Muscle Involvement in a Large Cohort of Pediatric Patients with Genetic Diagnosis of Mitochondrial Disease

 , ,
, , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Patients
2.2. Methods
2.2.1. Patient Recruitment
2.2.2. Clinical Data
2.2.3. Samples
2.2.4. Biochemical Analysis
2.2.5. Histopathological Analysis
2.2.6. Genetic Analysis
2.2.7. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Witters, P.; Saada, A.; Honzik, T.; Tesarova, M.; Kleinle, S.; Horvath, R.; Goldstein, A.; Morava, E. Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genet. Med. 2018, 20, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitocondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Phadke, R. Myopathology of adult and paediatric mitochondrial diseases. J. Clin. Med. 2017, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.J.; Riley, L.G.; Christodoulou, J. Mitochondrial respiratory chain disorders in childhood: Insights into diagnosis and management in the new era of genomic medicine. Biochim. Biophys. Acta 2014, 1840, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Cruz, S.; Taipa, R.; Nogueira, C.; Pereira, C.; Almeida, L.; Neiva, R.; Geraldes, T.; Guimarães, A.; Melo-Pires, M.; Vilarinho, L. Clinical, biochemical, molecular, and histological features of 65 Portuguese patients with mitochondrial disorders. Muscle Nerve 2017, 56, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Couser, N.; Gucsavas-Calikoglu, M. Mitochondrial disorders. In Biomarkers in Inborn Errors of Metabolism. Clinical Aspects and Laboratory Determination; Garg, U., Smith, L.D., Eds.; Elsevier: Cambridge, MA, USA, 2017; Volume 2, pp. 167–190. [Google Scholar]
