
Frozen Shoulder as a Metabolic and Immune Disorder: Potential Roles of Leptin Resistance, JAK-STAT Dysregulation, and Fibrosis


{kind=link}
Abstract
1. Introduction
2. Scoping the Role of JAK-STAT Signaling and MERTK+ Macrophages in Inflammatory Fibrosis Resolution
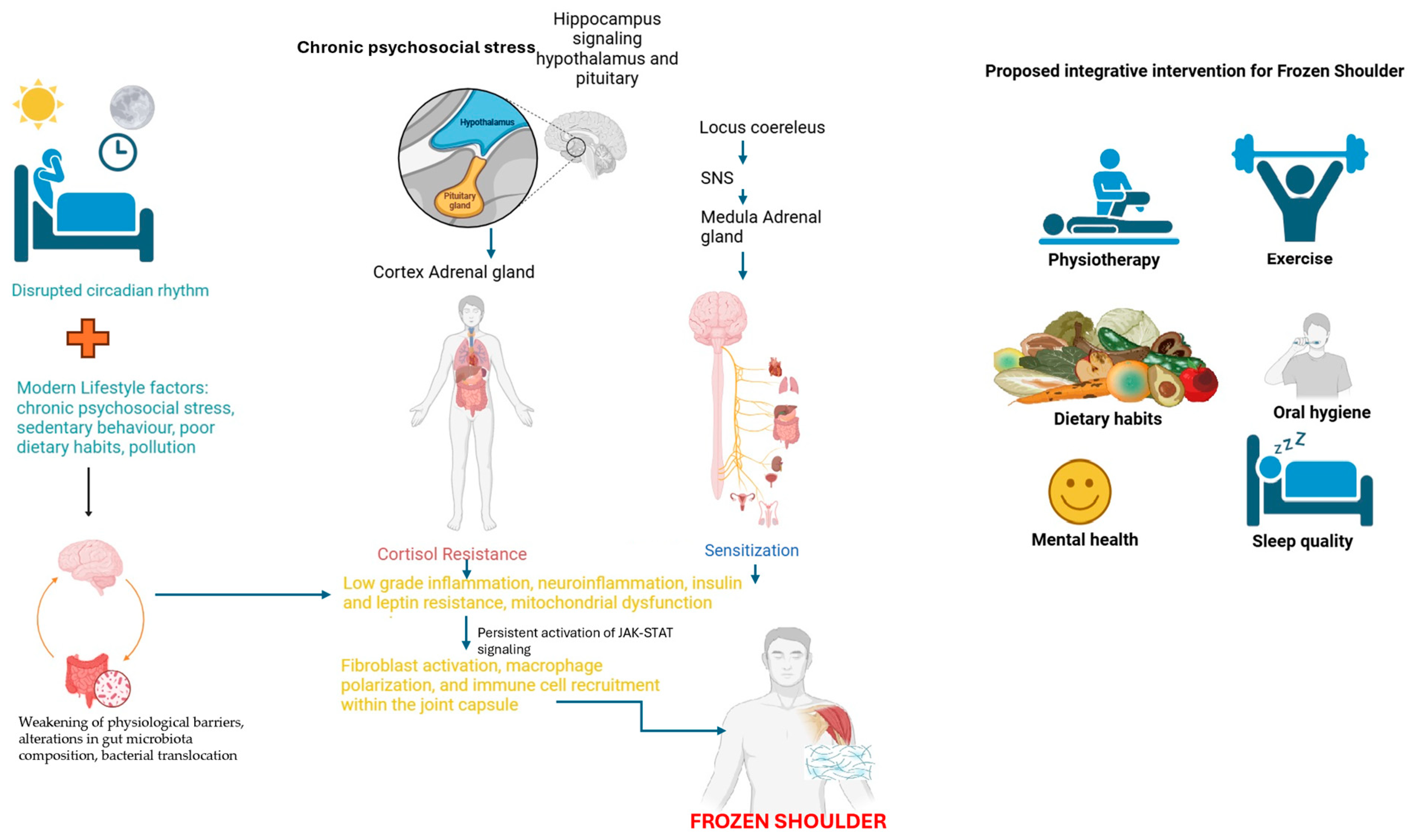
3. JAK-STAT, TGF-β, and Leptin Signaling in Frozen Shoulder
4. Chronic Neuroinflammation, Leptin Resistance, and JAK-STAT Signaling
5. Low-Grade Infections, Altered Microbiota, and Leptin Resistance
6. Discussion
7. Current Treatments and Future Directions
Conventional Therapies and Their Limitations
8. Emerging Therapeutic Approaches
8.1. Targeting Leptin Resistance and Metabolic Dysregulation
8.2. JAK-STAT Pathway as a Target
8.3. Gut Microbiota Modulation
9. Future Research Directions
9.1. Lifestyle Interventions as a Potential Therapeutic Approach in FS
9.2. Mitochondrial Dysfunction and FS Progression
9.3. Disruption of Circadian Rhythms
9.4. Leptin Resistance and JAK-STAT Pathway
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Abrassart, S.; Kolo, F.; Piotton, S.; Chih-Hao Chiu, J.; Stirling, P.; Hoffmeyer, P.; Lädermann, A. ‘Frozen shoulder’ is ill-defined. How can it be described better? EFORT Open Rev. 2020, 5, 273–279. [Google Scholar] [CrossRef]
- Brue, S.; Valentin, A.; Forssblad, M.; Werner, S.; Mikkelsen, C.; Cerulli, G. Idiopathic adhesive capsulitis of the shoulder: A review. Knee Surg. Sports Traumatol. Arthrosc. 2007, 15, 1048–1054. [Google Scholar] [CrossRef]
- Date, A.; Rahman, L. Frozen Shoulder: Overview of Clinical Presentation and Review of the Current Evidence Base for Management Strategies. Future Sci. OA 2020, 6, FSO647. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.G.; Struyf, F.; Verborgt, O.; Dueñas, L.; Balasch-Bernat, M.; Navarro-Ledesma, S.; Fernandez-Sanchez, M.; Luque-Suarez, A.; Lluch Girbes, E.; Meeus, M. Exploration of the clinical course and longitudinal correlations in frozen shoulder: The role of autonomic function, central pain processing, and psychological variables. A longitudinal multicenter prospective observational study. Musculoskelet. Sci. Pract. 2023, 67, 102857. [Google Scholar] [CrossRef]
- Mertens, M.G.; Meeus, M.; Verborgt, O.; Girbes, E.L.; Horno, S.M.-D.; Aguilar-Rodriguez, M.; Dueñas, L.; Navarro-Ledesma, S.; Fernandez-Sanchez, M.; Luque-Suarez, A.; et al. Exploration of the clinical course of frozen shoulder: A longitudinal multicenter prospective study of functional impairments. Braz. J. Phys. Ther. 2023, 27, 100539. [Google Scholar] [CrossRef] [PubMed]
- Lyne, S.A.; Goldblatt, F.M.; Shanahan, E.M. Living with a frozen shoulder—A phenomenological inquiry. BMC Musculoskelet. Disord. 2022, 23, 318. [Google Scholar] [CrossRef] [PubMed]
- Struyf, F.; Mertens, M.; Navarro-Ledesma, S. Causes of Shoulder Dysfunction in Diabetic Patients: A Review of Literature. Int. J. Environ. Res. Public Health 2022, 19, 6228. [Google Scholar] [CrossRef]
- De la Serna, D.; Navarro-Ledesma, S.; Alayón, F.; López, E.; Pruimboom, L. A Comprehensive View of Frozen Shoulder: A Mystery Syndrome. Front. Med. 2021, 8, 663703. [Google Scholar] [CrossRef]
