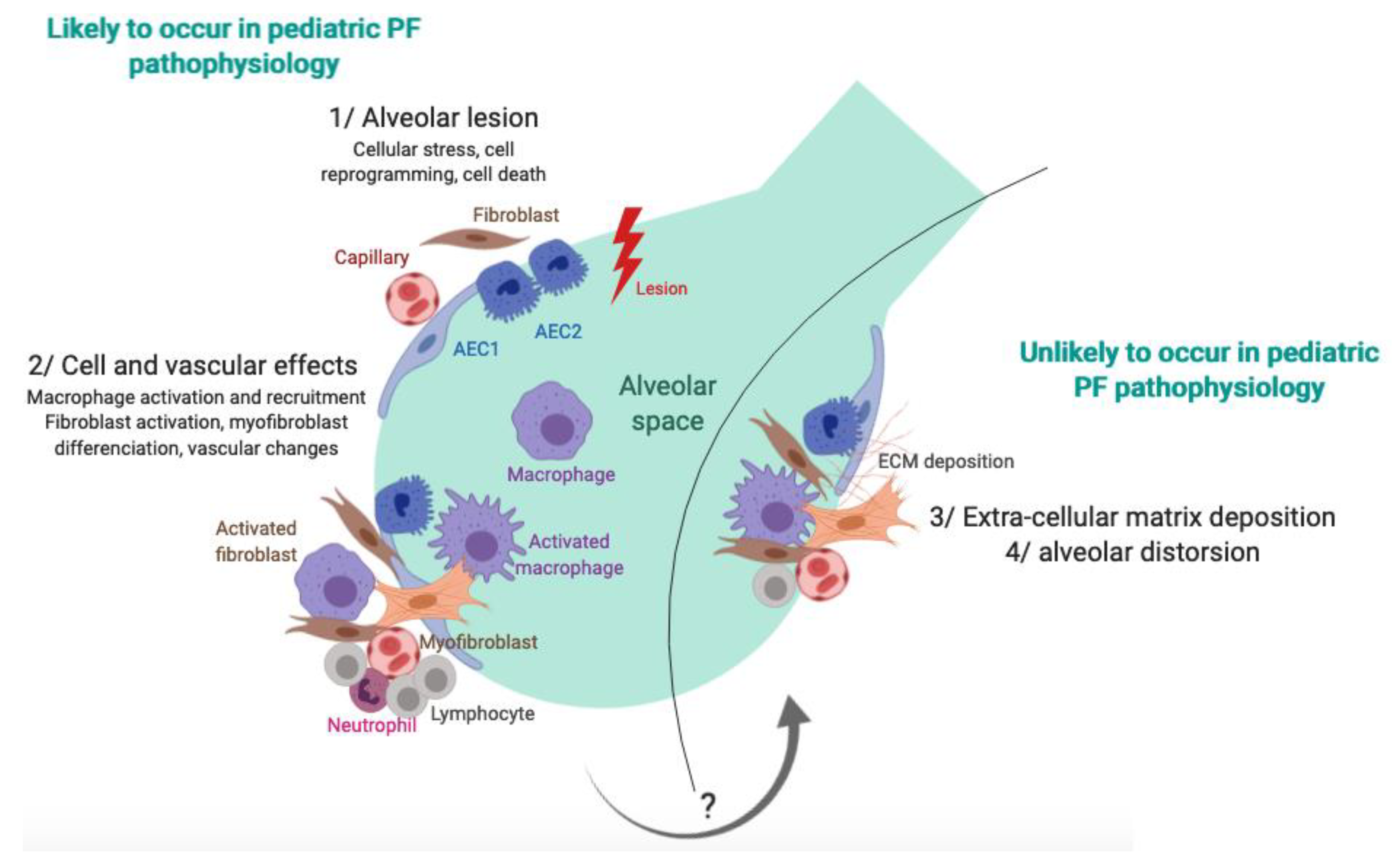
6. Pathophysiology
7. Treatments in Pediatric PF
8. Outcome of PF in Childhood
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deutsch, G.H.; Young, L.R.; Deterding, R.R.; Fan, L.L.; Dell, S.D.; Bean, J.A.; Brody, A.S.; Nogee, L.M.; Trapnell, B.C.; Langston, C.; et al. Diffuse lung disease in young children: Application of a novel classification scheme. Am. J. Respir. Crit. Care Med. 2007, 176, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.; Nathan, N.; Epaud, R.; Fauroux, B.; Corvol, H. Interstitial lung diseases in children. Orphanet J. Rare Dis. 2010, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Kurland, G.; Deterding, R.R.; Hagood, J.S.; Young, L.R.; Brody, A.S.; Castile, R.G.; Dell, S.; Fan, L.L.; Hamvas, A.; Hilman, B.C.; et al. An official American Thoracic Society clinical practice guideline: Classification, evaluation, and management of childhood interstitial lung disease in infancy. Am. J. Respir. Crit. Care Med. 2013, 188, 376–394. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.; Tran-Dang, M.-A.; Bush, A.; Nicholson, A.G. Diffuse lung disease in infancy and childhood: Expanding the chILD classification. Histopathology 2013, 63, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Griese, M.; Irnstetter, A.; Hengst, M.; Burmester, H.; Nagel, F.; Ripper, J.; Feilcke, M.; Pawlita, I.; Gothe, F.; Kappler, M.; et al. Categorizing diffuse parenchymal lung disease in children. Orphanet J. Rare Dis. 2015, 10, 122. [Google Scholar] [CrossRef]
- Nathan, N.; Berdah, L.; Borensztajn, K.; Clement, A. Chronic interstitial lung diseases in children: Diagnosis approaches. Expert Rev. Respir. Med. 2018, 12, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Mueller-Mang, C.; Grosse, C.; Schmid, K.; Stiebellehner, L.; Bankier, A.A. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics 2007, 27, 595–615. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Hashisako, M.; Fukuoka, J. Pathology of Idiopathic Interstitial Pneumonias. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 123–133. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nureki, S.-I.; Beers, M.F. Lost after translation: Insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L507–L525. [Google Scholar] [CrossRef] [PubMed]
- Dishop, M.K. Paediatric interstitial lung disease: Classification and definitions. Paediatr. Respir. Rev. 2011, 12, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Manali, E.D.; Legendre, M.; Nathan, N.; Kannengiesser, C.; Coulomb L’Hermine, A.; Tsiligiannis, T.; Tomos, P.; Griese, M.; Borie, R.; Clement, A.; et al. Bi-allelic missense ABCA3 mutations in a patient with children’s ILD who reached adulthood. ERJ Open Res. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidou, P.; Charocopos, E.; Anagnostopoulos, G.; Lazopoulou, D.; Kairis, M.; Lourida, A.; Tzoumakas, K.; Tsiligiannis, T. Cellular Interstitial Pneumonitis in Children: Response to Hydroxychloroquine Treatment in Two Cases. Pediatr. Asthma Allergy Immunol. 2003, 16, 45–51. [Google Scholar] [CrossRef]