- Batllori, M.; Molero-Luis, M.; Ormazabal, A.; Montero, R.; Sierra, C.; Ribes, A.; Montoya, J.; Ruiz-Pesini, E.; O’Callaghan, M.; Pias, L.; et al. Cerebrospinal fluid monoamines, pterins, and folate in patients with mitochondrial diseases: Systematic review and hospital experience. J. Inherit. Metab. Dis. 2018. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Vasta, V.; Merritt, J.L.; Saneto, R.P.; Hahn, S.H. Next-generation sequencing for mitochondrial diseases: A wide diagnostic spectrum: Mitochondrial disease and NGS technology. Pediatr. Int. 2012, 54, 585–601. [Google Scholar] [CrossRef]
- Tang, S.; Wang, J.; Zhang, V.W.; Li, F.Y.; Landsverk, M.; Cui, H.; Truong, C.K.; Wang, G.; Chen, L.C.; Graham, B.; et al. Transition to next generation analysis of the whole mitochondrial genome: A summary of molecular defects. Hum. Mutat. 2013, 34, 882–893. [Google Scholar] [CrossRef]
- DaRe, J.T.; Vasta, V.; Penn, J.; Tran, N.-T.B.; Hahn, S.H. Targeted exome sequencing for mitochondrial disorders reveals high genetic heterogeneity. BMC Med. Genet. 2013, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Pyle, A.; Griffin, H.; Blakely, E.L.; Duff, J.; He, L.; Smertenko, T.; Alston, C.L.; Neeve, V.C.; Best, A.; et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014, 312, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, A.; Murayama, K.; Mori, M.; Harashima, H.; Yamazaki, T.; Tamaru, S.; Yamashita, Y.; Kishita, Y.; Nakachi, Y.; Kohda, M.; et al. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: Exome sequencing for disease gene identification. Biochim. Biophys. Acta 2014, 1840, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucińska-Więckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosińska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta 2016, 1857, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Plutino, M.; Chaussenot, A.; Rouzier, C.; Ait-El-Mkadem, S.; Fragaki, K.; Paquis-Flucklinger, V.; Bannwarth, S. Targeted next generation sequencing with an extended gene panel does not impact variant detection in mitochondrial diseases. BMC Med. Genet. 2018, 19, 57. [Google Scholar] [CrossRef]