- Navarro-Ledesma, S.; Hamed-Hamed, D.; Pruimboom, L. A new perspective of frozen shoulder pathology; the interplay between the brain and the immune system. Front. Physiol. 2024, 15, 1248612. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Vilariño-García, T.; Fernández-Riejos, P.; Martín-González, J.; Segura-Egea, J.J.; Sánchez-Margalet, V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 2017, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Nataf, S. Evolution, immunity and the emergence of brain superautoantigens. F1000Research 2017, 6, 171. [Google Scholar] [CrossRef]
- Sohnlein, P.; Muller, M.; Syren, K.; Hartmann, U.; Bohm, B.O.; Meinck, H.M.; Knip, M.; Akerblom, H.K.; Richter, W. Epitope spreading and a varying but not disease-specific GAD65 antibody response in Type I diabetes. Diabetologia 2000, 43, 210–217. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPAR γ and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 818945. [Google Scholar] [CrossRef] [PubMed]
- Thon, M.; Hosoi, T.; Ozawa, K. Possible Integrative Actions of Leptin and Insulin Signaling in the Hypothalamus Targeting Energy Homeostasis. Front. Endocrinol. 2016, 7, 138. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Khorshidian, N.; Khanniri, E.; Koushki, M.R.; Sohrabvandi, S.; Yousefi, M. An Overview of Antimicrobial Activity of Lysozyme and Its Functionality in Cheese. Front. Nutr. 2022, 9, 833618. [Google Scholar] [CrossRef]
- Hand, G.C.R.; Athanasou, N.A.; Matthews, T.; Carr, A.J. The pathology of frozen shoulder. J. Bone Jt. Surg. Br. 2007, 89-B, 928–932. [Google Scholar] [CrossRef]
- Ng, M.T.H.; Borst, R.; Gacaferi, H.; Davidson, S.; Ackerman, J.E.; Johnson, P.A.; Machado, C.C.; Reekie, I.; Attar, M.; Windell, D.; et al. A single cell atlas of frozen shoulder capsule identifies features associated with inflammatory fibrosis resolution. Nat. Commun. 2024, 15, 1394. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 2022, 13, 977485. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Rodriguez-Cuenca, S.; Rouault, C.; Figueroa-Juarez, E.; Schilbert, H.; Virtue, S.; Moreno-Navarrete, J.M.; Bidault, G.; Vázquez-Borrego, M.C.; Dias, A.R.; et al. Dysregulation of macrophage PEPD in obesity determines adipose tissue fibro-inflammation and insulin resistance. Nat. Metab. 2022, 4, 476–494. [Google Scholar] [CrossRef]
- Pruimboom, L.; Rocio, F.A.; Navarro-Ledesma, S. Psychoneuroimmunology in the Daily Clinic is Only Possible Within a Contextual Frame. In PsychoNeuroImmunology: Volume 1: Integration of Psychology, Neurology, and Immunology; Springer Nature: Cham, Switzerland, 2024; pp. 515–563. [Google Scholar]
- Sarapultsev, A.; Gusev, E.; Komelkova, M.; Utepova, I.; Luo, S.; Hu, D. JAK-STAT signaling in inflammation and stress-related diseases: Implications for therapeutic interventions. Mol. Biomed. 2023, 4, 40. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Gurzov, E.N.; Stanley, W.J.; Pappas, E.G.; Thomas, H.E.; Gough, D.J. The JAK/STAT pathway in obesity and diabetes. FEBS J. 2016, 283, 3002–3015. [Google Scholar] [CrossRef]
- Yang, R.; Barouch, L.A. Leptin Signaling and Obesity. Circ. Res. 2007, 101, 545–559. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.; Wong, M.-L.; Licinio, J. Leptin therapy, insulin sensitivity, and glucose homeostasis. Indian J. Endocrinol. Metab. 2012, 16, 549. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Zhang, T.; Xiong, J.; Lu, W.; Duan, L.; Zhu, W.; Wang, D. RNA-sequence analysis of samples from patients with idiopathic adhesive capsulitis. Mol. Med. Rep. 2017, 16, 7665–7672. [Google Scholar] [CrossRef]
