iPS-Cell Technology and the Problem of Genetic Instability—Can It Ever Be Safe for Clinical Use?



Abstract
1. Introduction
1.1. Experiences with Human Embryonic Stem-Cell (hESC)-Derived Cells
- Ethical issues surround the source of hESC, which is usually the destruction of a 5–10-day-old blastocyst―a cluster of 100–200 cells [5].
- Therapeutic use of hESCs is inherently problematic. The cells are not only potentially immunogenic, but also the use of failed In Vitro Fertilization (IVF) embryos invites complications of abnormal development.
- hESC lines often show or develop karyotypic abnormalities associated with proliferative advantage or exhibit full teratomagenicity [6].
1.2. Human-Induced Pluripotent Stem Cells (hiPSC)
2. Reprogramming Leads to Genetic Dysregulation
3. iPSC Are Dangerous by Design?
3.1. Neoplasia Following Stem-Cell Therapies
3.2. The Challenge of Removal of Undifferentiated iPSC
3.3. Genetic Stability: iPSC Have a Good Safety Record
3.4. Clinical Trial History of MSC-Based Interventions Can Inform iPSC Safety Assessment
4. iPSC Are Inherently Unstable and Unreliable?
4.1. iPSC May Possess Overt Cancer Driver Mutations As Well As Cryptic Tumourigenic Genetic Changes
4.2. The Problem of iPS-Cell Evolution during Preparation of Therapeutic Product
4.3. Epigenetics
5. Translation into Standard of Care Will Be Problematic?
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ilic, D.; Devito, L.; Miere, C.; Codognotto, S. Human embryonic and induced pluripotent stem cells in clinical trials. Br. Med. Bull. 2015, 116, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Shroff, G.; Dhanda Titus, J.; Shroff, R. A review of the emerging potential therapy for neurological disorders: Human embryonic stem cell therapy. Am. J. Stem Cells 2017, 6, 1–12. [Google Scholar] [PubMed]
- Canham, M.A.; Van Deusen, A.; Brison, D.R.; De Sousa, P.A.; Downie, J.; Devito, L.; Hewitt, Z.A.; Ilic, D.; Kimber, S.J.; Moore, H.D.; et al. The Molecular Karyotype of 25 Clinical-Grade Human Embryonic Stem Cell Lines. Sci. Rep. 2015, 5, 17258. [Google Scholar] [CrossRef] [PubMed]
- Mitalipova, M.M.; Rao, R.R.; Hoyer, D.M.; Johnson, J.A.; Meisner, L.F.; Jones, K.L.; Dalton, S.; Stice, S.L. Preserving the genetic integrity of human embryonic stem cells. Nat. Biotechnol. 2005, 23, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, G.D.; Fischbach, R.L. Stem cells: Science, policy, and ethics. J. Clin. Investig. 2004, 114, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.A.; Klimanskaya, I.; McMahon, J.; Atienza, J.; Witmyer, J.; Zucker, J.P.; Wang, S.; Morton, C.C.; McMahon, A.P.; Powers, D.; et al. Derivation of Embryonic Stem-Cell Lines from Human Blastocysts. N. Engl. J. Med. 2004, 350, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, J.; Chen, T.; Wang, Y.; Xin, S.; Li, J.; Pei, G.; Kang, J. Yamanaka factors critically regulate the developmental signaling network in mouse embryonic stem cells. Cell Res. 2008, 18, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Kurimoto, Y.; Takahashi, M. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 377, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Harousseau, J.-L. Treatment of autologous stem cell transplant-eligible multiple myeloma patients: Ten questions and answers. Haematologica 2014, 99, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Wernig, M.; Markoulaki, S.; Sun, C.-W.; Meissner, A.; Cassady, J.P.; Beard, C.; Brambrink, T.; Wu, L.-C.; Townes, T.M.; et al. Treatment of Sickle Cell Anemia Mouse Model with iPS Cells Generated from Autologous Skin. Science 2007, 318, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. ‘Reprogrammed’ stem cells implanted into patient with Parkinson’s disease. Nature 2018, 563, 1–2. [Google Scholar] [CrossRef]
- Ahmed, R.P.; Haider, H.K.; Buccini, S.; Li, L.; Jiang, S.; Ashraf, M. Reprogramming of skeletal myoblasts for induction of pluripotency for tumor free cardiomyogenesis in the infarcted heart. Circ. Res. 2011, 109, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Abematsu, M.; Falk, A.; Tsujimura, K.; Sanosaka, T.; Juliandi, B.; Semi, K.; Namihira, M.; Komiya, S.; Smith, A.; et al. Treatment of a Mouse Model of Spinal Cord Injury by Transplantation of Human Induced Pluripotent Stem Cell-Derived Long-Term Self-Renewing Neuroepithelial-Like Stem Cells. Stem Cells 2012, 30, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.J.; Deveau, T.C.; Song, M.; Gu, X.; Chen, D.; Wei, L. iPSC Transplantation Increases Regeneration and Functional Recovery After Ischemic Stroke in Neonatal Rats. Stem Cells 2014, 32, 3075–3087. [Google Scholar] [CrossRef] [PubMed]
- Baum, C. Insertional mutagenesis in gene therapy and stem cell biology. Curr. Opin. Hematol. 2007, 14, 337. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of Defective and Persistent Sendai Virus Vector: A Unique Gene Delivery/Expression System Ideal For Cell Reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Ohtaka, M.; Takada, H.; Kurisaki, A.; Tran, N.V.K.; Tran, Y.T.H.; Hisatake, K.; Sano, M.; Nakanishi, M. Simple and effective generation of transgene-free induced pluripotent stem cells using an auto-erasable Sendai virus vector responding to microRNA-302. Stem Cell Res. 2017, 23, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2014, 33, nbt.3070. [Google Scholar] [CrossRef] [PubMed]