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial energy generation disorders: Genes, mechanisms and clues to pathology. J. Biol. Chem. 2017. [Google Scholar] [CrossRef]
- Palculict, M.E.; Zhang, V.W.; Wong, L.J.; Wang, J. comprehensive mitochondrial genome analysis by massively parallel sequencing. Methods Mol. Biol. 2016, 1351, 3–17. [Google Scholar] [PubMed]
- Abicht, A.; Scharf, F.; Kleinle, S.; Schön, U.; Holinski-Feder, E.; Horvath, R.; Benet-Pagès, A.; Diebold, I. Mitochondrial and nuclear disease panel (Mito-aND-Panel): Combined sequencing of mitochondrial and nuclear DNA by a cost-effective and sensitive NGS-based method. Mol. Genet. Genom. Med. 2018. [Google Scholar] [CrossRef]
- O’Callaghan, M.M.; Emperador, S.; Pineda, M.; López-Gallardo, E.; Montero, R.; Yubero, D.; Jou, C.; Jimenez-Mallebrera, C.; Nascimento, A.; Ferrer, I.; et al. Mutation loads in different tissues from six pathogenic mtDNA point mutations. Mitochondrion 2015, 22, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Montero, R.; Yubero, D.; Villarroya, J.; Henares, D.; Jou, C.; Rodríguez, M.A.; Ramos, F.; Nascimento, A.; Ortez, C.I.; Campistol, J.; et al. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS ONE 2016, 11, e0148709. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Brandi, N.; Ormazabal, A.; Garcia-Cazorla, A.; Pérez-Dueñas, B.; Campistol, J.; Ribes, A.; Palau, F.; Artuch, R.; Armstrong, J.; et al. Targeted next generation sequencing in patients with inborn errors of metabolism. PLoS ONE 2016, 11, e0156359. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Goldstein, A.C.; Bhatia, P.; Vento, J.M. Mitochondrial disease in childhood: Nuclear encoded. Neurotherapeutics 2013, 10, 212–226. [Google Scholar] [CrossRef]