- Burja, B.; Mertelj, T.; Frank-Bertoncelj, M. Hi-JAKi-ng Synovial Fibroblasts in Inflammatory Arthritis With JAK Inhibitors. Front. Med. 2020, 7, 124. [Google Scholar] [CrossRef]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.-S.; Zeng, J.; Mei, J.; Wang, P.-Y. JAK/STAT pathway: Extracellular signals, diseases, immunity, and therapeutic regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Xu, S.; Denton, C.P.; Abraham, D.J.; Ponticos, M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol. Biol. Cell 2018, 29, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Gibson, S.A.; Buckley, J.A.; Qin, H.; Benveniste, E.N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. 2018, 189, 4–13. [Google Scholar] [CrossRef]
- Kamal, N.; McGee, S.L.; Eng, K.; Brown, G.; Beattie, S.; Collier, F.; Gill, S.; Page, R.S. Transcriptomic analysis of adhesive capsulitis of the shoulder. J. Orthop. Res. 2020, 38, 2280–2289. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Qin, L.; Simons, M. TGFβ signaling pathways in human health and disease. Front. Mol. Biosci. 2023, 10, 1113061. [Google Scholar] [CrossRef]
- Giarratana, A.O.; Prendergast, C.M.; Salvatore, M.M.; Capaccione, K.M. TGF-β signaling: Critical nexus of fibrogenesis and cancer. J. Transl. Med. 2024, 22, 594. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Luo, F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules 2023, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef]
- Casado, M.E.; Collado-Pérez, R.; Frago, L.M.; Barrios, V. Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies. Int. J. Mol. Sci. 2023, 24, 1422. [Google Scholar] [CrossRef]
- Jain, M.; Budinger, G.R.S.; Lo, A.; Urich, D.; Rivera, S.E.; Ghosh, A.K.; Gonzalez, A.; Chiarella, S.E.; Marks, K.; Donnelly, H.K.; et al. Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-γ. Am. J. Respir. Crit. Care Med. 2011, 183, 1490–1498. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Syn, W.-K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef]
- Jain, M.; Singh, M.K.; Shyam, H.; Mishra, A.; Kumar, S.; Kumar, A.; Kushwaha, J. Role of JAK/STAT in the Neuroinflammation and its Association with Neurological Disorders. Ann. Neurosci. 2021, 28, 191–200. [Google Scholar] [CrossRef]
- Dragano, N.R.V.; Haddad-Tovolli, R.; Velloso, L.A. Leptin, Neuroinflammation and Obesity. Front. Horm. Res. 2017, 48, 84–96. [Google Scholar]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin Resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Philips, R.L.; Wang, Y.; Cheon, H.; Kanno, Y.; Gadina, M.; Sartorelli, V.; Horvath, C.M.; Darnell, J.E.; Stark, G.R.; O’Shea, J.J. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 2022, 185, 3857–3876. [Google Scholar] [CrossRef]
- Yang, H.; Cheng, H.; Dai, R.; Shang, L.; Zhang, X.; Wen, H. Macrophage polarization in tissue fibrosis. PeerJ 2023, 11, e16092. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Yao, Q.; Gu, X.; Shi, Q.; Yuan, X.; Chu, Q.; Bao, Z.; Lu, J.; Li, L. Evolving cognition of the JAK-STAT signaling pathway: Autoimmune disorders and cancer. Signal Transduct. Target. Ther. 2023, 8, 204. [Google Scholar] [CrossRef]
