Molecular Insights into miRNA-Driven Resistance to 5-Fluorouracil and Oxaliplatin Chemotherapy: miR-23b Modulates the Epithelial–Mesenchymal Transition of Colorectal Cancer Cells

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Establishment of Resistant Cell Lines
2.2. Cell Viability Evaluation in the 2D Cell Culture
2.3. RNA Isolation
2.4. cDNA Library Preparation for Illumina Sequencing
2.5. Quantitative Reverse Transcription PCR
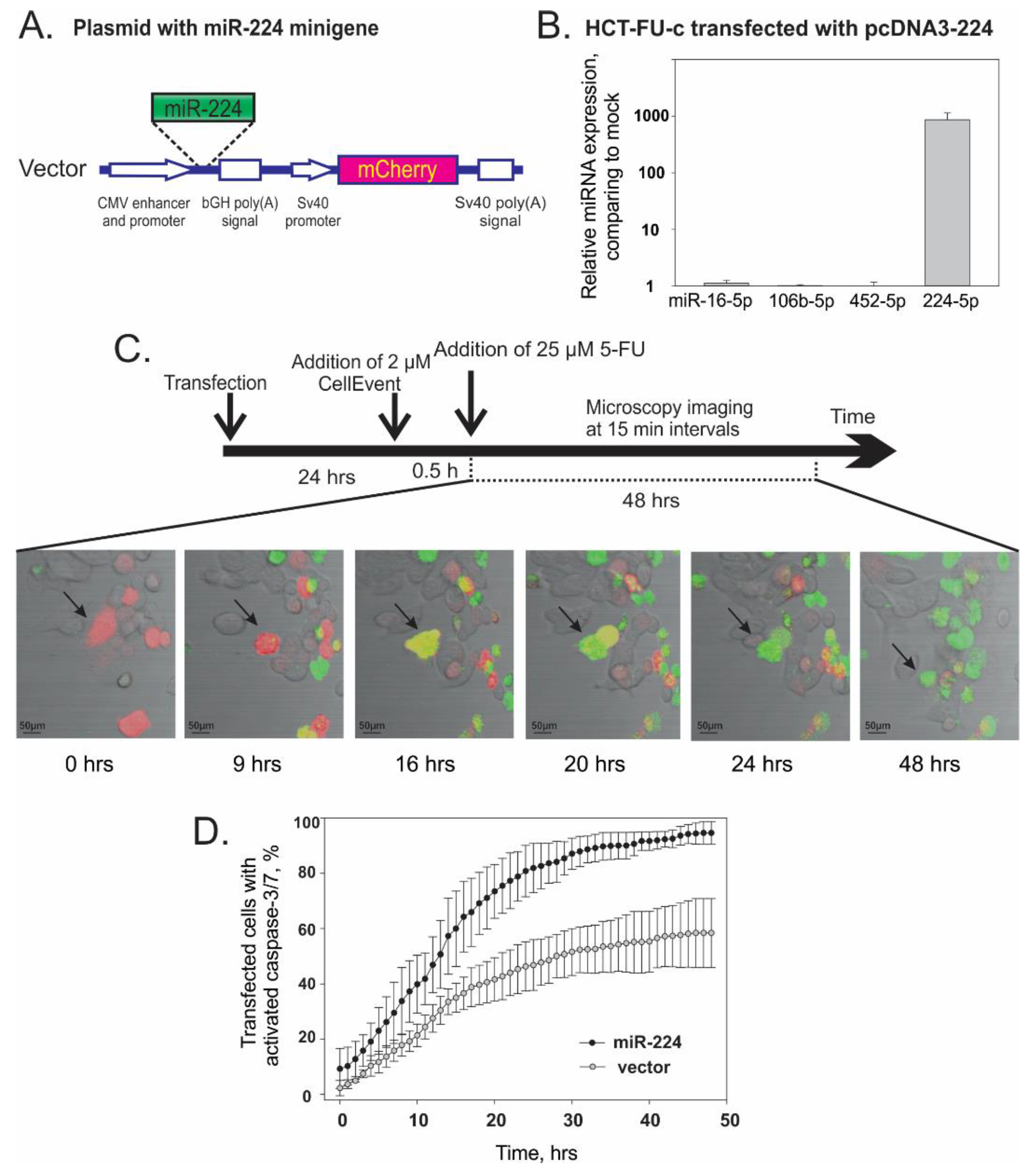
2.6. Analysis of miR-224 Overexpression Effect on 5-FU Resistance
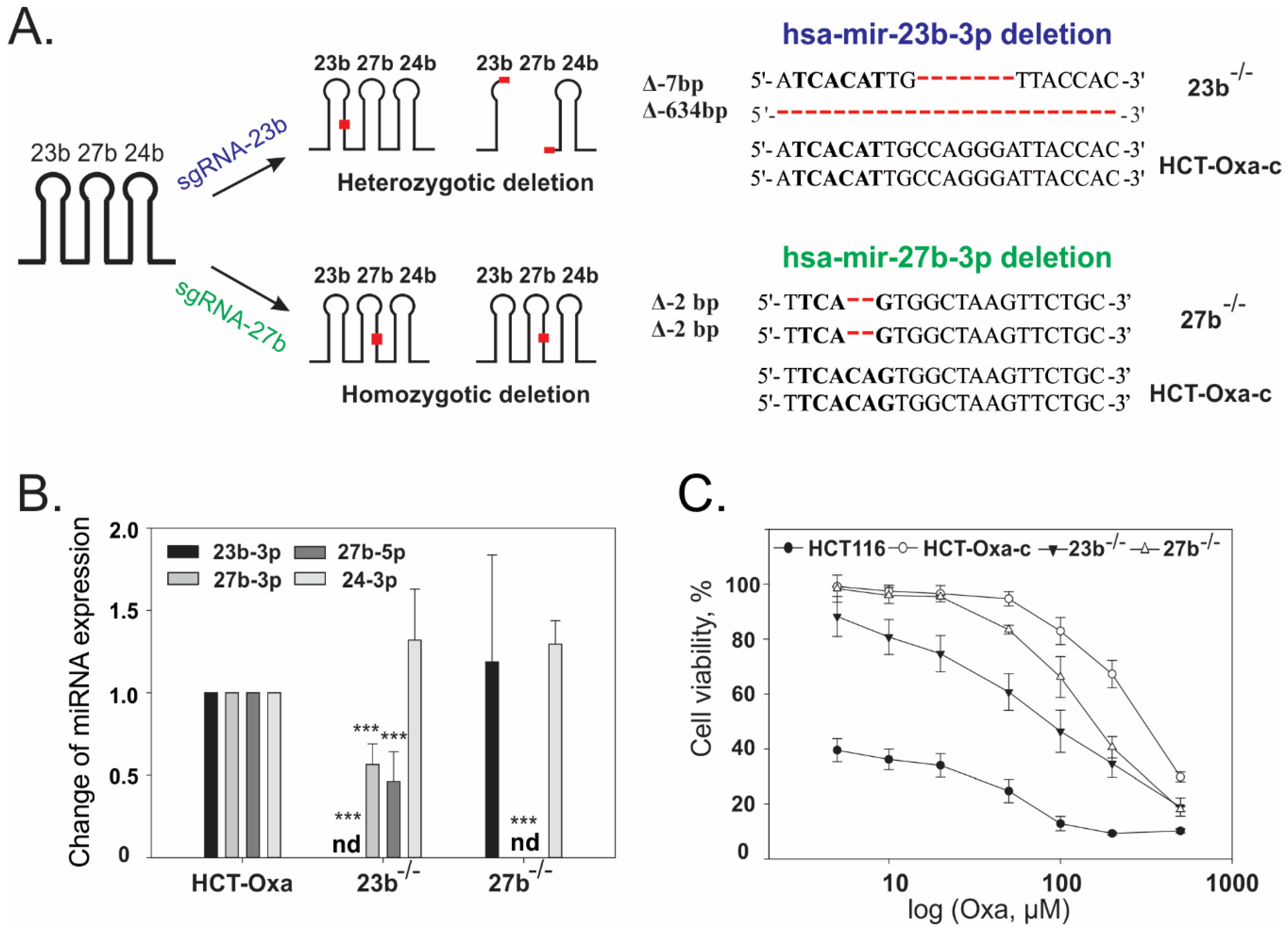
2.7. Generation of the Cell Lines with the Down-Regulated Expression of miR-23b or miR-27b Using CRISPR/Cas9
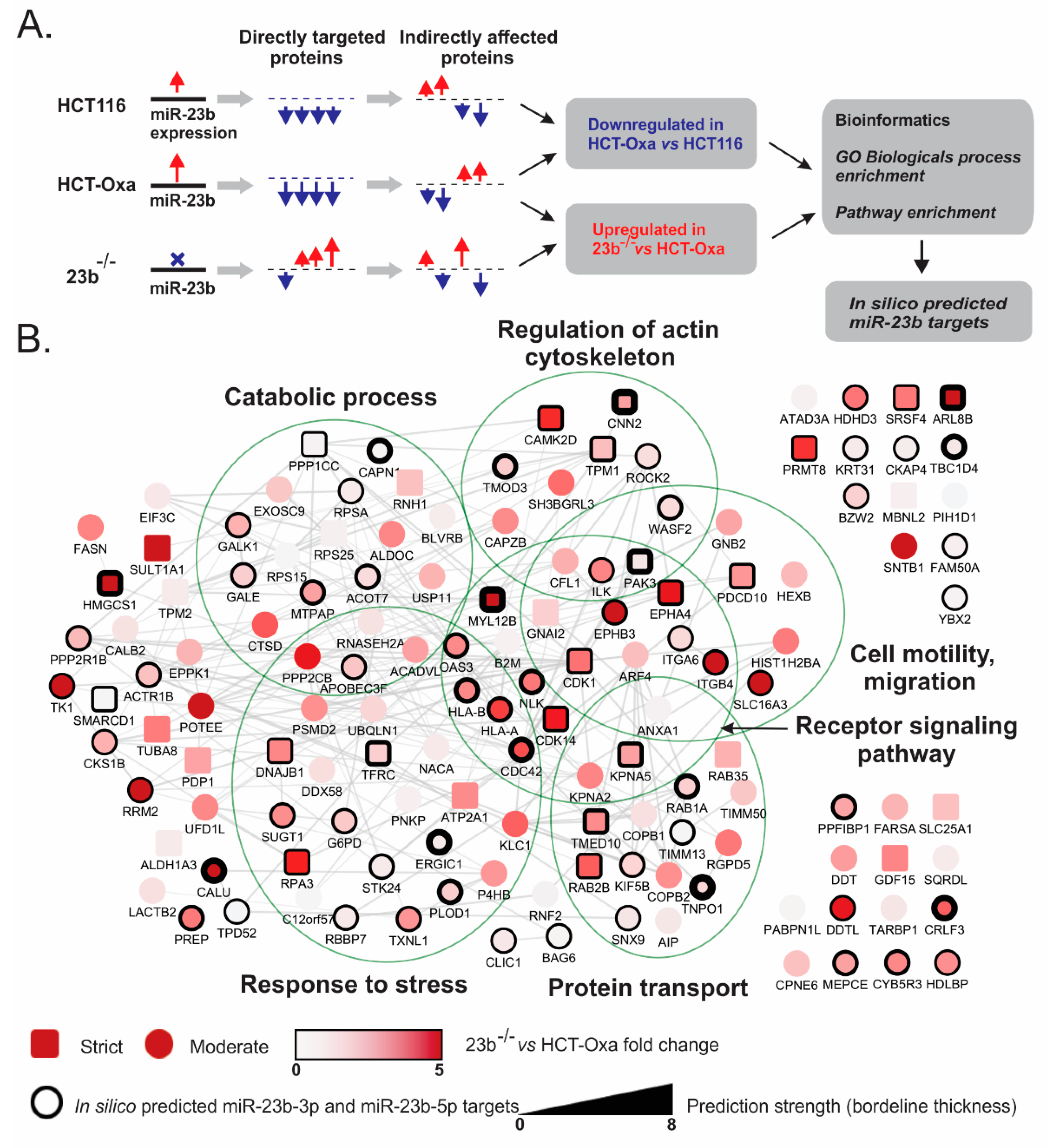
2.8. Proteomic Analysis
2.9. Confocal Microscopy
2.10. Statistical and Bioinformatics Analysis
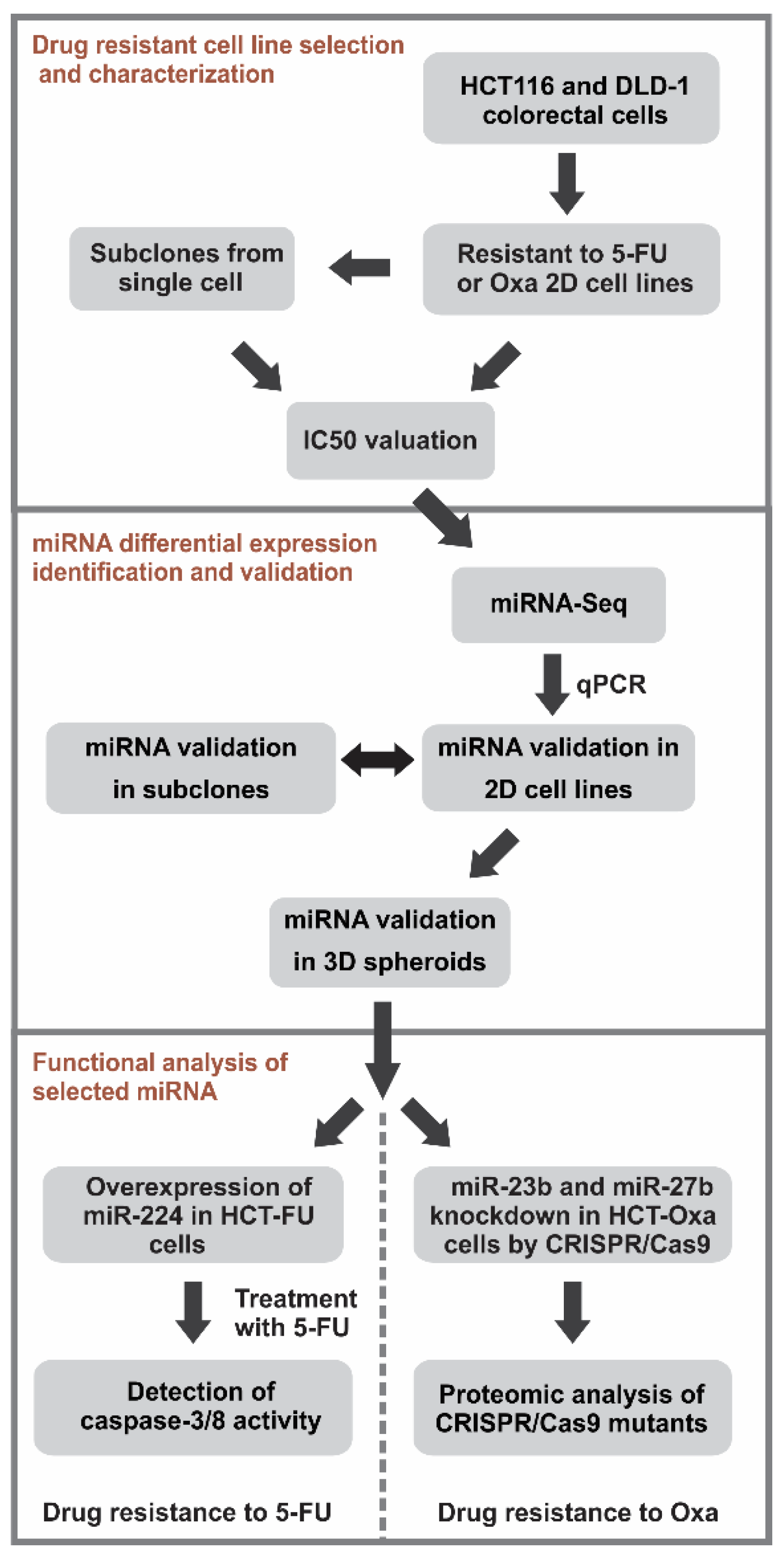
3. Results
3.1. In vitro Generation and Characterization of 5-Fluorouracil- or Oxaliplatin-Resistant Cell Sublines
3.2. High-Throughput Sequencing Analysis of miRNA Expression Profiles of the Drug-Resistant Sublines
3.3. Comparison of the Selected miRNA Expression in the 2D and 3D Cell Cultures of Drug-Resistant Sublines
3.4. Overexpression of miR-224-5p Promotes the 5-FU-Induced Apoptosis of CRC Cells
3.5. CRISPR/Cas9-Mediated Generation and Characterization of miR-23b or miR-27b Knockout Sublines in HCT-Oxa-c Cells
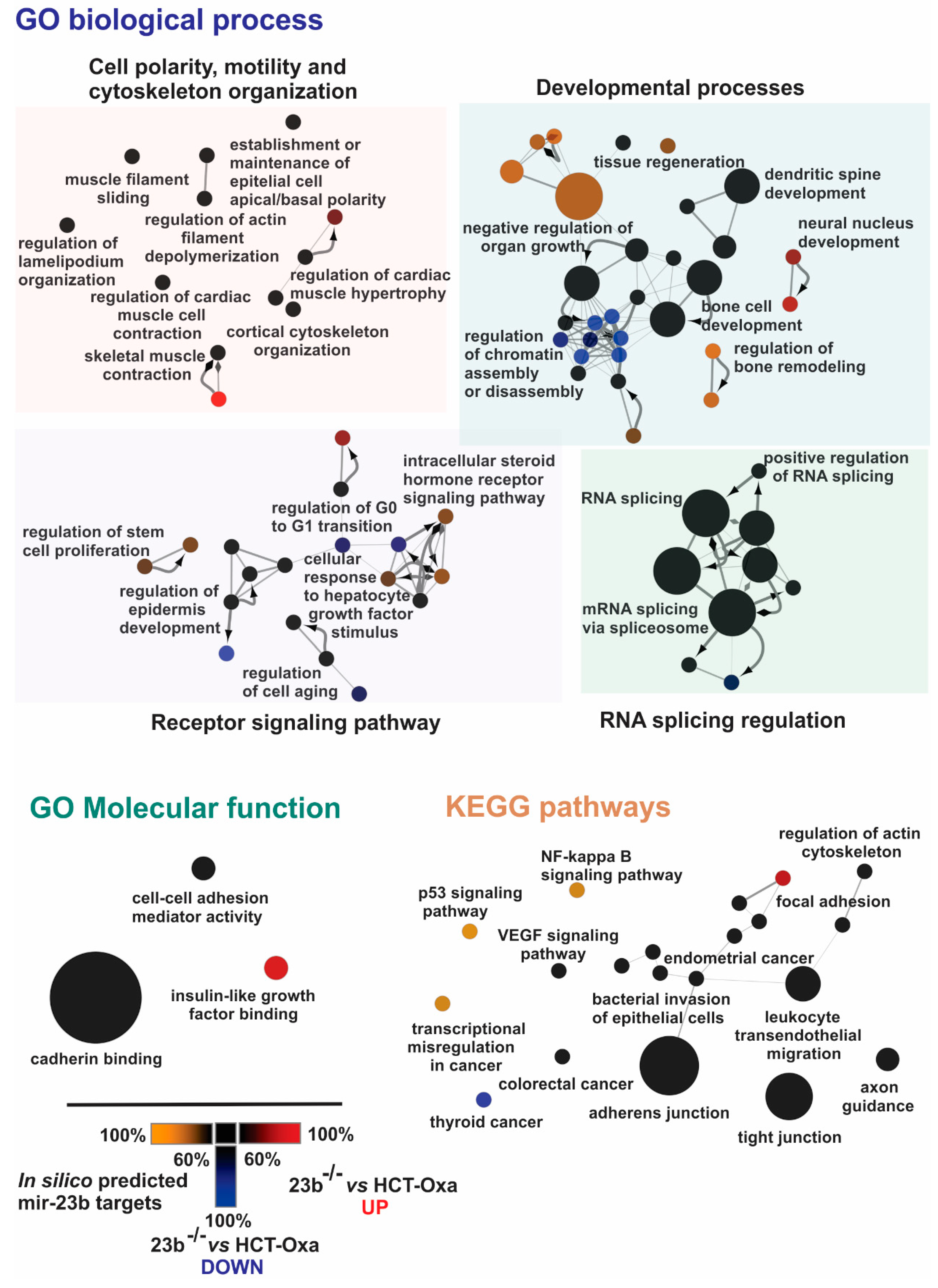
3.6. Identification of miR-23b Targets and Their Functional Bioinformatics Analysis
3.7. miR-23b Contributes to Epithelial Mesenchymal Transition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hyo-eun, C.B.; Ruddy, D.A.; Radhakrishna, V.K.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying Clonal Dynamics in Response to Cancer Therapy Using High-Complexity Barcoding. Nat. Med. 2015, 21, 440. [Google Scholar]
- Sharif, S.; O’Connell, M.J.; Yothers, G.; Lopa, S.; Wolmark, N. Folfox and Flox Regimens for the Adjuvant Treatment of Resected Stage Ii and Iii Colon Cancer. Cancer Investig. 2008, 26, 956–963. [Google Scholar] [CrossRef]
- André, T.; Afchain, P.; Barrier, A.; Blanchard, P.; Larsen, A.K.; Tournigand, C.; Louvet, C.; de Gramont, A. Current Status of Adjuvant Therapy for Colon Cancer. Gastrointest. Cancer Res. 2007, 1, 90–97. [Google Scholar]
- Degirmencioglu, S.; Tanrıverdi, O.; Demiray, A.G.; Senol, H.; Dogu, G.G.; Yaren, A. Retrospective Comparison of Efficacy and Safety of Capox and Folfox Regimens as Adjuvant Treatment in Patients with Stage Iii Colon Cancer. J. Int. Med. Res. 2019, 47, 2507–2515. [Google Scholar] [CrossRef]
- Marjaneh, R.M.; Khazaei, M.; Ferns, G.A.; Avan, A.; Aghaee-Bakhtiari, S.H. The Role of Micrornas in 5-Fu Resistance of Colorectal Cancer: Possible Mechanisms. J. Cell. Physiol. 2019, 234, 2306–2316. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Martínez-Cardús, A.; Ginés, A.; de Porras, V.R.; Moutinho, C.; Layos, L.; Manzano, J.L.; Buges, C.; Bystrup, S.; Esteller, M.; et al. Tumor-Related Molecular Mechanisms of Oxaliplatin Resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef]