- Rocha, M.C.; Grady, J.P.; Grünewald, A.; Vincent, A.; Dobson, P.F.; Taylor, R.W.; Turnbull, D.M.; Rygiel, K.A. A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: Understanding mechanisms and improving diagnosis. Sci. Rep. 2015, 5, 15037. [Google Scholar] [CrossRef]
- Alston, C.L.; Rocha, M.C.; Lax, N.; Turnbull, D.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Tulinius, M.; Olfors, A. Neonatal muscular manifestations in mitochondrial disorders. Semin. Fetal Neonatal Med. 2011, 16, 229–233. [Google Scholar] [CrossRef]
- Serrano, M.; García-Silva, M.T.; Martin-Hernandez, E.; del O’Callaghan, M.; Quijada, P.; Martinez-Aragón, A.; Ormazábal, A.; Blázquez, A.; Martín, M.A.; Briones, P.; et al. Kearns-Sayre syndrome: Cerebral folate deficiency, MRI findings and new cerebrospinal fluid biochemical features. Mitochondrion 2010, 10, 429–432. [Google Scholar] [CrossRef]
- Díez, H.; Cortès-Saladelafont, E.; Ormazábal, A.; Marmiese, A.F.; Armstrong, J.; Matalonga, L.; Bravo, M.; Briones, P.; Emperador, S.; Montoya, J.; et al. Severe infantile parkinsonism because of a de novo mutation on DLP1 mitochondrial-peroxisomal protein. Mov. Disord. 2017, 32, 1108–1110. [Google Scholar] [CrossRef]
- Brito, S.; Thompson, K.; Campistol, J.; Colomer, J.; Hardy, S.A.; He, L.; Fernández-Marmiesse, A.; Palacios, L.; Jou, C.; Jiménez-Mallebrera, C.; et al. Long-term survival in a child with severe encephalopathy, multiple respiratory chain deficiency and GFM1 mutations. Front. Genet. 2015, 23, 102. [Google Scholar] [CrossRef]