- Picard, C.; Thouvenin, G.; Kannengiesser, C.; Dubus, J.-C.; Jeremiah, N.; Rieux-Laucat, F.; Crestani, B.; Belot, A.; Thivolet-Béjui, F.; Secq, V.; et al. Severe Pulmonary Fibrosis as the First Manifestation of Interferonopathy (TMEM173 Mutation). Chest 2016, 150, e65–e71. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Nathan, N.; Giraud, V.; Picard, C.; Nunes, H.; Dastot-Le Moal, F.; Copin, B.; Galeron, L.; de Ligniville, A.; Kuziner, N.; Reynaud-Gaubert, M.; et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum. Mol. Genet. 2016, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Ota, C.; Kimura, M.; Kure, S. ABCA3 mutations led to pulmonary fibrosis and emphysema with pulmonary hypertension in an 8-year-old girl. Pediatr. Pulmonol. 2016, 51, E21–E23. [Google Scholar] [CrossRef] [PubMed]
- Thouvenin, G.; Nathan, N.; Epaud, R.; Clement, A. Diffuse parenchymal lung disease caused by surfactant deficiency: Dramatic improvement by azithromycin. BMJ Case Rep. 2013, 2013, bcr2013009988. [Google Scholar] [CrossRef]
- Thouvenin, G.; Abou Taam, R.; Flamein, F.; Guillot, L.; Le Bourgeois, M.; Reix, P.; Fayon, M.; Counil, F.; Depontbriand, U.; Feldmann, D.; et al. Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch. Dis. Child. 2010, 95, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Kröner, C.; Wittmann, T.; Reu, S.; Teusch, V.; Klemme, M.; Rauch, D.; Hengst, M.; Kappler, M.; Cobanoglu, N.; Sismanlar, T.; et al. Lung disease caused by ABCA3 mutations. Thorax 2017, 72, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.; van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.; van Es, H.W.; van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Reix, P.; Khouatra, C.; Thivolet-Béjui, F.; Feldmann, D.; Cordier, J.-F. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 2011, 66, 918–919. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Tanaka, T.; Ishida, M.; Kinoshita, A.; Fukuoka, J.; Takaki, M.; Sakamoto, N.; Ishimatsu, Y.; Kohno, S.; Hayashi, T.; et al. Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur. Respir. J. 2011, 38, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Epaud, R.; Delestrain, C.; Louha, M.; Simon, S.; Fanen, P.; Tazi, A. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur. Respir. J. 2014, 43, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, M.A.; Shifren, A.; Huang, H.J.; Russell, T.D.; Mitra, R.D.; Zhang, Q.; Wegner, D.J.; Cole, F.S.; Hamvas, A. Sequencing of idiopathic pulmonary fibrosis-related genes reveals independent single gene associations. BMJ Open Respir. Res. 2014, 1, e000057. [Google Scholar] [CrossRef] [PubMed]
- Doubková, M.; Staňo Kozubík, K.; Radová, L.; Pešová, M.; Trizuljak, J.; Pál, K.; Svobodová, K.; Réblová, K.; Svozilová, H.; Vrzalová, Z.; et al. A novel germline mutation of the SFTPA1 gene in familial interstitial pneumonia. Hum. Genome Var. 2019, 6, 12. [Google Scholar] [CrossRef]
- Van Moorsel, C.H.M.; Ten Klooster, L.; van Oosterhout, M.F.M.; de Jong, P.A.; Adams, H.; Wouter van Es, H.; Ruven, H.J.T.; van der Vis, J.J.; Grutters, J.C. SFTPA2 Mutations in Familial and Sporadic Idiopathic Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2015, 192, 1249–1252. [Google Scholar] [CrossRef]