- Beh-Pajooh, A.; Cantz, T. The Role of microRNAs in Embryonic and Induced Pluripotency. J. Stem Cells Regen. Med. 2018, 14, 3–9. [Google Scholar] [PubMed]
- Li, N.; Long, B.; Han, W.; Yuan, S.; Wang, K. microRNAs: Important regulators of stem cells. Stem Cell Res. Ther. 2017, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced Pluripotent Stem Cells Generated Without Viral Integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Rao, M.S. A Review of the Methods for Human iPSC Derivation. Methods Mol. Biol. Clifton NJ 2013, 997, 23–33. [Google Scholar]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Yin, X.; Yang, W.; Du, Y.; Hou, P.; Ge, J.; Liu, C.; Zhang, W.; Zhang, X.; et al. Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res. 2010, 21, cr2010142. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, N.; Gros, E.; Li, H.-R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.-D.; Yu, B.D.; et al. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, K.; Nazor, K.L.; Williams, R.; Tran, H.; Dai, H.; Džakula, Ž.; Cho, E.H.; Pang, A.W.C.; Rao, M.; Cao, H.; et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 2016, 7, 10536. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.-L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005, 122, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Woodard, L.E.; Wilson, M.H. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 2015, 33, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Yu, W.; Sun, Q.; Zhang, X.; Jiang, W.; Wu, C.; Chen, C.; Gu, J.; Zheng, Y.; Xu, C. High-level transgene expression mediated by the piggyBac transposon enhances transgenic therapeutic effects in cervical cancer xenografts. Oncol. Rep. 2010, 24, 897–907. [Google Scholar] [PubMed]
- Ghosh, Z.; Huang, M.; Hu, S.; Wilson, K.D.; Dey, D.; Wu, J.C. Dissecting the Oncogenic and Tumorigenic Potential of Differentiated Human Induced Pluripotent Stem Cells and Human Embryonic Stem Cells. Cancer Res. 2011, 71, 5030–5039. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.; Zhou, H.; Nadif Kasri, N. Choices for Induction of Pluripotency: Recent Developments in Human Induced Pluripotent Stem Cell Reprogramming Strategies. Stem Cell Rev. 2016, 12, 54–72. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Thier, M.; Münst, B.; Edenhofer, F. Exploring refined conditions for reprogramming cells by recombinant Oct4 protein. Int. J. Dev. Biol. 2010, 54, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Y.; Gu, J.; Liao, B.; Li, J.; Wong, J.; Jin, Y. Reprogramming of somatic cells via TAT-mediated protein transduction of recombinant factors. Biomaterials 2012, 33, 5047–5055. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Chen, R.; Teesalu, T.; Ruoslahti, E.; Clegg, D.O. Reprogramming human retinal pigmented epithelial cells to neurons using recombinant proteins. Stem Cells Transl. Med. 2014, 3, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Nemes, C.; Varga, E.; Polgar, Z.; Klincumhom, N.; Pirity, M.K.; Dinnyes, A. Generation of Mouse Induced Pluripotent Stem Cells by Protein Transduction. Tissue Eng. Part C Methods 2014, 20, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Borgohain, M.P.; Haridhasapavalan, K.K.; Dey, C.; Adhikari, P.; Thummer, R.P. An Insight into DNA-free Reprogramming Approaches to Generate Integration-free Induced Pluripotent Stem Cells for Prospective Biomedical Applications. Stem Cell Rev. Rep. 2018, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; De Los Angeles, A.; Zhang, J. The Art of Human Induced Pluripotent Stem Cells: The Past, the Present and the Future. Open Stem Cell J. 2010, 2, 2–7. [Google Scholar]
- Long, J.; Kim, H.; Kim, D.; Lee, J.B.; Kim, D.-H. A biomaterial approach to cell reprogramming and differentiation. J. Mater. Chem. B 2017, 5, 2375–2389. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Choi, H.W.; Cho, J.; Jung, S.; Seo, H.G.; Do, J.T. Activation of pluripotency genes by a nanotube-mediated protein delivery system. Mol. Reprod. Dev. 2013, 80, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Narayanan, K.; Lu, H.; Choo, Y.; Du, C.; Wiradharma, N.; Yang, Y.-Y.; Wan, A.C.A. Delivery of reprogramming factors into fibroblasts for generation of non-genetic induced pluripotent stem cells using a cationic bolaamphiphile as a non-viral vector. Biomaterials 2013, 34, 5336–5343. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; ISBN 3-900051-07-0. [Google Scholar]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.R.; Smith, J.D.; Vonesch, S.C.; Lin, G.; Tu, C.S.; Lederer, A.R.; Chu, A.; Suresh, S.; Nguyen, M.; Horecka, J.; et al. Multiplexed precision genome editing with trackable genomic barcodes in yeast. Nat. Biotechnol. 2018, 36, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Strohkendl, I.; Saifuddin, F.A.; Rybarski, J.R.; Finkelstein, I.J.; Russell, R. Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell 2018, 71, 816–824.e3. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, C. Fate Therapeutics Announces FDA Clearance of Landmark IND for FT500 iPSC-derived, Off-the-Shelf NK Cell Cancer Immunotherapy. 2018. Available online: https://ir.fatetherapeutics.com/news-releases/news-release-details/fate-therapeutics-announces-fda-clearance-landmark-ind-ft500 (accessed on 20 February 2019).