- Xie, T.; Huang, M.; Wang, Y.; Wang, L.; Chen, C.; Chu, X. Micrornas as Regulators, Biomarkers and Therapeutic Targets in the Drug Resistance of Colorectal Cancer. Cell. Physiol. Biochem. 2016, 40, 62–76. [Google Scholar] [CrossRef]
- Deng, J.; Wang, Y.; Lei, J.; Lei, W.; Xiong, J.P. Insights into the Involvement of Noncoding Rnas in 5-Fluorouracil Drug Resistance. Tumor Biol. 2017, 39, 1010428317697553. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, L.Y.; Talmon, G.; Wang, J. Microrna-520g Confers Drug Resistance by Regulating P21 Expression in Colorectal Cancer. J. Biol. Chem. 2015, 290, 6215–6225. [Google Scholar] [CrossRef]
- Chai, H.J.; Liu, M.; Tian, R.Q.; Li, X.; Tang, H. Mir-20a Targets Bnip2 and Contributes Chemotherapeutic Resistance in Colorectal Adenocarcinoma Sw480 and Sw620 Cell Lines. Acta Biochim. Biophys. Sin. 2011, 43, 217–225. [Google Scholar] [CrossRef]
- Zhang, L.; Pickard, K.; Jenei, V.; Bullock, M.D.; Bruce, A.; Mitter, R.; Kelly, G.; Paraskeva, C.; Strefford, J.; Primrose, J.; et al. Mir-153 Supports Colorectal Cancer Progression Via Pleiotropic Effects That Enhance Invasion and Chemotherapeutic Resistance. Cancer Res. 2013, 73, 6435–6447. [Google Scholar] [CrossRef]
- Zhou, Y.; Wan, G.; Spizzo, R.; Ivan, C.; Mathur, R.; Hu, X.; Ye, X.C.; Lu, J.; Fan, F.; Xia, L.; et al. Mir-203 Induces Oxaliplatin Resistance in Colorectal Cancer Cells by Negatively Regulating Atm Kinase. Mol. Oncol. 2014, 8, 83–92. [Google Scholar] [CrossRef]
- Mekenkamp, L.J.; Tol, J.; Dijkstra, J.R.; de Krijger, I.; Vink-Börger, M.E.; van Vliet, S.; Teerenstra, S.; Kamping, E.; Verwiel, E.; Koopman, M.; et al. Beyond Kras Mutation Status: Influence of Kras Copy Number Status and Micrornas on Clinical Outcome to Cetuximab in Metastatic Colorectal Cancer Patients. BMC Cancer 2012, 12, 292. [Google Scholar] [CrossRef]
- Du, B.W.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (Emt) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the Intermediate Stages of Epithelial-Mesenchymal Transition in Epithelial Stem Cells and Cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef]
- Leroy, P.; Mostov, K.E. Slug Is Required for Cell Survival During Partial Epithelial-Mesenchymal Transition of Hgf-Induced Tubulogenesis. Mol. Biol. Cell 2007, 18, 1943–1952. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.R.; Weberpals, J.; et al. Feilotter. Emt Transcription Factors Snail and Slug Directly Contribute to Cisplatin Resistance in Ovarian Cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Chang, T.H.; Tsai, M.F.; Su, K.Y.; Wu, S.G.; Huang, C.P.; Yu, S.L.; Yu, Y.L.; Lan, C.C.; Yang, C.H.; Lin, S.B.; et al. Slug Confers Resistance to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. Am. J. Respir. Crit. Care Med. 2011, 183, 1071–1079. [Google Scholar] [CrossRef]