- Abou Taam, R.; Jaubert, F.; Emond, S.; Le Bourgeois, M.; Epaud, R.; Karila, C.; Feldmann, D.; Scheinmann, P.; de Blic, J. Familial interstitial disease with I73T mutation: A mid- and long-term study. Pediatr. Pulmonol. 2009, 44, 167–175. [Google Scholar] [CrossRef]
- Thomas, A.Q.; Lane, K.; Phillips, J.; Prince, M.; Markin, C.; Speer, M.; Schwartz, D.A.; Gaddipati, R.; Marney, A.; Johnson, J.; et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 2002, 165, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Cameron, H.S.; Somaschini, M.; Carrera, P.; Hamvas, A.; Whitsett, J.A.; Wert, S.E.; Deutsch, G.; Nogee, L.M. A common mutation in the surfactant protein C gene associated with lung disease. J. Pediatr. 2005, 146, 370–375. [Google Scholar] [CrossRef]
- Guillot, L.; Epaud, R.; Thouvenin, G.; Jonard, L.; Mohsni, A.; Couderc, R.; Counil, F.; de Blic, J.; Taam, R.A.; Le Bourgeois, M.; et al. New surfactant protein C gene mutations associated with diffuse lung disease. J. Med. Genet. 2009, 46, 490–494. [Google Scholar] [CrossRef]
- Campo, I.; Zorzetto, M.; Mariani, F.; Kadija, Z.; Morbini, P.; Dore, R.; Kaltenborn, E.; Frixel, S.; Zarbock, R.; Liebisch, G.; et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir. Res. 2014, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Jovien, S.; Borie, R.; Doummar, D.; Clement, A.; Nathan, N. Respiratory Distress, Congenital Hypothyroidism and Hypotonia in a Newborn. Respiration 2016, 92, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Nattes, E.; Lejeune, S.; Carsin, A.; Borie, R.; Gibertini, I.; Balinotti, J.; Nathan, N.; Marchand-Adam, S.; Thumerelle, C.; Fauroux, B.; et al. Heterogeneity of lung disease associated with NK2 homeobox 1 mutations. Respir. Med. 2017, 129, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Jeremiah, N.; Neven, B.; Gentili, M.; Callebaut, I.; Maschalidi, S.; Stolzenberg, M.-C.; Goudin, N.; Frémond, M.-L.; Nitschke, P.; Molina, T.J.; et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Investig. 2014, 124, 5516–5520. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Montealegre Sanchez, G.A.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef]
- Watkin, L.B.; Jessen, B.; Wiszniewski, W.; Vece, T.J.; Jan, M.; Sha, Y.; Thamsen, M.; Santos-Cortez, R.L.P.; Lee, K.; Gambin, T.; et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat. Genet. 2015, 47, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Noorelahi, R.; Perez, G.; Otero, H.J. Imaging findings of Copa syndrome in a 12-year-old boy. Pediatr. Radiol. 2018, 48, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Tsui, J.L.; Estrada, O.A.; Deng, Z.; Wang, K.M.; Law, C.S.; Elicker, B.M.; Jones, K.D.; Dell, S.D.; Gudmundsson, G.; Hansdottir, S.; et al. Analysis of pulmonary features and treatment approaches in the COPA syndrome. ERJ Open Res. 2018, 4, 00017. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Nathan, N.; Corvol, H.; Amselem, S.; Clement, A. Biomarkers in Interstitial lung diseases. Paediatr. Respir. Rev. 2015, 16, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Cottin, V.; Kreuter, M.; Hilberg, O. IPF, comorbidities and management implications. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 32 (Suppl. S1), 17–23. [Google Scholar]