- Rossignol, J.; Crane, A.T.; Fink, K.D.; Dunbar, G.L. Will Undifferentiated Induced Pluripotent Stem Cells Ever have Clinical Utility? J. Stem Cell Res. Ther. 2014, 4, 189. [Google Scholar] [CrossRef]
- Nori, S.; Okada, Y.; Nishimura, S.; Sasaki, T.; Itakura, G.; Kobayashi, Y.; Renault-Mihara, F.; Shimizu, A.; Koya, I.; Yoshida, R.; et al. Long-Term Safety Issues of iPSC-Based Cell Therapy in a Spinal Cord Injury Model: Oncogenic Transformation with Epithelial-Mesenchymal Transition. Stem Cell Rep. 2015, 4, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.G.; Winkler, T.; Wu, C.; Guo, V.; Pittaluga, S.; Nicolae, A.; Donahue, R.E.; Metzger, M.E.; Price, S.D.; Uchida, N.; et al. Path to the clinic: Assessment of iPSC-based cell therapies in vivo in a non-human primate model. Cell Rep. 2014, 7, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.; Piedrahita, J.A. From “ES-like” cells to induced pluripotent stem cells: A historical perspective in domestic animals. Theriogenology 2014, 81, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Galat, V.; Galat, Y.; Perepitchka, M.; Jennings, L.J.; Iannaccone, P.M.; Hendrix, M.J.C. Transgene Reactivation in Induced Pluripotent Stem Cell Derivatives and Reversion to Pluripotency of Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Amariglio, N.; Hirshberg, A.; Scheithauer, B.W.; Cohen, Y.; Loewenthal, R.; Trakhtenbrot, L.; Paz, N.; Koren-Michowitz, M.; Waldman, D.; Leider-Trejo, L.; et al. Donor-Derived Brain Tumor Following Neural Stem Cell Transplantation in an Ataxia Telangiectasia Patient. PLOS Med. 2009, 6, e1000029. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, A.L.; Miller, M.B.; Mir, S.A.; Cagney, D.; Chavakula, V.; Guleria, I.; Aizer, A.; Ligon, K.L.; Chi, J.H. Glioproliferative Lesion of the Spinal Cord as a Complication of “Stem-Cell Tourism”. N. Engl. J. Med. 2016, 375, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.; Racke, M.; Kissel, J.; Imitola, J. Responsibilities of Health Care Professionals in Counseling and Educating Patients With Incurable Neurological Diseases Regarding “Stem Cell Tourism”: Caveat Emptor. JAMA Neurol. 2015, 72, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Lukovic, D.; Stojkovic, M.; Moreno-Manzano, V.; Bhattacharya, S.S.; Erceg, S. Perspectives and Future Directions of Human Pluripotent Stem Cell-Based Therapies: Lessons from Geron’s Clinical Trial for Spinal Cord Injury. Stem Cells Dev. 2013, 23, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet Lond. Engl. 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell-Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.E.; Garitaonandia, I.; Loring, J.F. The tumorigenic potential of pluripotent stem cells: What can we do to minimize it? BioEssays 2016, 38, S86–S95. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, M.A.; Pericuesta, E.; Fernández-González, R.; Pintado, B.; Gutiérrez-Adán, A. Inadvertent presence of pluripotent cells in monolayers derived from differentiated embryoid bodies. Int. J. Dev. Biol. 2007, 51, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Guan, Y.; Qu, Z.; Zhang, J.; Liao, B.; Ma, B.; Qian, J.; Li, D.; Li, W.; Xu, G.-T.; et al. WNT signaling determines tumorigenicity and function of ESC-derived retinal progenitors. J. Clin. Investig. 2013, 123, 1647–1661. [Google Scholar] [CrossRef] [PubMed]
- Schuldiner, M.; Itskovitz-Eldor, J.; Benvenisty, N. Selective Ablation of Human Embryonic Stem Cells Expressing a “Suicide” Gene. Stem Cells 2003, 21, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Biran, A.; Scaffidi, P.; Herold-Mende, C.; Boehringer, M.; Meshorer, E.; Benvenisty, N. Elimination of undifferentiated cancer cells by pluripotent stem cell inhibitors. J. Mol. Cell Biol. 2014, 6, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.L.; Altun, G.; Tran, H.T.; Garitaonandia, I.; Slavin, I.; Loring, J.F.; Nazor, K.L.; Laurent, L.C.; Nakagawa, M.; Lacharite, R.M.; et al. Specific lectin biomarkers for isolation of human pluripotent stem cells identified through array-based glycomic analysis. Cell Res. 2011, 21, 1551. [Google Scholar]
- Kuroda, T.; Yasuda, S.; Kusakawa, S.; Hirata, N.; Kanda, Y.; Suzuki, K.; Takahashi, M.; Nishikawa, S.-I.; Kawamata, S.; Sato, Y. Highly Sensitive In Vitro Methods for Detection of Residual Undifferentiated Cells in Retinal Pigment Epithelial Cells Derived from Human iPS Cells. PLoS ONE 2012, 7, e37342. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Cao, F.; Xie, X.; van der Bogt, K.; Hwang, A.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle 2009, 8, 2608–2612. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.E.; Loring, J.F. Genomic Instability in Pluripotent Stem Cells: Implications for Clinical Applications. J. Biol. Chem. 2014, 289, 4578–4584. [Google Scholar] [CrossRef] [PubMed]
- Down, T.A.; Rakyan, V.K.; Turner, D.J.; Flicek, P.; Li, H.; Kulesha, E.; Gräf, S.; Johnson, N.; Herrero, J.; Tomazou, E.M.; et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat. Biotechnol. 2008, 26, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Meneghini, V.; Frati, G.; Sala, D.; De Cicco, S.; Luciani, M.; Cavazzin, C.; Paulis, M.; Mentzen, W.; Morena, F.; Giannelli, S.; et al. Generation of Human Induced Pluripotent Stem Cell-Derived Bona Fide Neural Stem Cells for Ex Vivo Gene Therapy of Metachromatic Leukodystrophy. Stem Cells Transl. Med. 2017, 6, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Araki, R.; Uda, M.; Hoki, Y.; Sunayama, M.; Nakamura, M.; Ando, S.; Sugiura, M.; Ideno, H.; Shimada, A.; Nifuji, A.; et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 2013, 494, 100–104. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, P.E.; Meyer, E.H.; Kooreman, N.G.; Diecke, S.; Dey, D.; Sanchez-Freire, V.; Hu, S.; Ebert, A.; Odegaard, J.; Mordwinkin, N.M.; et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat. Commun. 2014, 5, 3903. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.; Alvarez-Palomo, A.B.; Codina-Pascual, M.; Delgado-Morales, R.; Moran, S.; Esteller, M.; Sal, M.; Juan-Otero, M.; Barado, A.B.; Consiglio, A.; et al. Global proteomic and methylome analysis in human induced pluripotent stem cells reveals overexpression of a human TLR3 affecting proper innate immune response signaling. Stem Cells 2019, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Knoepfler, P. Adverse Event in IPS Cell (ips細胞) Trial for Vision Loss in Japan: Initial Perspectives. The Niche, 16 January 2018. [Google Scholar]
- National Institutes of Health (NIH). A Study of CYP-001 for the Treatment of Steroid-Resistant Acute Graft Versus Host Disease. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02923375 (accessed on 29 December 2018).