- Xia, T.; Zhang, M.; Lei, W.; Yang, R.; Fu, S.; Fan, Z.; Yang, Y.; Zhang, T. Advances in the role of STAT3 in macrophage polarization. Front. Immunol. 2023, 14, 1160719. [Google Scholar] [CrossRef]
- Li, D.; Li, D.; Wang, Z.; Li, J.; Shahzad, K.A.; Wang, Y.; Tan, F. Signaling pathways activated and regulated by stem cell-derived exosome therapy. Cell Biosci. 2024, 14, 105. [Google Scholar] [CrossRef]
- Bjørbæk, C.; Lavery, H.J.; Bates, S.H.; Olson, R.K.; Davis, S.M.; Flier, J.S.; Myers, M.G. SOCS3 Mediates Feedback Inhibition of the Leptin Receptor via Tyr985. J. Biol. Chem. 2000, 275, 40649–40657. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, C.M.; Hövelmeyer, N.; Wunderlich, F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAK-STAT 2013, 2, e23878. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; O’Neill, H.M.; Sylow, L.; Honeyman, J.; Hewitt, K.A.; Palanivel, R.; Fullerton, M.D.; Öberg, L.; Balendran, A.; Galic, S.; et al. Deletion of Skeletal Muscle SOCS3 Prevents Insulin Resistance in Obesity. Diabetes 2013, 62, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Brindisino, F.; Minnucci, S.; Sergi, G.; Lorusso, M.; Struyf, F.; Innocenti, T. Does the psychological profile of a patient with frozen shoulder predict future outcome? A systematic review. Physiother. Res. Int. 2024, 29, e2056. [Google Scholar] [CrossRef]
- Perry, R.J.; Shulman, G.I. The Role of Leptin in Maintaining Plasma Glucose During Starvation. Postdoc J. 2018, 6, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin Functions in Infectious Diseases. Front. Immunol. 2018, 9, 2741. [Google Scholar] [CrossRef] [PubMed]
- Alti, D.; Sambamurthy, C.; Kalangi, S.K. Emergence of Leptin in Infection and Immunity: Scope and Challenges in Vaccines Formulation. Front. Cell. Infect. Microbiol. 2018, 8, 147. [Google Scholar] [CrossRef]
- Faggioni, R.; Moser, A.; Feingold, K.R.; Grunfeld, C. Reduced Leptin Levels in Starvation Increase Susceptibility to Endotoxic Shock. Am. J. Pathol. 2000, 156, 1781–1787. [Google Scholar] [CrossRef]
- Oishi, K.; Hashimoto, C. Short-term time-restricted feeding during the resting phase is sufficient to induce leptin resistance that contributes to development of obesity and metabolic disorders in mice. Chronobiol. Int. 2018, 35, 1576–1594. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, F.; García-Macedo, R.; Alarcón-Aguilar, F.; Cruz, M. Adipocitokines, adipose tissue and its relationship with immune system cells. Gac. Med. Mex. 2005, 141, 505–512. [Google Scholar] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Birlutiu, V.; Boicean, L.C. Serum leptin level as a diagnostic and prognostic marker in infectious diseases and sepsis. Medicine 2021, 100, e25720. [Google Scholar] [CrossRef]
- Kiernan, K.; MacIver, N.J. The Role of the Adipokine Leptin in Immune Cell Function in Health and Disease. Front. Immunol. 2021, 11, 622468. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Franco, J.-S.; Amaya-Amaya, J.; Anaya, J.-M. Thyroid disease and autoimmune diseases. In Autoimmunity: From Bench to Bedside; Anaya, J.-M., Shoenfeld, Y., Rojas-Villarraga, A., Levy, R.A., Cervera, R., Eds.; El Rosario University Press: Bogotá, Colombia, 2013. [Google Scholar]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, T.; Liu, H. Leptin signaling and its central role in energy homeostasis. Front. Neurosci. 2023, 17, 1238528. [Google Scholar] [CrossRef] [PubMed]