- Kumar, A.; Cherian, S.V.; Vassallo, R.; Yi, E.S.; Ryu, J.H. Current Concepts in Pathogenesis, Diagnosis, and Management of Smoking-Related Interstitial Lung Diseases. Chest 2018, 154, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Mendez, M.; Choi, K.; Subbotina, N.; Courey, A.; Cunningham, A.; Dave, A.; Engelhardt, J.F.; Liu, X.; White, E.S.; et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 287–301. [Google Scholar] [CrossRef]
- Ahluwalia, N.; Shea, B.S.; Tager, A.M. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am. J. Respir. Crit. Care Med. 2014, 190, 867–878. [Google Scholar] [CrossRef]
- Maguire, J.A.; Mulugeta, S.; Beers, M.F. Multiple ways to die: Delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int. J. Biochem. Cell Biol. 2012, 44, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Brandenberger, C.; Mühlfeld, C. Mechanisms of lung aging. Cell Tissue Res. 2017, 367, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Callaghan, M.J.; Longaker, M.T. Progress and potential for regenerative medicine. Annu. Rev. Med. 2007, 58, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Komi-Kuramochi, A.; Kawano, M.; Oda, Y.; Asada, M.; Suzuki, M.; Oki, J.; Imamura, T. Expression of fibroblast growth factors and their receptors during full-thickness skin wound healing in young and aged mice. J. Endocrinol. 2005, 186, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Lemischka, I.R. Stem cells and their niches. Science 2006, 311, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Chapman, M.A. Regenerative Medicine: Charting a New Course in Wound Healing. Adv. Wound Care (N. Rochelle) 2016, 5, 314–328. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Fine, A. Stem Cells in Lung Injury and Repair. Am. J. Pathol. 2016, 186, 2544–2550. [Google Scholar] [CrossRef]
- Schilders, K.A.A.; Eenjes, E.; van Riet, S.; Poot, A.A.; Stamatialis, D.; Truckenmüller, R.; Hiemstra, P.S.; Rottier, R.J. Regeneration of the lung: Lung stem cells and the development of lung mimicking devices. Respir. Res. 2016, 17, 44. [Google Scholar] [CrossRef]
- Pomerantz, J.; Blau, H.M. Nuclear reprogramming: A key to stem cell function in regenerative medicine. Nat. Cell Biol. 2004, 6, 810–816. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Huang, S.X.; de Carvalho, A.L.R.T.; Ho, S.-H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.-L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yin, C.; Lu, X.I.; Jiang, H.; Jin, F. Bone marrow mesenchymal stem cells protect lungs from smoke inhalation injury by differentiating into alveolar epithelial cells via Notch signaling. J. Biosci. 2019, 44, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Gonzalez De Los Santos, F.; Zhao, Y.; Wu, Z.; Rinke, A.E.; Kim, K.K.; Phan, S.H. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J. Biol. Chem. 2019, 294, 8861–8871. [Google Scholar] [CrossRef] [PubMed]