- Dabkeviciene, D.; Jonusiene, V.; Zitkute, V.; Zalyte, E.; Grigaitis, P.; Kirveliene, V.; Sasnauskiene, A. The Role of Interleukin-8 (Cxcl8) and Cxcr2 in Acquired Chemoresistance of Human Colorectal Carcinoma Cells Hct116. Med. Oncol. 2015, 32, 258. [Google Scholar] [CrossRef]
- Kukcinaviciute, E.; Jonusiene, V.; Sasnauskiene, A.; Dabkeviciene, D.; Eidenaite, E.; Laurinavicius, A. Significance of Notch and Wnt Signaling for Chemoresistance of Colorectal Cancer Cells Hct116. J. Cell Biochem. 2018, 119, 5913–5920. [Google Scholar] [CrossRef]
- Butkytė, S.; Čiupas, L.; Jakubauskienė, E.; Vilys, L.; Mocevicius, P.; Kanopka, A.; Vilkaitis, G. Splicing-Dependent Expression of Micrornas of Mirtron Origin in Human Digestive and Excretory System Cancer Cells. Clin. Epigenet. 2016, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the Crispr-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, Y.; Yu, S.; Lu, L.; Ding, M.; Cheng, J.; Song, G.X.; Gao, X.; Yao, L.M.; Fan, D.D.; et al. An Efficient Genotyping Method for Genome-Modified Animals and Human Cells Generated with Crispr/Cas9 System. Sci. Rep. 2014, 4, 6420. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6, 359. [Google Scholar] [CrossRef]
- Ger, M.; Kaupinis, A.; Nemeikaite-Ceniene, A.; Sarlauskas, J.; Cicenas, J.; Cenas, N.; Valius, M. Quantitative Proteomic Analysis of Anticancer Drug Rh1 Resistance in Liver Carcinoma. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 219–232. [Google Scholar] [CrossRef]
- Kuharev, J.; Navarro, P.; Distler, U.; Jahn, O.; Tenzer, S. In-Depth Evaluation of Software Tools for Data-Independent Acquisition Based Label-Free Quantification. Proteomics 2015, 15, 3140–3151. [Google Scholar] [CrossRef]
- Varemo, L.; Nielsen, J.; Nookaew, I. Enriching the Gene Set Analysis of Genome-Wide Data by Incorporating Directionality of Gene Expression and Combining Statistical Hypotheses and Methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Wang, Y.R.; Luo, J.J.; Zhang, H.; Lu, J. Micrornas in the Same Clusters Evolve to Coordinately Regulate Functionally Related Genes. Mol. Biol. Evol. 2016, 33, 2232–2247. [Google Scholar] [CrossRef]
- Zhou, P.; Ma, L.; Zhou, J.; Jiang, M.; Rao, E.; Zhao, Y.; Guo, F. Mir-17-92 Plays an Oncogenic Role and Conveys Chemo-Resistance to Cisplatin in Human Prostate Cancer Cells. Int. J. Oncol. 2016, 48, 1737–1748. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y. The Mir-17-92 Cluster Counteracts Quiescence and Chemoresistance in a Distinct Subpopulation of Pancreatic Cancer Stem Cells. Gut 2016, 65, 1224. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; Van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Non-Coding Rna Towards a Molecular Understanding of Microrna-Mediated Gene Silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The String Database in 2017: Quality-Controlled Protein-Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, 362–368. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Yu, J.; Chan, M.T.; Wu, W.K. Micrornas Predict and Modulate Responses to Chemotherapy in Colorectal Cancer. Cell Prolif. 2015, 48, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, M.; Delforge, Y.; Deward, A.; Habraken, Y.; Hennuy, B.; Piette, J.; Klinck, R.; Chabot, B.; Colige, A.; Lambert, C. Role of the Splicing Factor Srsf4 in Cisplatin-Induced Modifications of Pre-Mrna Splicing and Apoptosis. BMC Cancer 2015, 15, 227. [Google Scholar] [CrossRef]
- Urfali-Mamatoglu, C.; Kazan, H.H.; Gündüz, U. Dual Function of Programmed Cell Death 10 (Pdcd10) in Drug Resistance. Biomed. Pharmacother. 2018, 101, 129–136. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. Cluego: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. Cluepedia Cytoscape Plugin: Pathway Insights Using Integrated Experimental and in Silico Data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Sun, B.O.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of Cellular Cytoskeleton in Epithelial-Mesenchymal Transition Process During Cancer Progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Shankar, J.; Nabi, I.R. Actin Cytoskeleton Regulation of Epithelial Mesenchymal Transition in Metastatic Cancer Cells. PLoS ONE 2015, 10, e0119954. [Google Scholar]
- Friedl, P.; Gilmour, D. Collective Cell Migration in Morphogenesis, Regeneration and Cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-Mesenchymal Transitions: The Importance of Changing Cell State in Development and Disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef]
- Kerosuo, L.; Bronner-Fraser, M. What Is Bad in Cancer Is Good in the Embryo: Importance of Emt in Neural Crest Development. Semin. Cell Dev. Biol. 2012, 23, 320–332. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An Emt-Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype. PLoS Genet 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Carstens, R.P. Complex Changes in Alternative Pre-Mrna Splicing Play a Central Role in the Epithelial-to-Mesenchymal Transition (Emt). Semin. Cancer Biol. 2012, 22, 417–427. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling Mechanisms of the Epithelial-Mesenchymal Transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of Cadherin-Mediated Cell Adhesion and Canonical Wnt Signaling. Cold Spring Harbor Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The Regulation of Cell-Cell Adhesion During Epithelial-Mesenchymal Transition, Motility and Tumor Progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. Nf-Kappa B Is Essential for Epithelial-Mesenchymal Transition and Metastasis in a Model of Breast Cancer Progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef]