- Czaja-Stolc, S.; Potrykus, M.; Stankiewicz, M.; Kaska, Ł.; Małgorzewicz, S. Pro-Inflammatory Profile of Adipokines in Obesity Contributes to Pathogenesis, Nutritional Disorders, and Cardiovascular Risk in Chronic Kidney Disease. Nutrients 2022, 14, 1457. [Google Scholar] [CrossRef]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A. V The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.-S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Islam, M.R.; Arthur, S.; Haynes, J.; Butts, M.R.; Nepal, N.; Sundaram, U. The Role of Gut Microbiota and Metabolites in Obesity-Associated Chronic Gastrointestinal Disorders. Nutrients 2022, 14, 624. [Google Scholar] [CrossRef]
- Vetrani, C.; Di Nisio, A.; Paschou, S.A.; Barrea, L.; Muscogiuri, G.; Graziadio, C.; Savastano, S.; Colao, A. From Gut Microbiota through Low-Grade Inflammation to Obesity: Key Players and Potential Targets. Nutrients 2022, 14, 2103. [Google Scholar] [CrossRef] [PubMed]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef] [PubMed]
- Beam, A.; Clinger, E.; Hao, L. Effect of Diet and Dietary Components on the Composition of the Gut Microbiota. Nutrients 2021, 13, 2795. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Tohumcu, E.; Raoul, P.; Fiorani, M.; Cintoni, M.; Mele, M.C.; Cammarota, G.; Gasbarrini, A.; Ianiro, G. The role of diet in shaping human gut microbiota. Best Pract. Res. Clin. Gastroenterol. 2023, 62–63, 101828. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Valentini, M.; Piermattei, A.; Di Sante, G.; Migliara, G.; Delogu, G.; Ria, F. Immunomodulation by Gut Microbiota: Role of Toll-Like Receptor Expressed by T Cells. J. Immunol. Res. 2014, 2014, 586939. [Google Scholar] [CrossRef]
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the Intricate Interaction among Toll-Like Receptors, Microbiota, and Intestinal Immunity Can Influence Gastrointestinal Pathology. J. Immunol. Res. 2015, 2015, 489821. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M.; et al. Responses of Gut Microbiota and Glucose and Lipid Metabolism to Prebiotics in Genetic Obese and Diet-Induced Leptin-Resistant Mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef]
- Piggott, D.A.; Higgins, Y.M.; Melia, M.T.; Ellis, B.; Carroll, K.C.; McFarland, E.G.; Auwaerter, P.G. Characteristics and Treatment Outcomes of Propionibacterium acnes Prosthetic Shoulder Infections in Adults. Open Forum Infect. Dis. 2015, 3, ofv191. [Google Scholar] [CrossRef] [PubMed]
- Bunker, T.D.; Boyd, M.; Gallacher, S.; Auckland, C.R.; Kitson, J.; Smith, C.D. Association between Propionibacterium acnes and frozen shoulder: A pilot study. Shoulder Elb. 2014, 6, 257–261. [Google Scholar] [CrossRef]
- Booker, S.J.; Boyd, M.; Gallacher, S.; Evans, J.P.; Auckland, C.; Kitson, J.; Thomas, W.; Smith, C.D. The colonisation of the glenohumeral joint by Propionibacterium acnes is not associated with frozen shoulder but is more likely to occur after an injection into the joint. Bone Jt. J. 2017, 99-B, 1067–1072. [Google Scholar] [CrossRef]