- Koliakos, G. Stem Cells and Aging. Rejuvenation Res. 2017, 20, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.-L.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.; ERS Task Force. Task force on chronic interstitial lung disease in immunocompetent children. Eur. Respir. J. 2004, 24, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.; Eber, E. Interstitial lung diseases in infants and children. Eur. Respir. J. 2008, 31, 658–666. [Google Scholar] [CrossRef]
- Bush, A.; Cunningham, S.; de Blic, J.; Barbato, A.; Clement, A.; Epaud, R.; Hengst, M.; Kiper, N.; Nicholson, A.G.; Wetzke, M.; et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax 2015, 70, 1078–1084. [Google Scholar] [CrossRef]
- Roberts, D.; Brown, J.; Medley, N.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2017, 3, CD004454. [Google Scholar] [CrossRef]
- Hime, N.J.; Zurynski, Y.; Fitzgerald, D.; Selvadurai, H.; Phu, A.; Deverell, M.; Elliott, E.J.; Jaffe, A. Childhood interstitial lung disease: A systematic review. Pediatr. Pulmonol. 2015, 50, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Nathan, N.; Borensztajn, K.; Clement, A. Genetic causes and clinical management of pediatric interstitial lung diseases. Curr. Opin. Pulm. Med. 2018, 24, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Taytard, J.; Nathan, N.; de Blic, J.; Fayon, M.; Epaud, R.; Deschildre, A.; Troussier, F.; Lubrano, M.; Chiron, R.; Reix, P.; et al. New insights into pediatric idiopathic pulmonary hemosiderosis: The French RespiRare (®) cohort. Orphanet J. Rare Dis. 2013, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Alimi, A.; Taytard, J.; Abou Taam, R.; Houdouin, V.; Forgeron, A.; Lubrano Lavadera, M.; Cros, P.; Gibertini, I.; Derelle, J.; Deschildre, A.; et al. Pulmonary hemosiderosis in children with Down syndrome: A national experience. Orphanet J. Rare Dis. 2018, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.A.; Jaroszewski, D.E.; Helmers, R.A.; Colby, T.V.; Patel, B.M.; Mookadam, F. Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia: A systematic overview. Am. J. Respir. Crit. Care Med. 2011, 184, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Ferner, M.; Kronfeld, K.; Griese, M. Hydroxychloroquine in children with interstitial (diffuse parenchymal) lung diseases. Pediatr. Pulmonol. 2015, 50, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Klay, D.; Hoffman, T.W.; Harmsze, A.M.; Grutters, J.C.; van Moorsel, C.H.M. Systematic review of drug effects in humans and models with surfactant-processing disease. Eur. Respir. Rev. 2018, 27, 170135. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.; Wallis, C. Ten-year follow up of hydroxychloroquine treatment for ABCA3 deficiency. Pediatr. Pulmonol. 2014, 49, 299–301. [Google Scholar] [CrossRef]
- Vece, T.J.; Fan, L.L. Diagnosis and management of diffuse lung disease in children. Paediatr. Respir. Rev. 2011, 12, 238–242. [Google Scholar] [CrossRef]
- Guillot, L.; Tabary, O.; Nathan, N.; Corvol, H.; Clement, A. Macrolides: New therapeutic perspectives in lung diseases. Int. J. Biochem. Cell Biol. 2011, 43, 1241–1246. [Google Scholar] [CrossRef]