- Hong, J.P.; Li, X.M.; Li, M.X.; Zheng, F.L. Vegf Suppresses Epithelial-Mesenchymal Transition by Inhibiting the Expression of Smad3 and Mir-192, a Smad3-Dependent Microrna. Int. J. Mol. Med. 2013, 31, 1436–1442. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.I.; Goldman, R.D. Vimentin Induces Changes in Cell Shape, Motility, and Adhesion During the Epithelial to Mesenchymal Transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin Regulates Emt Induction by Slug and Oncogenic H-Ras and Migration by Governing Axl Expression in Breast Cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef]
- Li, P.L.; Zhang, X.; Wang, L.L.; Du, L.T.; Yang, Y.M.; Li, J.; Wang, C.X. Microrna-218 Is a Prognostic Indicator in Colorectal Cancer and Enhances 5-Fluorouracil-Induced Apoptosis by Targeting Birc5. Carcinogenesis 2015, 36, 1484–1493. [Google Scholar]
- Offer, S.M.; Butterfield, G.L.; Jerde, C.R.; Fossum, C.C.; Wegner, N.J.; Diasio, R.B. Micrornas Mir-27a and Mir-27b Directly Regulate Liver Dihydropyrimidine Dehydrogenase Expression through Two Conserved Binding Sites. Mol. Cancer Ther. 2014, 13, 742–751. [Google Scholar] [CrossRef]
- Poel, D.; Boyd, L.N.; Beekhof, R.; Schelfhorst, T.; Pham, T.V.; Piersma, S.R.; Knol, J.C.; Jimenez, C.R.; Verheul, H.M.W.; Buffart, T.E. Proteomic Analysis of Mir-195 and Mir-497 Replacement Reveals Potential Candidates That Increase Sensitivity to Oxaliplatin in Msi/P53wt Colorectal Cancer Cells. Cells 2019, 8, 1111. [Google Scholar] [CrossRef]
- Jeppesen, M.; Hagel, G.; Glenthoj, A.; Vainer, B.; Ibsen, P.; Harling, H.; Thastrup, O.; Jorgensen, L.N.; Thastrup, J. Short-Term Spheroid Culture of Primary Colorectal Cancer Cells as an in Vitro Model for Personalizing Cancer Medicine. PLoS ONE 2017, 12, e1083074. [Google Scholar] [CrossRef]
- Amankwatia, E.B.; Chakravarty, P.; Carey, F.A.; Weidlich, S.; Steele, R.J.C.; Munro, A.J.; Wolf, C.R.; Smith, G. Microrna-224 Is Associated with Colorectal Cancer Progression and Response to 5-Fluorouracil-Based Chemotherapy by Kras-Dependent and -Independent Mechanisms. Br. J. Cancer 2015, 112, 1480–1490. [Google Scholar] [CrossRef]
- Kwak, M.S.; Cha, J.M.; Yoon, J.Y.; Jeon, J.W.; Shin, H.P.; Chang, H.J.; Kim, H.K.; Joo, K.R.; Lee, J.I. Prognostic Value of Kras Codon 13 Gene Mutation for Overall Survival in Colorectal Cancer Direct and Indirect Comparison Meta-Analysis. Medicine 2017, 96, e7882. [Google Scholar] [CrossRef]
- Olaru, A.V.; Yamanaka, S.; Vazquez, C.; Mori, Y.; Cheng, Y.; Abraham, J.M.; Bayless, T.M.; Harpaz, N.; Selaru, F.M.; Meltzer, S.J. Microrna-224 Negatively Regulates P21 Expression During Late Neoplastic Progression in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2013, 19, 471–480. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.J.; Chen, X.B. Examination of the Expanding Pathways for the Regulation of P21 Expression and Activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef]