- Cher, J.Z.B.; Akbar, M.; Kitson, S.; Crowe, L.A.N.; Garcia-Melchor, E.; Hannah, S.C.; McLean, M.; Fazzi, U.G.; Kerr, S.C.; Murrell, G.A.C.; et al. Alarmins in Frozen Shoulder: A Molecular Association Between Inflammation and Pain. Am. J. Sports Med. 2018, 46, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Portillo, M.E.; Corvec, S.; Borens, O.; Trampuz, A. Propionibacterium acnes: An Underestimated Pathogen in Implant-Associated Infections. BioMed Res. Int. 2013, 2013, 804391. [Google Scholar] [CrossRef]
- Corvec, S.; Portillo, M.E.; Pasticci, B.M.; Borens, O.; Trampuz, A. Epidemiology and New Developments in the Diagnosis of Prosthetic Joint Infection. Int. J. Artif. Organs 2012, 35, 923–934. [Google Scholar] [CrossRef]
- Capoor, M.N.; Ruzicka, F.; Schmitz, J.E.; James, G.A.; Machackova, T.; Jancalek, R.; Smrcka, M.; Lipina, R.; Ahmed, F.S.; Alamin, T.F.; et al. Propionibacterium acnes biofilm is present in intervertebral discs of patients undergoing microdiscectomy. PLoS ONE 2017, 12, e0174518. [Google Scholar] [CrossRef]
- Lavergne, V.; Malo, M.; Gaudelli, C.; Laprade, M.; Leduc, S.; Laflamme, P.; Rouleau, D.M. Clinical impact of positive Propionibacterium acnes cultures in orthopedic surgery. Orthop. Traumatol. Surg. Res. 2017, 103, 307–314. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia Associated with Toothbrushing and Dental Extraction. Circulation 2008, 117, 3118–3125. [Google Scholar] [CrossRef]
- Tang, G.; Wang, Z.; Chen, J.; Zhang, Z.; Qian, H.; Chen, Y. Latent infection of low-virulence anaerobic bacteria in degenerated lumbar intervertebral discs. BMC Musculoskelet. Disord. 2018, 19, 445. [Google Scholar] [CrossRef]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef]
- Pérez, S.; Rius-Pérez, S. Macrophage Polarization and Reprogramming in Acute Inflammation: A Redox Perspective. Antioxidants 2022, 11, 1394. [Google Scholar] [CrossRef] [PubMed]
- Lis-López, L.; Bauset, C.; Seco-Cervera, M.; Cosín-Roger, J. Is the Macrophage Phenotype Determinant for Fibrosis Development? Biomedicines 2021, 9, 1747. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.d.B.; Prodonoff, J.S.; de Aguiar, C.F.; Correa-Da-Silva, F.; Castoldi, A.; Bakker, N.v.T.; Davanzo, G.G.; Castelucci, B.; Pereira, J.A.d.S.; Curtis, J.; et al. Leptin Signaling Suppression in Macrophages Improves Immunometabolic Outcomes in Obesity. Diabetes 2022, 71, 1546–1561. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.; Pereira, J.A.d.S.; Palhinha, L.; Moraes-Vieira, P.M.M. Leptin in the regulation of the immunometabolism of adipose tissue-macrophages. J. Leukoc. Biol. 2019, 106, 703–716. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Pan, D.; Li, G.; Jiang, C.; Hu, J.; Hu, X. Regulatory mechanisms of macrophage polarization in adipose tissue. Front. Immunol. 2023, 14, 1149366. [Google Scholar] [CrossRef]
- Sun, J.-X.; Xu, X.-H.; Jin, L. Effects of Metabolism on Macrophage Polarization Under Different Disease Backgrounds. Front. Immunol. 2022, 13, 880286. [Google Scholar] [CrossRef]
- Matarese, G.; Moschos, S.; Mantzoros, C.S. Leptin in Immunology. J. Immunol. 2005, 174, 3137–3142. [Google Scholar] [CrossRef] [PubMed]