- Aravena, C.; Labarca, G.; Venegas, C.; Arenas, A.; Rada, G. Pirfenidone for Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0136160. [Google Scholar]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lauby, C.; Boelle, P.-Y.; Abou Taam, R.; Bessaci, K.; Brouard, J.; Dalphin, M.-L.; Delacourt, C.; Delestrain, C.; Deschildre, A.; Dubus, J.-C.; et al. Health-related quality of life in infants and children with interstitial lung disease. Pediatr. Pulmonol. 2019, 54, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Breuer, O.; Schultz, A. Side effects of medications used to treat childhood interstitial lung disease. Paediatr. Respir. Rev. 2018, 28, 68–79. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| chILD Condition | Number of Patients | Number of Patients with Available Lung Samples | Number of Cases with Suspected PF |
|---|---|---|---|
| Surfactant disorders | 17 | 5 | 2 |
| Autoinflammatory and systemic disorders | 6 | 6 | 1 |
| Developmental disorders | 8 | 8 | 0 |
| Others | 88 | 25 | 7 |
| Total | 119 | 44 | 10 |
| Patient Number | Clinical Presentation | Treatment | Outcome |
|---|---|---|---|
| 1 [14,15] | 6-year-old girl, ABCA3-related disease | HCQ, azithromycin | Diffuse fibrosing ILD at age 26 |
| 2 [16] | 8-year-old boy, TMEM173-related disease | Corticosteroid pulses, oral corticosteroids, ruxolitinib at age 13. | Lung transplantation at age 14, died at age 16 after second lung transplantation |
| 3 | 3-year-old boy, undefined chILD | Corticosteroids | Died at age 3 from respiratory failure |
| 4 | 2-year-old girl, undefined chILD | Corticosteroid pulses, oral corticosteroids, azithromycin, immunosuppressive drugs | Died at age 2 from respiratory failure |
| 5 | 2-year-old girl, undefined chILD | Corticosteroid pulses, oral corticosteroids, azithromycin | Asymptomatic at age 8 |
| Pediatric PF | Adult IPF/Probable IPF | |
|---|---|---|
| Parenchymal distortion | + | +++ |
| Cellular recruitment | +++ | + |
| Extracellular matrix deposition | + | +++ |
| Fibroblast foci | +/− | +++ |
| Honeycombing | +/− | +++ |
| Global pattern | Predominant NSIP mixed with alveolar proteinosis, DIP, and follicular bronchiolitis | Predominant UIP pattern |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nathan, N.; Sileo, C.; Thouvenin, G.; Berdah, L.; Delestrain, C.; Manali, E.; Papiris, S.; Léger, P.-L.; Ducou le Pointe, H.; Coulomb l’Hermine, A.; et al. Pulmonary Fibrosis in Children. J. Clin. Med. 2019, 8, 1312. https://doi.org/10.3390/jcm8091312
Nathan N, Sileo C, Thouvenin G, Berdah L, Delestrain C, Manali E, Papiris S, Léger P-L, Ducou le Pointe H, Coulomb l’Hermine A, et al. Pulmonary Fibrosis in Children. Journal of Clinical Medicine. 2019; 8(9):1312. https://doi.org/10.3390/jcm8091312
Chicago/Turabian StyleNathan, Nadia, Chiara Sileo, Guillaume Thouvenin, Laura Berdah, Céline Delestrain, Effrosyne Manali, Spyros Papiris, Pierre-Louis Léger, Hubert Ducou le Pointe, Aurore Coulomb l’Hermine, and et al. 2019. "Pulmonary Fibrosis in Children" Journal of Clinical Medicine 8, no. 9: 1312. https://doi.org/10.3390/jcm8091312
APA StyleNathan, N., Sileo, C., Thouvenin, G., Berdah, L., Delestrain, C., Manali, E., Papiris, S., Léger, P.-L., Ducou le Pointe, H., Coulomb l’Hermine, A., & Clement, A. (2019). Pulmonary Fibrosis in Children. Journal of Clinical Medicine, 8(9), 1312. https://doi.org/10.3390/jcm8091312