- Maiuthed, A.; Ninsontia, C.; Erlenbach-Wuensch, K.; Ndreshkjana, B.; Muenzner, J.; Caliskan, A.; Ahmed, P.H.; Chaotham, C.; Hartmann, A.; Roehe, A.V.; et al. Cytoplasmic P21 Mediates 5-Fluorouracil Resistance by Inhibiting Pro-Apoptotic Chk2. Cancers 2018, 10, 373. [Google Scholar] [CrossRef]
- Mencia, N.; Selga, E.; Noe, V.; Ciudad, C.J. Underexpression of Mir-224 in Methotrexate Resistant Human Colon Cancer Cells. Biochem. Pharmacol. 2011, 82, 1572–1582. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y.; Liu, B.; Shan, Y.; Li, Y.; Zhao, L.; Jia, L. Downregulation of Mir-224 and Let-7i Contribute to Cell Survival and Chemoresistance in Chronic Myeloid Leukemia Cells by Regulating St3gal Iv Expression. Gene 2017, 626, 106–118. [Google Scholar] [CrossRef]
- Kristensen, H.; Haldrup, C.; Strand, S.; Mundbjerg, K.; Mortensen, M.M.; Thorsen, K.; Ostenfeld, M.S.; Wild, P.J.; Arsov, C.; Goering, W.; et al. Hypermethylation of the Gabre Similar to Mir-452 Similar to Mir-224 Promoter in Prostate Cancer Predicts Biochemical Recurrence after Radical Prostatectomy. Clin. Cancer Res. 2014, 20, 2169–2181. [Google Scholar] [CrossRef]
- Wang, Y.; Toh, H.C.; Chow, P.; Chung, A.Y.; Meyers, D.J.; Cole, P.A.; Ooi, L.L.P.J.; Lee, C.G.L. Microrna-224 Is up-Regulated in Hepatocellular Carcinoma through Epigenetic Mechanisms. FASEB J. 2012, 26, 3032–3041. [Google Scholar] [CrossRef]
- Laddha, S.V.; Nayak, S.; Paul, D.; Reddy, R.; Sharma, C.; Jha, P.; Hariharan, M.; Agrawal, A.; Chowdhury, S.; Sarkar, C.A. Mukhopadhyay. Genome-Wide Analysis Reveals Downregulation of Mir-379/Mir-656 Cluster in Human Cancers. Biol. Direct 2013, 8, 10. [Google Scholar] [CrossRef]
- Laddha, S.V.; Nayak, S.; Paul, D.; Reddy, R.; Sharma, C.; Jha, P. Dysregulation of Microrna-543 Expression in Colorectal Cancer Promotes Tumor Migration and Invasion. Mol. Carcinog. 2017, 56, 250–257. [Google Scholar]
- Gu, X.; Jin, R.; Mao, X.; Wang, J.; Yuan, J.; Zhao, G. Prognostic Value of Mirna-181a/B in Colorectal Cancer: A Meta-Analysis. Biomark. Med. 2018, 12, 299–308. [Google Scholar] [CrossRef]
- Haier, J.; Strose, A.; Matuszcak, C.; Hummel, R. Mir Clusters Target Cellular Functional Complexes by Defining Their Degree of Regulatory Freedom. Cancer Metastasis Rev. 2016, 35, 289–322. [Google Scholar] [CrossRef]
- Deng, Y.H.; Deng, Z.H.; Hao, H.; Wu, X.L.; Gao, H.; Tang, S.H.; Tang, H. Microrna-23a Promotes Colorectal Cancer Cell Survival by Targeting Pdk4. Exp. Cell Res. 2018, 373, 171–179. [Google Scholar] [CrossRef]
- Peng, F.S.; Zhang, H.L.; Du, Y.H.; Tan, P.Q. Mir-23a Promotes Cisplatin Chemoresistance and Protects against Cisplatin-Induced Apoptosis in Tongue Squamous Cell Carcinoma Cells through Twist. Oncol. Rep. 2015, 33, 942–950. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Song, Y.; Fu, Z.; Yu, W. Mir-27a Regulates Cisplatin Resistance and Metastasis by Targeting Rkip in Human Lung Adenocarcinoma Cells. Mol. Cancer 2014, 13, 193. [Google Scholar] [CrossRef]