- Da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, K. First iPS Cell Trial for Heart Disease Raises Excitement, Concern. The Scientist, 31 August 2018. [Google Scholar]
- Ben-David, U.; Mayshar, Y.; Benvenisty, N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell 2011, 9, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Rehen, S.K.; Yung, Y.C.; McCreight, M.P.; Kaushal, D.; Yang, A.H.; Almeida, B.S.V.; Kingsbury, M.A.; Cabral, K.M.S.; McConnell, M.J.; Anliker, B.; et al. Constitutional aneuploidy in the normal human brain. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.; Hall, I.M.; et al. Mosaic Copy Number Variation in Human Neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, D.; Contos, J.J.A.; Treuner, K.; Yang, A.H.; Kingsbury, M.A.; Rehen, S.K.; McConnell, M.J.; Okabe, M.; Barlow, C.; Chun, J. Alteration of gene expression by chromosome loss in the postnatal mouse brain. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 5599–5606. [Google Scholar] [CrossRef]
- Young, M.A.; Larson, D.E.; Sun, C.-W.; George, D.R.; Ding, L.; Miller, C.A.; Lin, L.; Pawlik, K.M.; Chen, K.; Fan, X.; et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell 2012, 10, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.A.; Muotri, A.R. LINE-1: Creators of neuronal diversity. Front. Biosci. Elite Ed. 2012, 4, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Wissing, S.; Muñoz-Lopez, M.; Macia, A.; Yang, Z.; Montano, M.; Collins, W.; Garcia-Perez, J.L.; Moran, J.V.; Greene, W.C. Reprogramming somatic cells into iPS cells activates LINE-1 retroelement mobility. Hum. Mol. Genet. 2012, 21, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Varela, C.; Denis, J.A.; Polentes, J.; Feyeux, M.; Aubert, S.; Champon, B.; Piétu, G.; Peschanski, M.; Lefort, N. Recurrent genomic instability of chromosome 1q in neural derivatives of human embryonic stem cells. J. Clin. Investig. 2012, 122, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, M.; Araki, R.; Kasama, Y.; Sunayama, M.; Abe, M.; Nishida, K.; Kawaji, H.; Hayashizaki, Y.; Murakawa, Y. Hotspots of De novo Point Mutations in Induced Pluripotent Stem Cells. Cell Rep. 2017, 21, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018, 47, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Růžička, M.; Kulhánek, P.; Radová, L.; Čechová, A.; Špačková, N.; Fajkusová, L.; Réblová, K. DNA mutation motifs in the genes associated with inherited diseases. PLOS ONE 2017, 12, e0182377. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Georgieva, D.; Vanoli, F.; Shi, Z.-D.; Stadtfeld, M.; Ludwig, T.; Jasin, M.; Huangfu, D. Homologous Recombination DNA Repair Genes Play a Critical Role in Reprogramming to a Pluripotent State. Cell Rep. 2013, 3, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kim, D.-K.; Ko, J.-J.; Kim, K.P.; Park, K.-S. Rad51 Regulates Reprogramming Efficiency through DNA Repair Pathway. Dev. Reprod. 2016, 20, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Salci, K.R.; Reid, J.C.; Orlando, L.; Tanasijevic, B.; Shapovalova, Z.; Bhatia, M. Brief Report: Human Acute Myeloid Leukemia Reprogramming to Pluripotency Is a Rare Event and Selects for Patient Hematopoietic Cells Devoid of Leukemic Mutations. Stem Cells 2017, 35, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of iPSCs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Sakaguchi, H.; Sakamoto-Abutani, R.; Nakanishi, M.; Nishimura, K.; Yamazaki-Inoue, M.; Ohtaka, M.; Periasamy, V.S.; Alshatwi, A.A.; Higuchi, A.; et al. Distinctive features of single nucleotide alterations in induced pluripotent stem cells with different types of DNA repair deficiency disorders. Sci. Rep. 2016, 6, 26342. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.J.; Nik-Zainal, S.; Wuster, A.; Li, Y.; Conte, N.; Koike-Yusa, H.; Kumasaka, N.; Vallier, L.; Yusa, K.; Bradley, A. Mutational History of a Human Cell Lineage from Somatic to Induced Pluripotent Stem Cells. PLoS Genet. 2016, 12, e1005932. [Google Scholar] [CrossRef] [PubMed]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human embryonic stem cells fail to activate CHK1 and commit to apoptosis in response to DNA replication stress. Stem Cells Dayt. Ohio 2012, 30, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.M.; Connelly, J.P.; Hansen, N.F.; Donovan, F.X.; Winkler, T.; Davis, B.W.; Alkadi, H.; Chandrasekharappa, S.C.; Dunbar, C.E.; Mullikin, J.C.; et al. iPSCs and fibroblast subclones from the same fibroblast population contain comparable levels of sequence variations. Proc. Natl. Acad. Sci. USA 2017, 114, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Kasama, Y.; Araki, R.; Hoki, Y.; Sunayama, M.; Uda, M.; Nakamura, M.; Ando, S.; Abe, M. Induced pluripotent stem cell generation-associated point mutations arise during the initial stages of the conversion of these cells. Stem Cell Rep. 2014, 2, 52–63. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, M.; Benaglio, P.; Jakubosky, D.; Greenwald, W.W.; Matsui, H.; Donovan, M.K.R.; Li, H.; Smith, E.N.; D’Antonio-Chronowska, A.; Frazer, K.A. Insights into the Mutational Burden of Human Induced Pluripotent Stem Cells from an Integrative Multi-Omics Approach. Cell Rep. 2018, 24, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Orlando, L.; Giachino, C. Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process. Int. J. Mol. Sci. 2017, 18, 1952. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Bester, A.C.; Kerem, B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012, 28, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Sipp, D.; Robey, P.G.; Turner, L. Clear up this stem-cell mess. Nature 2018, 561, 455. [Google Scholar] [CrossRef] [PubMed]
- Steens, J.; Klein, D. Current Strategies to Generate Human Mesenchymal Stem Cells In Vitro. Stem Cells Int. 2018, 2018, 6726185. [Google Scholar] [CrossRef] [PubMed]
- Bloor, A.; Patel, A.; Griffin, J.E.; Gilleece, M.H.; Radia, R.; Yeung, D.T.; Slukvin, I.; Kelly, K.; Rasko, J.E.J. A Phase I Trial of iPSC-Derived MSCs (CYP-001) in Steroid-Resistant Acute GvHD. Blood 2018, 132, 4562. [Google Scholar]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Lalu, M.M.; McIntyre, L.; Pugliese, C.; Fergusson, D.; Winston, B.W.; Marshall, J.C.; Granton, J.; Stewart, D.J.; Canadian Critical Care Trials Group. Safety of cell therapy with mesenchymal stromal cells (SafeCell): A systematic review and meta-analysis of clinical trials. PLoS ONE 2012, 7, e47559. [Google Scholar] [CrossRef] [PubMed]
- Peeters, C.M.M.; Leijs, M.J.C.; Reijman, M.; van Osch, G.J.V.M.; Bos, P.K. Safety of intra-articular cell-therapy with culture-expanded stem cells in humans: A systematic literature review. Osteoarthritis Cartilage 2013, 21, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Zong, C.; Zhang, H.; Yang, X.; Gao, L.; Hou, J.; Ye, F.; Jiang, J.; Yang, Y.; Li, R.; Han, Z.; et al. The distinct roles of mesenchymal stem cells in the initial and progressive stage of hepatocarcinoma. Cell Death Dis. 2018, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.R.; Abadeh, A.; Connelly, K.A. Concise Review: Rational Use of Mesenchymal Stem Cells in the Treatment of Ischemic Heart Disease. Stem Cells Transl. Med. 2018, 7, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, A.; Swiech, K. Mesenchymal Stromal Cells: From Discovery to Manufacturing and Commercialization. Stem Cells Int. 2018, 2018, 4083921. [Google Scholar] [CrossRef] [PubMed]
- Heathman, T.R.J.; Rafiq, Q.A.; Chan, A.K.C.; Coopman, K.; Nienow, A.W.; Kara, B.; Hewitt, C.J. Characterization of human mesenchymal stem cells from multiple donors and the implications for large scale bioprocess development. Biochem. Eng. J. 2016, 108, 14–23. [Google Scholar] [CrossRef]
- Tarte, K.; Gaillard, J.; Lataillade, J.-J.; Fouillard, L.; Becker, M.; Mossafa, H.; Tchirkov, A.; Rouard, H.; Henry, C.; Splingard, M.; et al. Clinical-grade production of human mesenchymal stromal cells: Occurrence of aneuploidy without transformation. Blood 2010, 115, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.K.; Ogando, C.R.; Wang See, C.; Chang, T.-Y.; Barabino, G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Blanc, K.L.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Hansen, N.F.; Zhao, L.; Du, Y.; Zou, C.; Donovan, F.X.; Chou, B.-K.; Zhou, G.; Li, S.; Dowey, S.N.; et al. Low Incidence of DNA Sequence Variation in Human Induced Pluripotent Stem Cells Generated by Nonintegrating Plasmid Expression. Cell Stem Cell 2012, 10, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Lo Sardo, V.; Ferguson, W.; Erikson, G.A.; Topol, E.J.; Baldwin, K.K.; Torkamani, A. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 2017, 35, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, H.; Go, M.J.; Shikamura, M.; Nishishita, N.; Sakai, N.; Kamao, H.; Mandai, M.; Morinaga, C.; Takahashi, M.; Kawamata, S. Tumorigenicity Studies of Induced Pluripotent Stem Cell (iPSC)-Derived Retinal Pigment Epithelium (RPE) for the Treatment of Age-Related Macular Degeneration. PLOS ONE 2014, 9, e85336. [Google Scholar] [CrossRef] [PubMed]