- Ortigoza-Escobar, J.D.; Oyarzabal, A.; Montero, R.; Artuch, R.; Jou, C.; Jiménez, C.; Gort, L.; Briones, P.; Muchart, J.; López-Gallardo, E.; et al. Ndufs4 related Leigh syndrome: A case report and review of the literature. Mitochondrion 2016, 28, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Cortès, X.; Font, A.; Bujan, N.; Navarro-Sastre, A.; Matalonga, L.; Arranz, J.A.; Riudor, E.; del Toro, M.; Garcia-Cazorla, A.; Campistol, J.; et al. Protein expression profiles in patients carrying NFU1 mutations. Contribution to the pathophysiology of the disease. J. Inherit. Metab. Dis. 2013, 36, 841–847. [Google Scholar] [CrossRef]
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.E.; Stroud, D.A.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Asencio, C.; Rodríguez-Hernandez, M.A.; Briones, P.; Montoya, J.; Cortés, A.; Emperador, S.; Gavilán, A.; Ruiz-Pesini, E.; Yubero, D.; Montero, R.; et al. Severe encephalopathy associated to pyruvate dehydrogenase mutations and unbalanced coenzyme Q10 content. Eur. J. Hum. Genet. 2016, 24, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Ortigoza-Escobar, J.D.; Molero-Luis, M.; Arias, A.; Martí-Sánchez, L.; Rodriguez-Pombo, P.; Artuch, R.; Pérez-Dueñas, B. Treatment of genetic defects of thiamine transport and metabolism. Expert Rev Neurother. 2016, 16, 755–763. [Google Scholar] [CrossRef]
- Navarro-Sastre, A.; Tort, F.; Garcia-Villoria, J.; Pons, M.R.; Nascimento, A.; Colomer, J.; Campistol, J.; Yoldi, M.E.; López-Gallardo, E.; Montoya, J.; et al. Mitochondrial DNA depletion syndrome: New descriptions and the use of citrate synthase as a helpful tool to better characterise the patients. Mol. Genet. Metab. 2012, 107, 409–415. [Google Scholar] [CrossRef]
- Kalko, S.G.; Paco, S.; Jou, C.; Rodríguez, M.A.; Meznaric, M.; Rogac, M.; Jekovec-Vrhovsek, M.; Sciacco, M.; Moggio, M.; Fagiolari, G.; et al. Transcriptomic profiling of TK2 deficient human skeletal muscle suggests a role for the p53 signalling pathway and identifies growth and differentiation factor-15 as a potential novel biomarker for mitochondrial myopathies. BMC Genom. 2014, 1, 91. [Google Scholar] [CrossRef]