- Rasmussen, M.H.; Jensen, N.F.; Tarpgaard, L.S.; Qvortrup, C.; Rømer, M.U.; Stenvang, J.; Hansen, T.P.; Christensen, L.L.; Lindebjerg, J.; Hansen, F.; et al. High Expression of Microrna-625-3p Is Associated with Poor Response to First-Line Oxaliplatin Based Treatment of Metastatic Colorectal Cancer. Mol. Oncol. 2013, 7, 637–646. [Google Scholar] [CrossRef]
- Kjersem, J.B.; Ikdahl, T.; Lingjaerde, O.C.; Guren, T.; Tveit, K.M.; Kure, E.H. Plasma Micrornas Predicting Clinical Outcome in Metastatic Colorectal Cancer Patients Receiving First-Line Oxaliplatin-Based Treatment. Mol. Oncol. 2014, 8, 59–67. [Google Scholar] [CrossRef]
- Wu, S.; Bai, L.; Li, Z.F.; Li, Q.G.; Xie, J.; Jian, B. Clinical Significance and Prognostic Value of Microrna-23b Expression Level in Colon Cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 10587–10592. [Google Scholar]
- Kou, C.H.; Zhou, T.; Han, X.L.; Zhuang, H.J.; Qian, H.X. Downregulation of Mir-23b in Plasma Is Associated with Poor Prognosis in Patients with Colorectal Cancer. Oncol. Lett. 2016, 12, 4838–4844. [Google Scholar] [CrossRef]
- Zhang, H.; Hao, Y.; Yang, J.; Zhou, Y.; Li, J.; Yin, S.; Sun, C.H.; Ma, M.; Huang, Y.Y.; Xi, J.J. Genome-Wide Functional Screening of Mir-23b as a Pleiotropic Modulator Suppressing Cancer Metastasis. Nat. Commun. 2011, 2, 554. [Google Scholar] [CrossRef]
- Ling, H.; Pickard, K.; Ivan, C.; Isella, C.; Ikuo, M.; Mitter, R.; Spizzo, R.; Bullock, M.D.; Braicu, C.; Pileczki, V.; et al. The Clinical and Biological Significance of Mir-224 Expression in Colorectal Cancer Metastasis. Gut 2016, 65, 977–989. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073. [Google Scholar] [CrossRef]
- Zhang, C.M.; Lv, J.F.; Gong, L.; Yu, L.Y.; Chen, X.P.; Zhou, H.H.; Fan, L. Role of Deficient Mismatch Repair in the Personalized Management of Colorectal Cancer. Int. J. Environ. Res. Public Health 2016, 13, 892. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Oxaliplatin-Resistant Sublines | 5-Fluorouracil-Resistant Sublines | Spheroids | |||||
|---|---|---|---|---|---|---|---|---|
| HCT-Oxa-c | HCT-Oxa-p | HCT-FU-c | DLD-FU-p | HCT-Oxa-c | HCT-FU-c | |||
| RNA-Seq | qPCR | qPCR | RNA-Seq | qPCR | qPCR | qPCR | qPCR | |
| Oxaliplatin-resistance related miRNAs | ||||||||
| 23b-3p | up | 5.2 ± 0.9 | 9.5 ± 2.5 | constant | 1.7 ± 0.1 | −2.1 ± 0.6 | 7.3 ± 3.8 | 1.0 ± 0.3 |
| 27b-3p | up | 5.9 ± 1.2 | 6.6 ± 1.4 | constant | −1.0 ± 0.2 | −1.1 ± 0.1 | 5.6 ± 0.4 | 1.2 ± 0.3 |
| 27b-5p | up | 9.1 ± 1.0 | 5.3 ± 2.2 | constant | n.d. | n.d. | 6.7 ± 1.3 | n.d. |
| 24-3p | constant | −1.1 ± 0.0 | 1.6 ± 0.1 | constant | n.d. | n.d. | 1.5 ± 0.1 | n.d. |
| 23a-3p | down | −1.9 ± 0.4 | −1.2 ± 0.2 | constant | −1.1 ± 0.1 | 1.4 ± 0.3 | 1.0 ± 0.3 | −1.1 ± 0.1 |