- DeBoever, C.; Li, H.; Jakubosky, D.; Benaglio, P.; Reyna, J.; Olson, K.M.; Huang, H.; Biggs, W.; Sandoval, E.; D’Antonio, M.; et al. Large-Scale Profiling Reveals the Influence of Genetic Variation on Gene Expression in Human Induced Pluripotent Stem Cells. Cell Stem Cell 2017, 20, 533–546.e7. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Ng, S.H.; Sharma, V.; Neculai, D.; Hussein, S.; Sam, M.; Trinh, Q.; Church, G.M.; McPherson, J.D.; Nagy, A.; et al. Elevated coding mutation rate during the reprogramming of human somatic cells into induced pluripotent stem cells. Stem Cells Dayt. Ohio 2012, 30, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Chen, K.; Gao, Y.-X.; Arzigian, M.; Xie, Y.-L.; Malcosky, C.; Yang, Y.-G.; Wu, W.-S.; Wang, Z.Z. Bcl-xL enhances single-cell survival and expansion of human embryonic stem cells without affecting self-renewal. Stem Cell Res. 2012, 8, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Spits, C.; Mateizel, I.; Geens, M.; Mertzanidou, A.; Staessen, C.; Vandeskelde, Y.; Van der Elst, J.; Liebaers, I.; Sermon, K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 2008, 26, 1361–1363. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Colman, A.; Robins, A.; Hampl, A.; Bosman, A.; Fraga, A.M.; Nagy, A.; Choo, A.B.H.; Laslett, A.L.; Ford, A.; Feki, A.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132. [Google Scholar]
- Skotheim, R.I.; Monni, O.; Mousses, S.; Fosså, S.D.; Kallioniemi, O.-P.; Lothe, R.A.; Kallioniemi, A. New insights into testicular germ cell tumorigenesis from gene expression profiling. Cancer Res. 2002, 62, 2359–2364. [Google Scholar] [PubMed]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Yang, J.-Y.; Xia, W.; Chen, C.-T.; Xie, X.; Chao, C.-H.; Woodward, W.A.; Hsu, J.-M.; Hortobagyi, G.N.; Hung, M.-C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS Transcription Factors Control Transcription of EZH2 and Epigenetic Silencing of the Tumor Suppressor Gene Nkx3.1 in Prostate Cancer. PLoS ONE 2010, 5, e10547. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Shilatifard, A. Chromatin signatures of cancer. Genes Dev. 2015, 29, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hung, M.-C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2014, 46, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, N.R.; Musio, A.; Vezzoni, P.; Simpson, A.H.R.W.; Noble, B.S.; McWhir, J. Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 2006, 8, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.G.; Ryan, S.D.; Ramnarine, K.; Rosen, S.A.; Mann, S.E.; Kulick, A.; De Stanchina, E.; Müller, F.-J.; Kacmarczyk, T.J.; Zhang, C.; et al. CryoPause: A New Method to Immediately Initiate Experiments after Cryopreservation of Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Morey, R.; O’Neil, R.C.; He, Y.; Daughtry, B.; Schultz, M.D.; Hariharan, M.; Nery, J.R.; Castanon, R.; Sabatini, K.; et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 2014, 511, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.P.; Morey, R.; Kang, E.; Ma, H.; Hayama, T.; Laurent, L.C.; Mitalipov, S. Concise Review: Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer: A Horse in the Race? Stem Cells 2017, 35, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.G.; Eum, J.H.; Lee, J.E.; Shim, S.H.; Sepilian, V.; Hong, S.W.; Lee, Y.; Treff, N.R.; Choi, Y.H.; Kimbrel, E.A.; et al. Human Somatic Cell Nuclear Transfer Using Adult Cells. Cell Stem Cell 2014, 14, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Johannesson, B.; Sagi, I.; Burnett, L.C.; Kort, D.H.; Prosser, R.W.; Paull, D.; Nestor, M.W.; Freeby, M.; Greenberg, E.; et al. Human oocytes reprogram adult somatic nuclei of a type 1 diabetic to diploid pluripotent stem cells. Nature 2014, 510, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Jóhannesson, B.; Sagi, I.; Gore, A.; Paull, D.J.; Yamada, M.; Golan-lev, T.; Li, Z.; LeDuc, C.A.; Shen, Y.; Stern, S.; et al. Comparable frequencies of coding mutations and loss of imprinting in human pluripotent cells derived by nuclear transfer and defined factors. Cell Stem Cell 2014, 15, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-T.; Chen, H.; Liu, Q.; Shao, N.-Y.; Sayed, N.; Wo, H.-T.; Zhang, J.Z.; Ong, S.-G.; Liu, C.; Kim, Y.; et al. Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc. Natl. Acad. Sci. USA 2017, 114, E11111–E11120. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Gutierrez, N.M.; Tippner-Hedges, R.; Ma, H.; Kang, E.; Fulati, A.; Lee, H.-S.; Sritanaudomchai, H.; et al. Human Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer. Cell 2013, 153, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Zost, S.J.; Parkhouse, K.; Gumina, M.E.; Kim, K.; Perez, S.D.; Wilson, P.C.; Treanor, J.J.; Sant, A.J.; Cobey, S.; Hensley, S.E. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 2017, 114, 12578–12583. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. A Fresh Look at iPS Cells. Cell 2009, 137, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.; Gulbranson, D.R.; George, N.; Siniscalchi, L.I.; Jones, J.; Thomson, J.A.; Chen, G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012, 7, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Geens, M.; Spits, C. Genetic and epigenetic instability in human pluripotent stem cells. Hum. Reprod. Update 2013, 19, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Catalina, P.; Montes, R.; Ligero, G.; Sanchez, L.; de la Cueva, T.; Bueno, C.; Leone, P.E.; Menendez, P. Human ESCs predisposition to karyotypic instability: Is a matter of culture adaptation or differential vulnerability among hESC lines due to inherent properties? Mol. Cancer 2008, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D.; Mineev, K. Mutation rate in stem cells: An underestimated barrier on the way to therapy. Trends Mol. Med. 2013, 19, 273–280. [Google Scholar] [CrossRef] [PubMed]
- MySQL. MySQL 5.7 Reference Manual. 2019. Available online: https://dev.mysql.com/doc/refman/5.7/en/ (accessed on 8 January 2019).
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Nachman, I.; Regev, A.; Meissner, A. Dynamic single-cell imaging of direct reprogramming reveals an early specifying event. Nat. Biotechnol. 2010, 28, 521. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. On the Mathematical Foundations of Theoretical Statistics. Philos. Trans. R. Soc. A 1922, 222, 309–368. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [PubMed]
- Kingman, J.F. On the genealogy of large populations. J. Appl. Probab. 1982, 19A, 27–43. [Google Scholar] [CrossRef]
- Campbell, R.B. Coalescent Size versus Coalescent Time with Strong Selection. Bull. Math. Biol. 2007, 69, 2249–2259. [Google Scholar] [CrossRef] [PubMed]
- Haldane, J.B.S. A Mathematical Theory of Natural and Artificial Selection, Part V: Selection and Mutation. Math. Proc. Camb. Philos. Soc. 1927, 23, 838–844. [Google Scholar] [CrossRef]
- Miller, C.A.; McMichael, J.; Dang, H.X.; Maher, C.A.; Ding, L.; Ley, T.J.; Mardis, E.R.; Wilson, R.K. Visualizing tumor evolution with the fishplot package for R. BMC Genomics 2016, 17, 880. [Google Scholar] [CrossRef] [PubMed]
- Churko, J.M.; Lee, J.; Ameen, M.; Gu, M.; Venkatasubramanian, M.; Diecke, S.; Sallam, K.; Im, H.; Wang, G.; Gold, J.D.; et al. Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat. Biomed. Eng. 2017, 1, 826. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Närvä, E.; Ng, S.; Sourour, M.; Hämäläinen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, H.; Loring, J.; Hormuzdi, S.; Disteche, C.M.; Bornstein, P.; Jaenisch, R. Trisomy eight in ES cells is a common potential problem in gene targeting and interferes with germ line transmission. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1997, 209, 85–91. [Google Scholar] [CrossRef]
- Gao, S.; Zheng, C.; Chang, G.; Liu, W.; Kou, X.; Tan, K.; Tao, L.; Xu, K.; Wang, H.; Cai, J.; et al. Unique features of mutations revealed by sequentially reprogrammed induced pluripotent stem cells. Nat. Commun. 2015, 6, 6318. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Park, I.-H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7, e1002085. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Feinberg, A.P.; Takeuchi, A.; Yabuuchi, A.; Wen, B.; Daley, G.Q.; Hongguang, H.; Ji, H.; Weissman, I.L.; Hanna, J.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar]
- Mekhoubad, S.; Bock, C.; de Boer, A.S.; Kiskinis, E.; Meissner, A.; Eggan, K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012, 10, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Pick, M.; Stelzer, Y.; Bar-Nur, O.; Mayshar, Y.; Eden, A.; Benvenisty, N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells Dayt. Ohio 2009, 27, 2686–2690. [Google Scholar] [CrossRef] [PubMed]
- Altun, G.; Loring, J.F.; Laurent, L.C. DNA methylation in embryonic stem cells. J. Cell. Biochem. 2010, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Weissbein, U.; Benvenisty, N.; Ben-David, U. Genome maintenance in pluripotent stem cells. J. Cell Biol. 2014, 204, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. High prevalence of evolutionarily conserved and species-specific genomic aberrations in mouse pluripotent stem cells. Stem Cells Dayt. Ohio 2012, 30, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Induced Pluripotent Stem Cells: Past, Present, and Future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Cavagnaro, J.; Cavanagro, J.; Deans, R.; Feigal, E.; Feigel, E.; Horowitz, E.; Keating, A.; Rao, M.; Turner, M.; et al. Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat. Biotechnol. 2014, 32, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tang, Y.; Lü, S.; Zhou, J.; Du, Z.; Duan, C.; Li, Z.; Wang, C. The tumourigenicity of iPS cells and their differentiated derivates. J. Cell. Mol. Med. 2013, 17, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Boyette, L.B.; Tuan, R.S. Adult Stem Cells and Diseases of Aging. J. Clin. Med. 2014, 3, 88–134. [Google Scholar] [CrossRef] [PubMed]