- Challoumas, D.; Biddle, M.; McLean, M.; Millar, N.L. Comparison of Treatments for Frozen Shoulder: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2029581. [Google Scholar] [CrossRef] [PubMed]
- de Luxán-Delgado, B.; Potes, Y.; Rubio-González, A.; Solano, J.J.; Boga, J.A.; Antuña, E.; Cachán-Vega, C.; Bermejo-Millo, J.C.; Menéndez-Coto, N.; García-González, C.; et al. Melatonin Alleviates Liver Mitochondrial Dysfunction in Leptin-Deficient Mice. Int. J. Mol. Sci. 2024, 25, 8677. [Google Scholar] [CrossRef]
- Wang, W.; Shi, M.; Zhou, C.; Shi, Z.; Cai, X.; Lin, T.; Yan, S. Effectiveness of corticosteroid injections in adhesive capsulitis of shoulder: A meta-analysis. Medicine 2017, 96, e7529. [Google Scholar] [CrossRef]
- Gaujoux-Viala, C.; Dougados, M.; Gossec, L. Efficacy and safety of steroid injections for shoulder and elbow tendonitis: A meta-analysis of randomised controlled trials. Ann. Rheum. Dis. 2009, 68, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, L.; Balasch-Bernat, M.; Aguilar-Rodríguez, M.; Struyf, F.; Meeus, M.; Lluch, E. A Manual Therapy and Home Stretching Program in Patients With Primary Frozen Shoulder Contracture Syndrome: A Case Series. J. Orthop. Sport. Phys. Ther. 2019, 49, 192–201. [Google Scholar] [CrossRef]
- Page, M.J.; Green, S.; Kramer, S.; Johnston, R.V.; McBain, B.; Chau, M.; Buchbinder, R. Manual therapy and exercise for adhesive capsulitis (frozen shoulder). Cochrane Database Syst. Rev. 2014, 2014, CD011275. [Google Scholar] [CrossRef]
- Çelik, D.; Kaya Mutlu, E. Does adding mobilization to stretching improve outcomes for people with frozen shoulder? A randomized controlled clinical trial. Clin. Rehabil. 2016, 30, 786–794. [Google Scholar] [CrossRef]
- Wagner, E.R.; Farley, K.X.; Higgins, I.; Wilson, J.M.; Daly, C.A.; Gottschalk, M.B. The incidence of shoulder arthroplasty: Rise and future projections compared with hip and knee arthroplasty. J. Shoulder Elb. Surg. 2020, 29, 2601–2609. [Google Scholar] [CrossRef]
- Kraal, T.; The, B.; Boer, R.; van den Borne, M.P.; Koenraadt, K.; Goossens, P.; Eygendaal, D. Manipulation under anesthesia versus physiotherapy treatment in stage two of a frozen shoulder: A study protocol for a randomized controlled trial. BMC Musculoskelet. Disord. 2017, 18, 412. [Google Scholar] [CrossRef]
- Zabeau, L.; Lavens, D.; Peelman, F.; Eyckerman, S.; Vandekerckhove, J.; Tavernier, J. The ins and outs of leptin receptor activation. FEBS Lett. 2003, 546, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Lee, E.B.; Kaplan, I.V.; Kwok, K.; Geier, J.; Benda, B.; Soma, K.; Wang, L.; Riese, R. Tofacitinib, an oral Janus kinase inhibitor: Analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann. Rheum. Dis. 2016, 75, 831–841. [Google Scholar] [CrossRef]
- Reyes Diaz, R.A.; Cruz Lara, N.M. Papel de la microbiota intestinal en el desarrollo del síndrome metabólico: Revisión narrativa. Rev. Nutr. Clínica y Metab. 2024, 7, 45–54. [Google Scholar] [CrossRef]
- García-Ríos, A.; Camargo Garcia, A.; Perez-Jimenez, F.; Perez-Martinez, P. Microbiota intestinal: ¿un nuevo protagonista en el riesgo de enfermedad cardiovascular? Clínica e Investig. en Arterioscler. 2019, 31, 178–185. [Google Scholar] [CrossRef]