| 27a-3p | down | −1.6 ± 0.3 | −1.6 ± 0.2 | constant | n.d. | n.d. | −1.3 ± 0.1 | n.d. |
| 27a-5p | down | −1.6 ± 0.6 | −1.8 ± 0.2 | down | −1.7 ± 0.2 | 1.1 ± 0.1 | −2.0 ± 0.3 | 1.2 ± 0.4 |
| 24-2-5p | down | −1.5 ± 0.1 | −1.6 ± 0.3 | constant | n.d. | n.d. | −2.0 ± 0.4 | n.d. |
| 181b-5p | up | 2.5 ± 0.4 | 4.9 ± 0.4 | constant | −1.6 ± 0.1 | 1.3 ± 0.2 | 2.5 ± 0.5 | −1.5 ± 0.6 |
| 5-FU-resistance related miRNAs | ||||||||
| 203b-3p | constant | −1.3 ± 0.1 | 1.9 ± 0.3 | down | −4.6 ± 1.0 | 1.8 ± 0.2 | n.d. | −2.5 ± 1.6 |
| 195-5p | constant | −1.5 ± 0.1 | 1.2 ± 0.2 | down | −2.3 ± 0.3 | −1.3 ± 0.1 | n.d. | −1.7 ± 0.8 |
| 224-5p | constant | 1.2 ± 0.2 | 1.2 ± 0.3 | down | −2.3 ± 0.7 | −57 ± 16 | −1.2 ± 0.0 | −1.5 ± 0.1 |
| 452-5p | constant | −1.1 ± 0.1 | 1.1 ± 0.1 | down | −2.9 ± 0.3 | −3.8 ± 1.0 | −1.2 ± 0.3 | −1.5 ± 0.3 |
| Unbiased miRNAs | ||||||||
| 17-5p | constant | n.d. | n.d. | constant | 1.6 ± 0.3 | 1.3 ± 0.1 | n.d. | 1.4 ± 0.2 |
| 19b-3p | constant | n.d. | n.d. | constant | 1.9 ± 0.5 | 1.5 ± 0.1 | n.d. | 1.0 ± 0.4 |
| 20a-5p | constant | n.d. | n.d. | constant | 1.6 ± 0.1 | −1.5 ± 0.3 | n.d. | 1.3 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasiulė, S.; Dreize, N.; Kaupinis, A.; Ražanskas, R.; Čiupas, L.; Stankevičius, V.; Kapustina, Ž.; Laurinavičius, A.; Valius, M.; Vilkaitis, G. Molecular Insights into miRNA-Driven Resistance to 5-Fluorouracil and Oxaliplatin Chemotherapy: miR-23b Modulates the Epithelial–Mesenchymal Transition of Colorectal Cancer Cells. J. Clin. Med. 2019, 8, 2115. https://doi.org/10.3390/jcm8122115
Gasiulė S, Dreize N, Kaupinis A, Ražanskas R, Čiupas L, Stankevičius V, Kapustina Ž, Laurinavičius A, Valius M, Vilkaitis G. Molecular Insights into miRNA-Driven Resistance to 5-Fluorouracil and Oxaliplatin Chemotherapy: miR-23b Modulates the Epithelial–Mesenchymal Transition of Colorectal Cancer Cells. Journal of Clinical Medicine. 2019; 8(12):2115. https://doi.org/10.3390/jcm8122115
Chicago/Turabian StyleGasiulė, Stasė, Nadezda Dreize, Algirdas Kaupinis, Raimundas Ražanskas, Laurynas Čiupas, Vaidotas Stankevičius, Žana Kapustina, Arvydas Laurinavičius, Mindaugas Valius, and Giedrius Vilkaitis. 2019. "Molecular Insights into miRNA-Driven Resistance to 5-Fluorouracil and Oxaliplatin Chemotherapy: miR-23b Modulates the Epithelial–Mesenchymal Transition of Colorectal Cancer Cells" Journal of Clinical Medicine 8, no. 12: 2115. https://doi.org/10.3390/jcm8122115
APA StyleGasiulė, S., Dreize, N., Kaupinis, A., Ražanskas, R., Čiupas, L., Stankevičius, V., Kapustina, Ž., Laurinavičius, A., Valius, M., & Vilkaitis, G. (2019). Molecular Insights into miRNA-Driven Resistance to 5-Fluorouracil and Oxaliplatin Chemotherapy: miR-23b Modulates the Epithelial–Mesenchymal Transition of Colorectal Cancer Cells. Journal of Clinical Medicine, 8(12), 2115. https://doi.org/10.3390/jcm8122115