- Ruhnke, M.; Ungefroren, H.; Nussler, A.; Martin, F.; Brulport, M.; Schormann, W.; Hengstler, J.G.; Klapper, W.; Ulrichs, K.; Hutchinson, J.A.; et al. Differentiation of in vitro-modified human peripheral blood monocytes into hepatocyte-like and pancreatic islet-like cells. Gastroenterology 2005, 128, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Nam, D.; Choi, J.-K.; Araúzo-Bravo, M.J.; Kwon, S.-Y.; Zaehres, H.; Lee, T.; Park, C.Y.; Kang, H.-W.; Schöler, H.R.; et al. Establishment of feeder-free culture system for human induced pluripotent stem cell on DAS nanocrystalline graphene. Sci. Rep. 2016, 6, 20708. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Gore, A.; Li, Z.; Panopoulos, A.D.; Montserrat, N.; Fung, H.-L.; Giorgetti, A.; Bilic, J.; Batchelder, E.M.; Zaehres, H.; et al. Analysis of protein coding mutations in hiPSCs and their possible role during somatic cell reprogramming. Nat. Commun. 2013, 4, 1382. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lv, W.; Ye, X.; Wang, L.; Zhang, M.; Yang, H.; Okuka, M.; Zhou, C.; Zhang, X.; Liu, L.; et al. Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of iPS cells as demonstrated by tetraploid complementation. Cell Res. 2013, 23, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Prakash Bangalore, M.; Adhikarla, S.; Mukherjee, O.; Panicker, M.M. Genotoxic Effects of Culture Media on Human Pluripotent Stem Cells. Sci. Rep. 2017, 7, 42222. [Google Scholar] [CrossRef] [PubMed]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
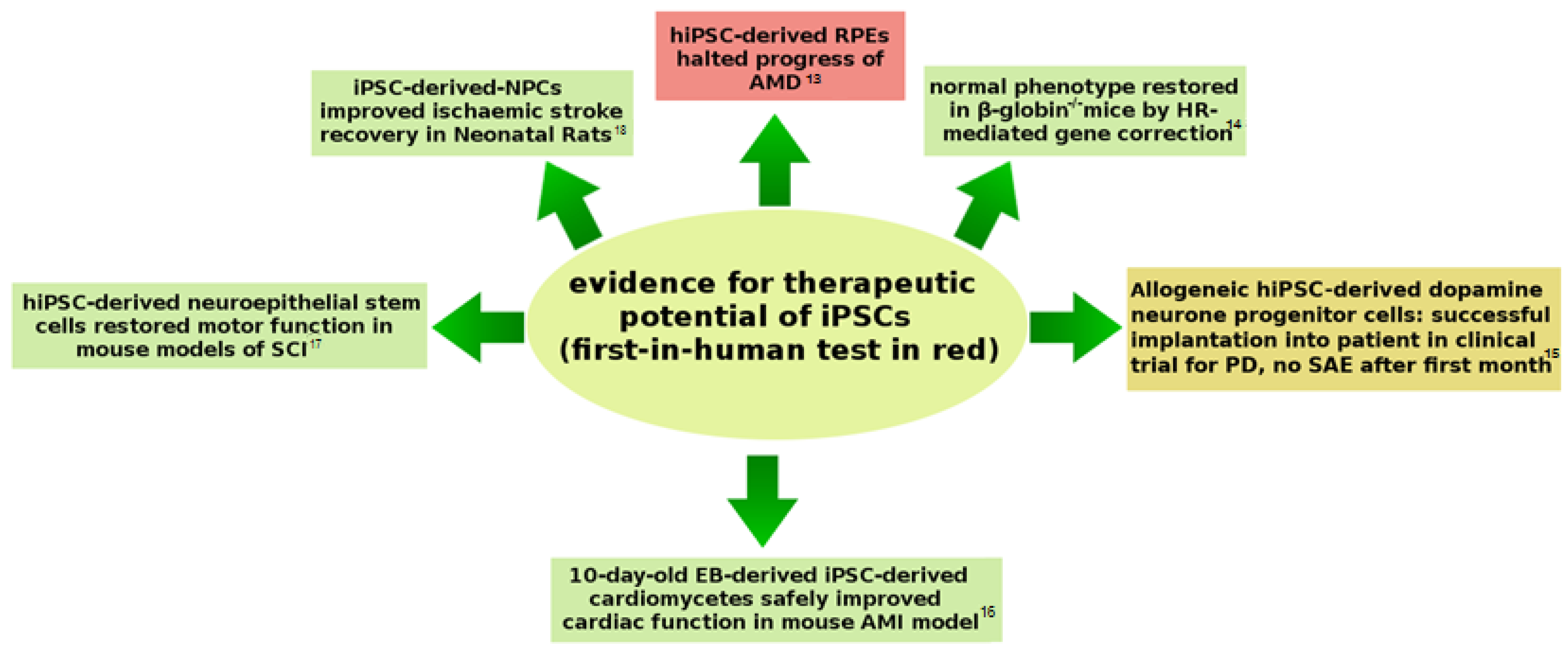{kind=link}
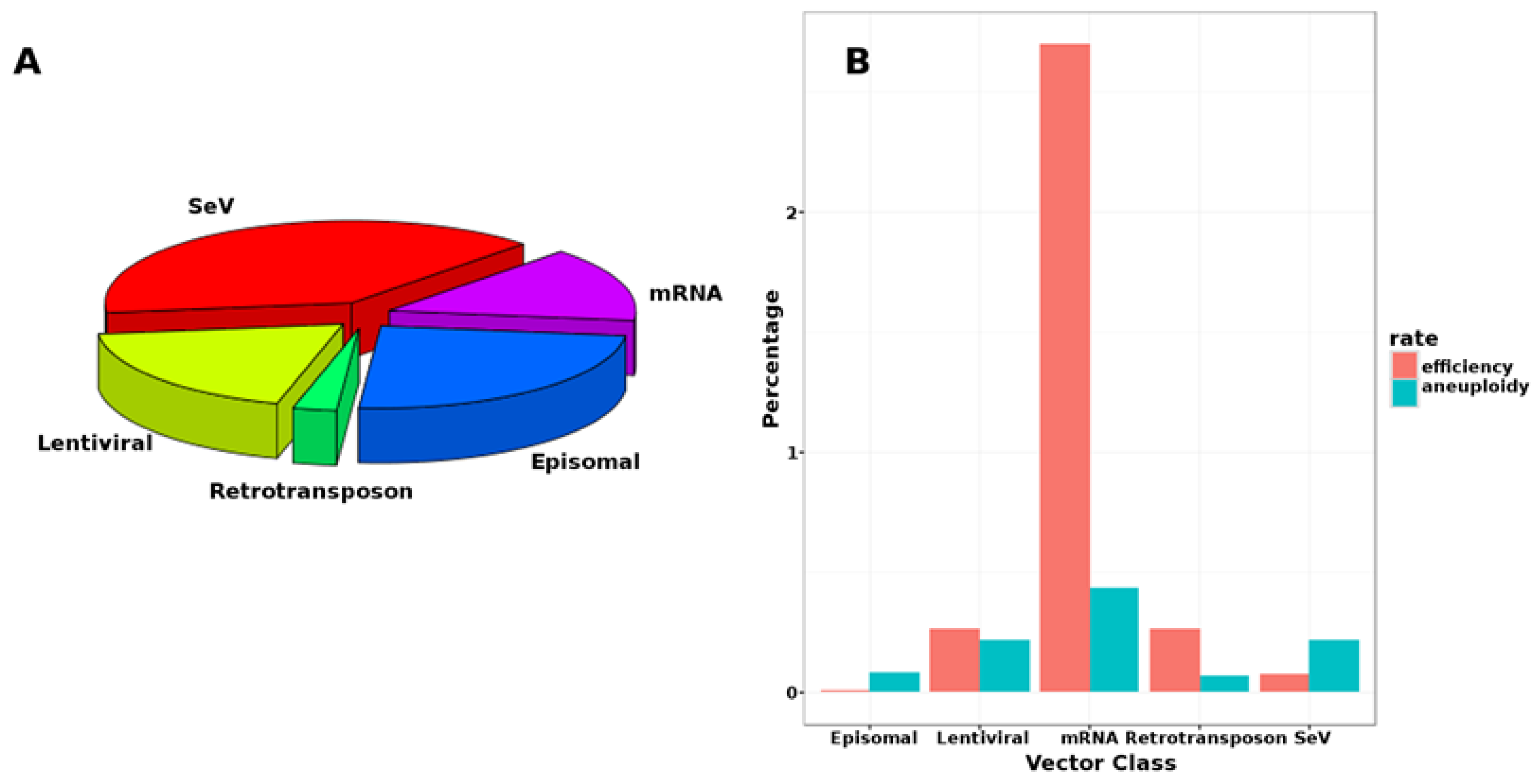{kind=link}
{kind=link}
| Procedure | Caveats | Source |
|---|---|---|
| mir-200c, 302s and 369s (direct) | efficiency 0.01% cf. 0.02% adenovirus and 0.27% retroviral | [26] |
| mRNAs (direct transfection) | 1.4–4.4% efficiency, but high in vitro cytotoxicity, fails with hematopoietic cells | [27] |
| non-integrating adenoviruses | transfected hepatocytes (show high permissivity to adenovirus) | [28] |
| OriP/EBNA episomal plasmids | 0.006–0.1% (with EBNA mRNA coexpression and hypoxia) cGMP | [29] |
| Sendai-viral (SeV) | efficiency 0.077%, but complex protocols | [30] |
| Small molecules (e.g., epigenetic regulators) | usually require one transgene (e.g., VPA, CHIR99021 and 616452 + Oct4), non-persistent | [31] |
| Date | Agent (N) | Condition | Derivate | Comments |
|---|---|---|---|---|
| 2011 AU | hESC (4) | ASCI | OPCs | Geron: effect remyelination; no SAE; early termination on financial grounds or futility; not reproducible; contains xeno-derived components (e.g., Matrigel) of potential immunogenicity [70] |
| 2013 AU | Hesc (?) | ASCI | OPCs | NCT01217008 (Asterias Biotherapeutics): continuation of Geron’s phase 1 trial; completed but unpublished |
| 2017 AU | hiPSC (FIH) | AMD | RPE | RIKEN: RPE engraftment to effect photoreceptor rescue; no SAE at 27 months; degeneration only halted; costly $930,000 [11] |
| 2015 AL | hESC (9) | AMD | RPE | NCT01344993: RPE engraftment to effect photoreceptor rescue; no SAE at 12–37 months; visual acuity gain in 6 eyes at 6 months [71] |
| 2015 AL | hESC (9) | SMD | RPE | NCT01345006: RPE engraftment to effect photoreceptor rescue; no SAE at 12–37 months; visual acuity gain in 3 eyes at 6 months [71] |
| 2018 AL | hESC(6) | IHD | CVP | NCT02057900: Epicardial delivery of hESC-derivates to improve systolic motion in severe ischemic left ventricular dysfunction; no SAE at 18 months [72] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attwood, S.W.; Edel, M.J. iPS-Cell Technology and the Problem of Genetic Instability—Can It Ever Be Safe for Clinical Use? J. Clin. Med. 2019, 8, 288. https://doi.org/10.3390/jcm8030288
Attwood SW, Edel MJ. iPS-Cell Technology and the Problem of Genetic Instability—Can It Ever Be Safe for Clinical Use? Journal of Clinical Medicine. 2019; 8(3):288. https://doi.org/10.3390/jcm8030288
Chicago/Turabian StyleAttwood, Stephen W., and Michael J. Edel. 2019. "iPS-Cell Technology and the Problem of Genetic Instability—Can It Ever Be Safe for Clinical Use?" Journal of Clinical Medicine 8, no. 3: 288. https://doi.org/10.3390/jcm8030288
APA StyleAttwood, S. W., & Edel, M. J. (2019). iPS-Cell Technology and the Problem of Genetic Instability—Can It Ever Be Safe for Clinical Use? Journal of Clinical Medicine, 8(3), 288. https://doi.org/10.3390/jcm8030288