- Mallappa, R.; Rokana, N.; Duary, R.; Panwar, H.; Batish, V.; Grover, S. Management of metabolic syndrome through probiotic and prebiotic interventions. Indian J. Endocrinol. Metab. 2012, 16, 20. [Google Scholar] [CrossRef]
- Uchida, F.; Oh, S.; Shida, T.; Suzuki, H.; Yamagata, K.; Mizokami, Y.; Bukawa, H.; Tanaka, K.; Shoda, J. Effects of Exercise on the Oral Microbiota and Saliva of Patients with Non-Alcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2021, 18, 3470. [Google Scholar] [CrossRef]
- Hamed Hamed, D.; Struyf, F.; Pruimboom, L.; Navarro-Ledesma, S. Efficacy of combined strategies of physical activity, diet and sleep disorders as treatment in patients with chronic shoulder pain. A systematic review. Front. Physiol. 2023, 14, 1221807. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Ledesma, S.; Hamed-Hamed, D.; González-Muñoz, A.; Pruimboom, L. Physical Activity, Insulin Resistance and Cancer: A Systematic Review. Cancers 2024, 16, 656. [Google Scholar] [CrossRef]
- Navarro-Ledesma, S.; Hamed-Hamed, D.; Gonzalez-Muñoz, A.; Pruimboom, L. Impact of physical therapy techniques and common interventions on sleep quality in patients with chronic pain: A systematic review. Sleep Med. Rev. 2024, 76, 101937. [Google Scholar] [CrossRef]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef] [PubMed]
- Ansarin, A.; Mahdavi, A.M.; Javadivala, Z.; Shanehbandi, D.; Zarredar, H.; Ansarin, K. The cross-talk between leptin and circadian rhythm signaling proteins in physiological processes: A systematic review. Mol. Biol. Rep. 2023, 50, 10427–10443. [Google Scholar] [CrossRef]
- Maury, E. Off the Clock: From Circadian Disruption to Metabolic Disease. Int. J. Mol. Sci. 2019, 20, 1597. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Civelek, E.; Ozturk Civelek, D.; Akyel, Y.K.; Kaleli Durman, D.; Okyar, A. Circadian Dysfunction in Adipose Tissue: Chronotherapy in Metabolic Diseases. Biology 2023, 12, 1077. [Google Scholar] [CrossRef]
- Dibner, C.; Gachon, F. Circadian Dysfunction and Obesity: Is Leptin the Missing Link? Cell Metab. 2015, 22, 359–360. [Google Scholar] [CrossRef]
- Dessie, G.; Ayelign, B.; Akalu, Y.; Shibabaw, T.; Molla, M.D. Effect of Leptin on Chronic Inflammatory Disorders: Insights to Therapeutic Target to Prevent Further Cardiovascular Complication. Diabetes. Metab. Syndr. Obes. 2021, 14, 3307–3322. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro-Ledesma, S. Frozen Shoulder as a Metabolic and Immune Disorder: Potential Roles of Leptin Resistance, JAK-STAT Dysregulation, and Fibrosis. J. Clin. Med. 2025, 14, 1780. https://doi.org/10.3390/jcm14051780
Navarro-Ledesma S. Frozen Shoulder as a Metabolic and Immune Disorder: Potential Roles of Leptin Resistance, JAK-STAT Dysregulation, and Fibrosis. Journal of Clinical Medicine. 2025; 14(5):1780. https://doi.org/10.3390/jcm14051780
Chicago/Turabian StyleNavarro-Ledesma, Santiago. 2025. "Frozen Shoulder as a Metabolic and Immune Disorder: Potential Roles of Leptin Resistance, JAK-STAT Dysregulation, and Fibrosis" Journal of Clinical Medicine 14, no. 5: 1780. https://doi.org/10.3390/jcm14051780
APA StyleNavarro-Ledesma, S. (2025). Frozen Shoulder as a Metabolic and Immune Disorder: Potential Roles of Leptin Resistance, JAK-STAT Dysregulation, and Fibrosis. Journal of Clinical Medicine, 14(5), 1780. https://doi.org/10.3390/jcm14051780