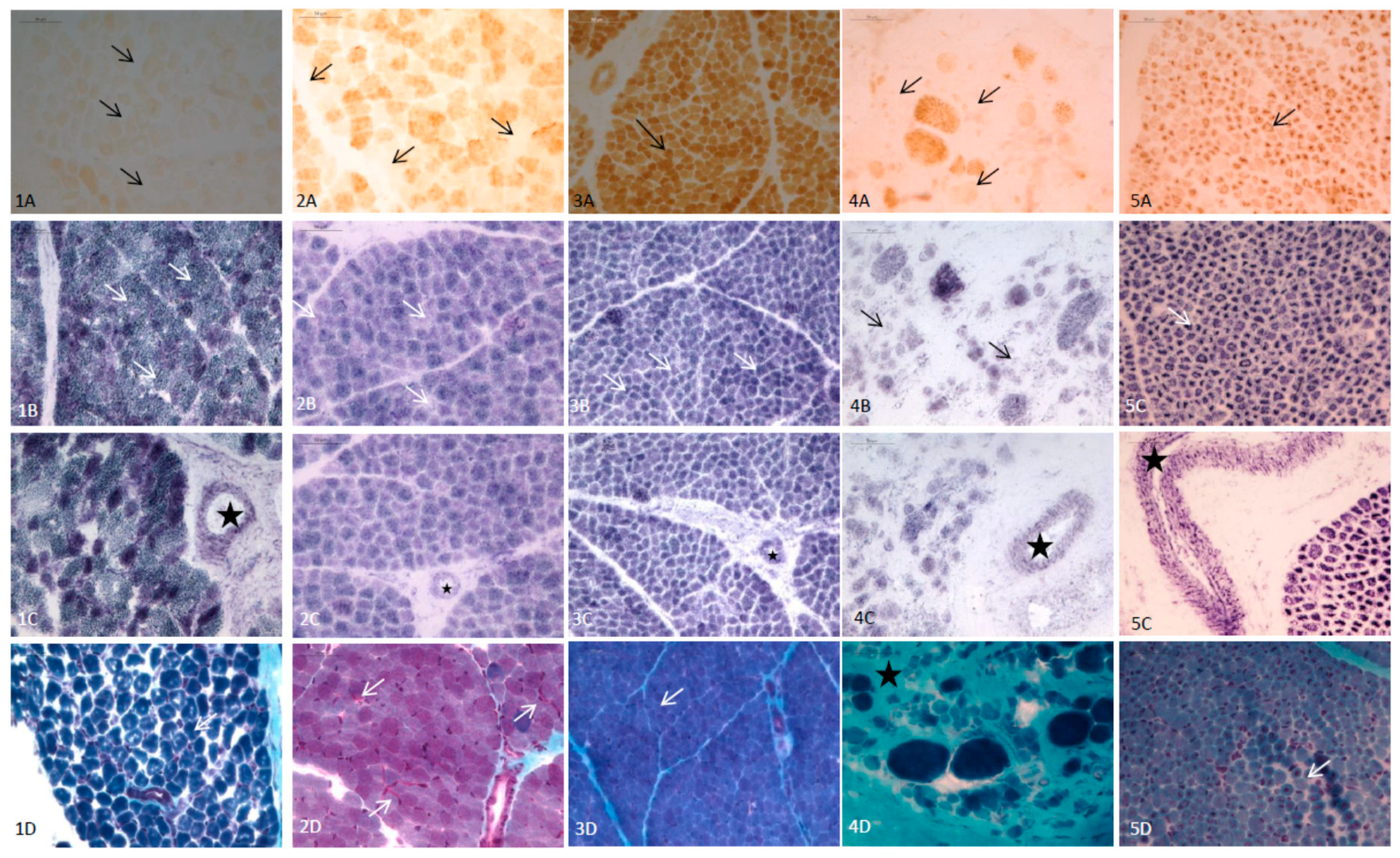

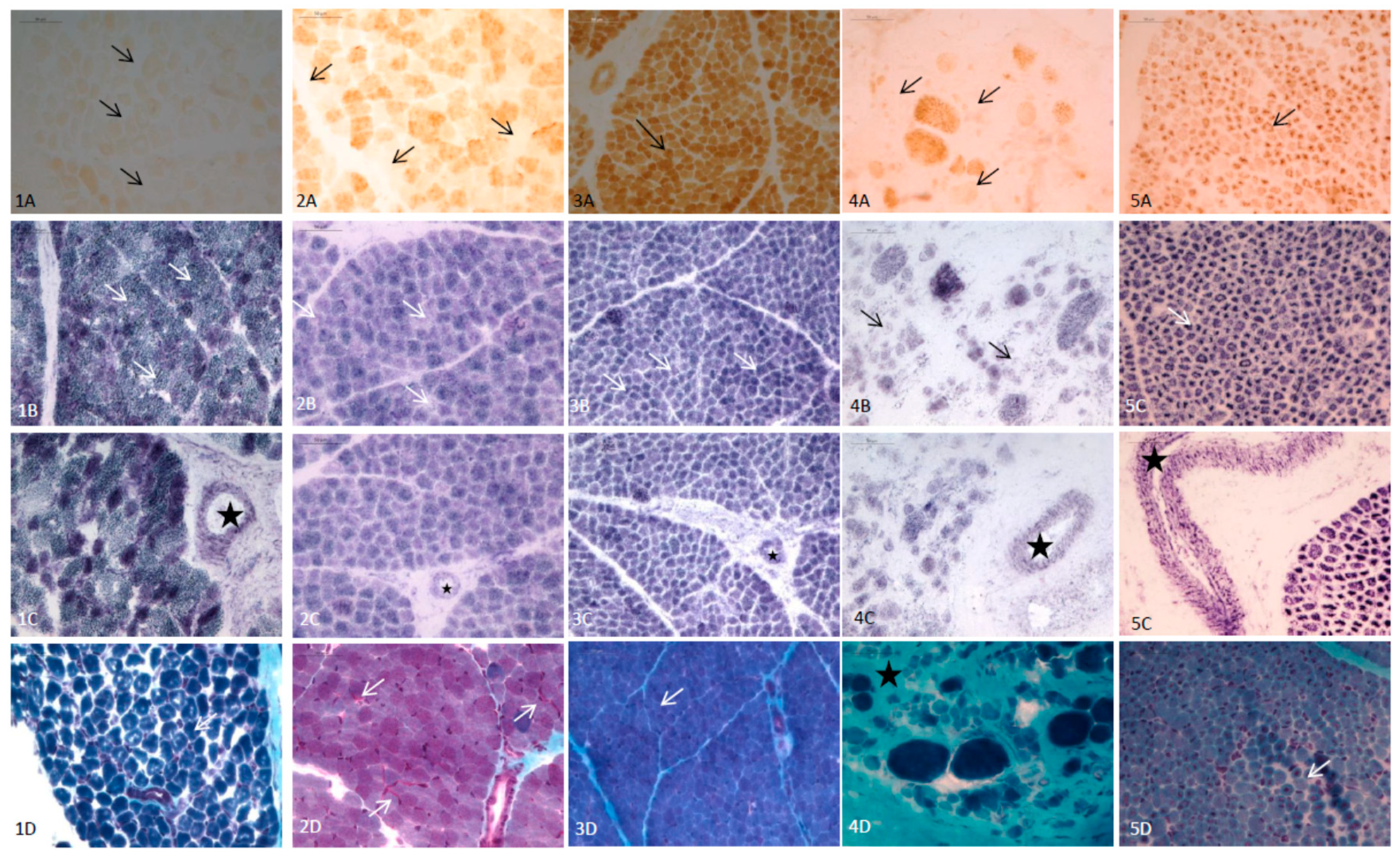
{kind=link}
{kind=link}
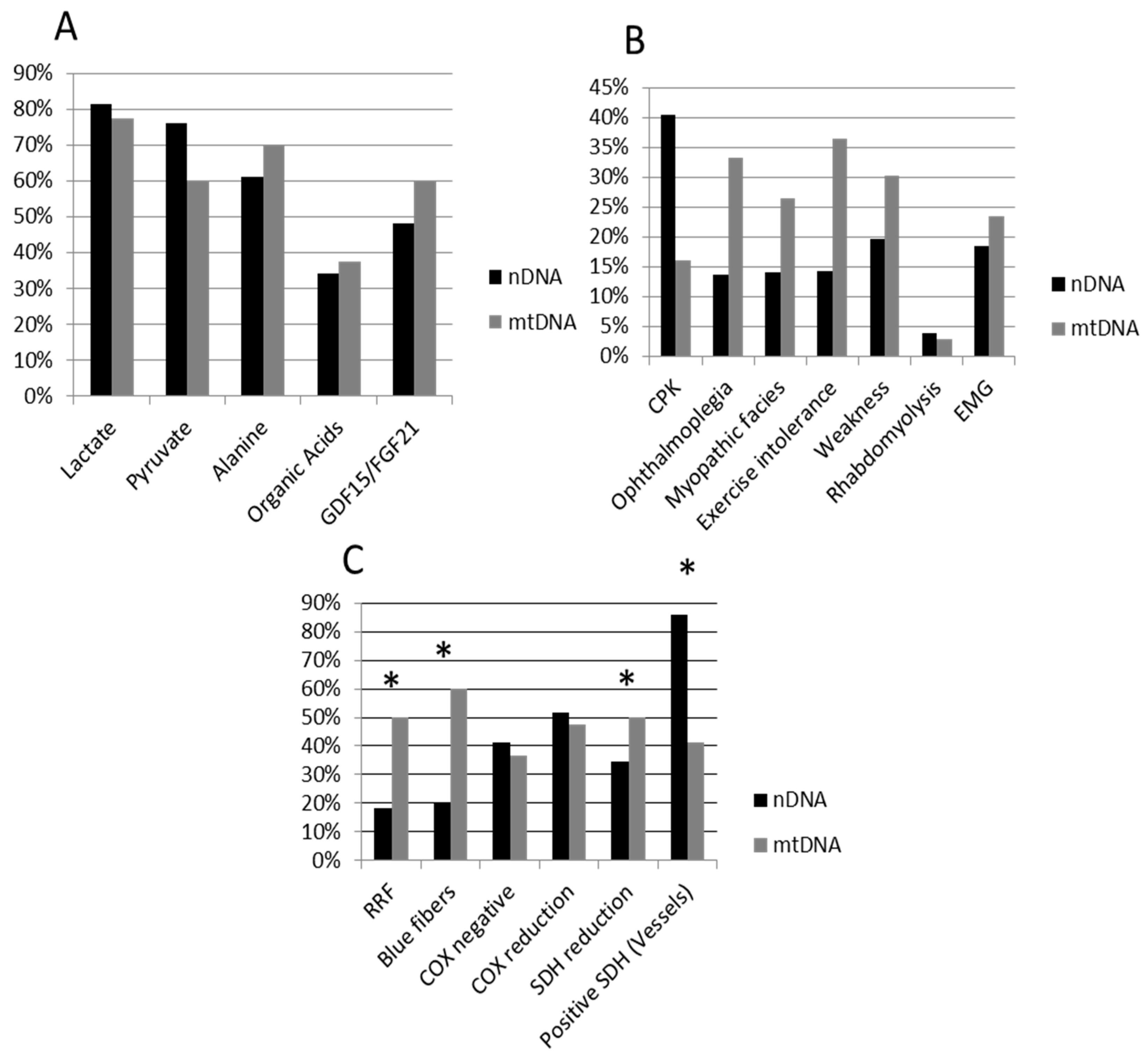
| Percentage (%) | Total Group (n = 103) | nDNA Patients (n = 59) | mtDNA Patients (n = 44) | Chi-Square |
|---|---|---|---|---|
| Sex (M/F) | 58.3/41.7 | 57.6/42.4 | 59.1/40.9 | n.s. |
| Exitus | 41.7 | 40.6 | 44.4 | n.s. |
| Survival (years) | 6.7 | 11.9 | n.s. | |
| Biomarkers | ||||
| Lactate | 79.8 | 81.5 | 77.5 | n.s |
| Pyruvate | 69.1 | 76.1 | 60.0 | n.s. |
| Alanine | 64.9 | 61.1 | 70.0 | n.s |
| Organic acids | 35.3 | 34.0 | 37.5 | n.s |
| GDF-15/FGF-21 | 51.4 | 48.0 | 60.0 | n.s |
| Myopathy | ||||
| CPK | 28.3 | 37.1 | 16.0 | n.s. |
| Ophthalmoplegia | 23.4 | 15.9 | 33.3 | n.s. |
| Myopathic facies | 20.8 | 16.3 | 26.5 | n.s. |
| Exercise intolerance | 25.3 | 16.7 | 36.4 | n.s. |
| Weakness | 26.0 | 22.7 | 30.3 | n.s. |
| Rhabdomyolysis | 3.9 | 4.7 | 2.9 | n.s. |
| EMG (myopathic) | 19.9 | 18.5 | 23.5 | n.s. * |
| Histopathology | ||||
| RRF | 32.8 | 18.2 | 50.0 | 6.959 (p = 0.008) |
| Blue fibers | 36.0 | 20.0 | 60.0 | 8.333 (p = 0.004) |
| COX negative | 39.6 | 41.4 | 36.8 | n.s. |
| COX reduction | 50.0 | 51.7 | 47.4 | n.s. |
| SDH reduction | 22.4 | 34.5 | 5.0 | 5.910 (p = 0.015) |
| Positive SDH (Vessels) | 65.8 | 85.7 | 41.2 | 8.828 (p = 0.004) |
| Histopathology | Chi-Square | ||
| Positive | Negative | ||
| Myopathy | |||
| Ophthalmoplegia | 44.0 | 8.3 | 7.991 (p = 0.005) |
| Myopathic facies | 33.3 | 8.3 | 4.547 (p = 0.033) |
| Exercise intolerance | 45.8 | 4.5 | 10.148 (p = 0.001) |
| Weakness | 48.0 | 0 | 14.720 (p < 0.0001) |
| Muscular Involvement | Chi-Square | ||
| Yes | No | ||
| Histopathology | |||
| RRF | 63.6 | 13.8 | 13.609 (p < 0.0001) |
| Blue fibers | 65.0 | 18.5 | 10.505 (p = 0.001) |
| COX negative | 63.2 | 16.0 | 10.375 (p = 0.001) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jou, C.; Ortigoza-Escobar, J.D.; O’Callaghan, M.M.; Nascimento, A.; Darling, A.; Pias-Peleteiro, L.; Perez-Dueñas, B.; Pineda, M.; Codina, A.; Arjona, C.; et al. Muscle Involvement in a Large Cohort of Pediatric Patients with Genetic Diagnosis of Mitochondrial Disease. J. Clin. Med. 2019, 8, 68. https://doi.org/10.3390/jcm8010068
Jou C, Ortigoza-Escobar JD, O’Callaghan MM, Nascimento A, Darling A, Pias-Peleteiro L, Perez-Dueñas B, Pineda M, Codina A, Arjona C, et al. Muscle Involvement in a Large Cohort of Pediatric Patients with Genetic Diagnosis of Mitochondrial Disease. Journal of Clinical Medicine. 2019; 8(1):68. https://doi.org/10.3390/jcm8010068
Chicago/Turabian StyleJou, Cristina, Juan D. Ortigoza-Escobar, Maria M. O’Callaghan, Andres Nascimento, Alejandra Darling, Leticia Pias-Peleteiro, Belén Perez-Dueñas, Mercedes Pineda, Anna Codina, César Arjona, and et al. 2019. "Muscle Involvement in a Large Cohort of Pediatric Patients with Genetic Diagnosis of Mitochondrial Disease" Journal of Clinical Medicine 8, no. 1: 68. https://doi.org/10.3390/jcm8010068
APA StyleJou, C., Ortigoza-Escobar, J. D., O’Callaghan, M. M., Nascimento, A., Darling, A., Pias-Peleteiro, L., Perez-Dueñas, B., Pineda, M., Codina, A., Arjona, C., Armstrong, J., Palau, F., Ribes, A., Gort, L., Tort, F., Navas, P., Ruiz-Pesini, E., Emperador, S., Lopez-Gallardo, E., ... Artuch, R. (2019). Muscle Involvement in a Large Cohort of Pediatric Patients with Genetic Diagnosis of Mitochondrial Disease. Journal of Clinical Medicine, 8(1), 68. https://doi.org/10.3390/jcm8010068