
The Role of MCM9 in the Etiology of Sertoli Cell-Only Syndrome and Premature Ovarian Insufficiency

 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Characteristics in Females
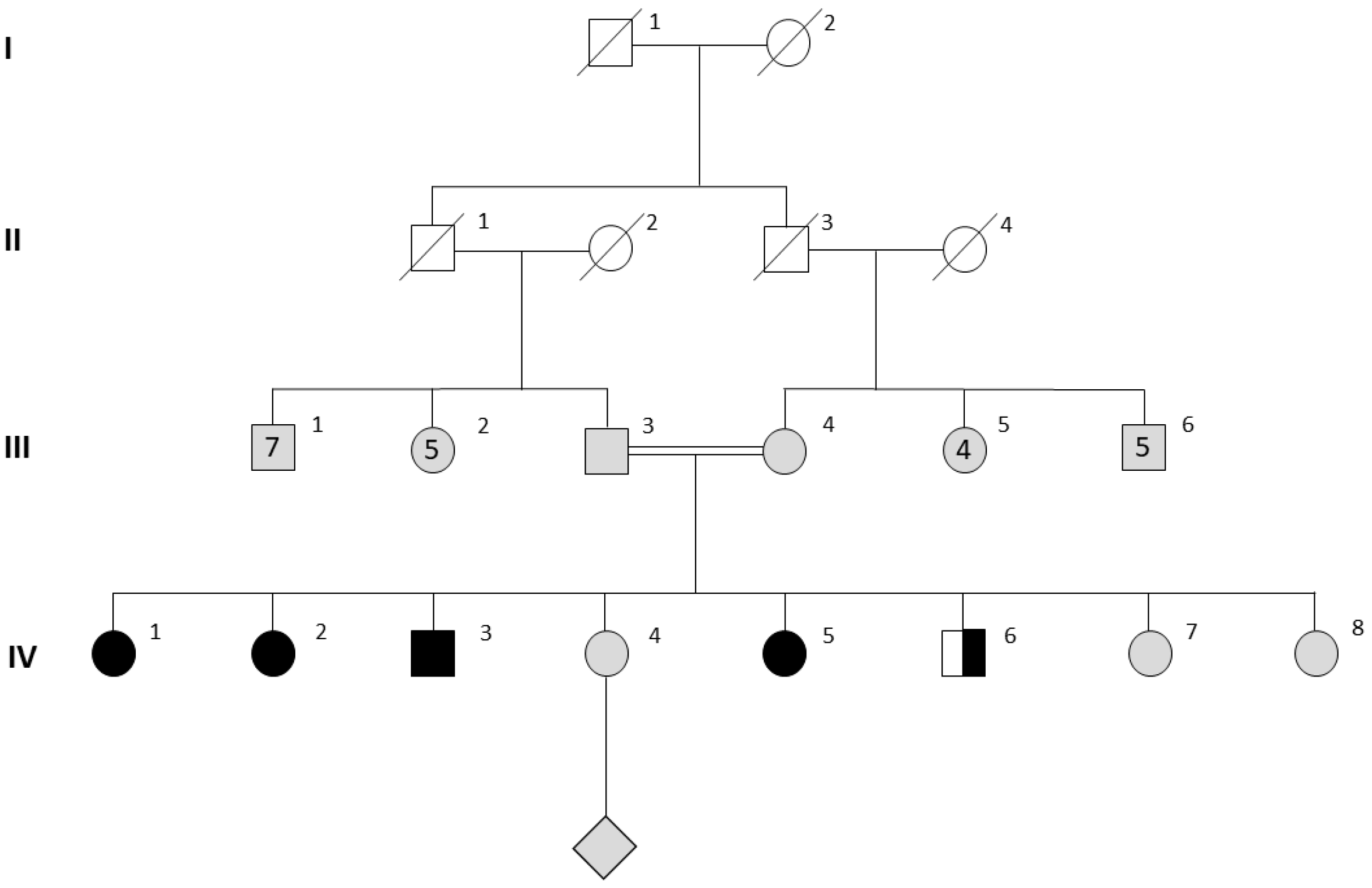
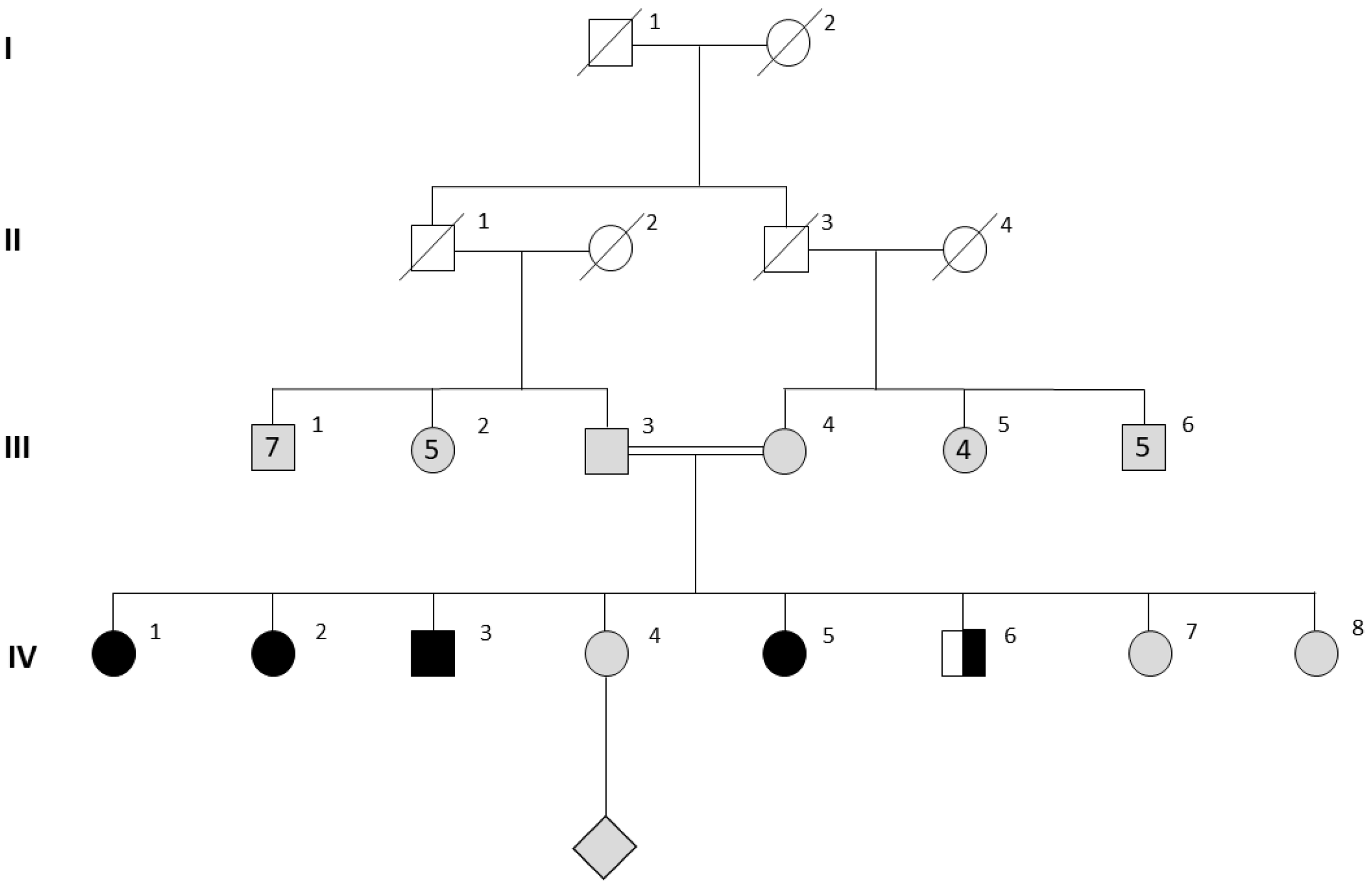
3.2. Genetic Investigations
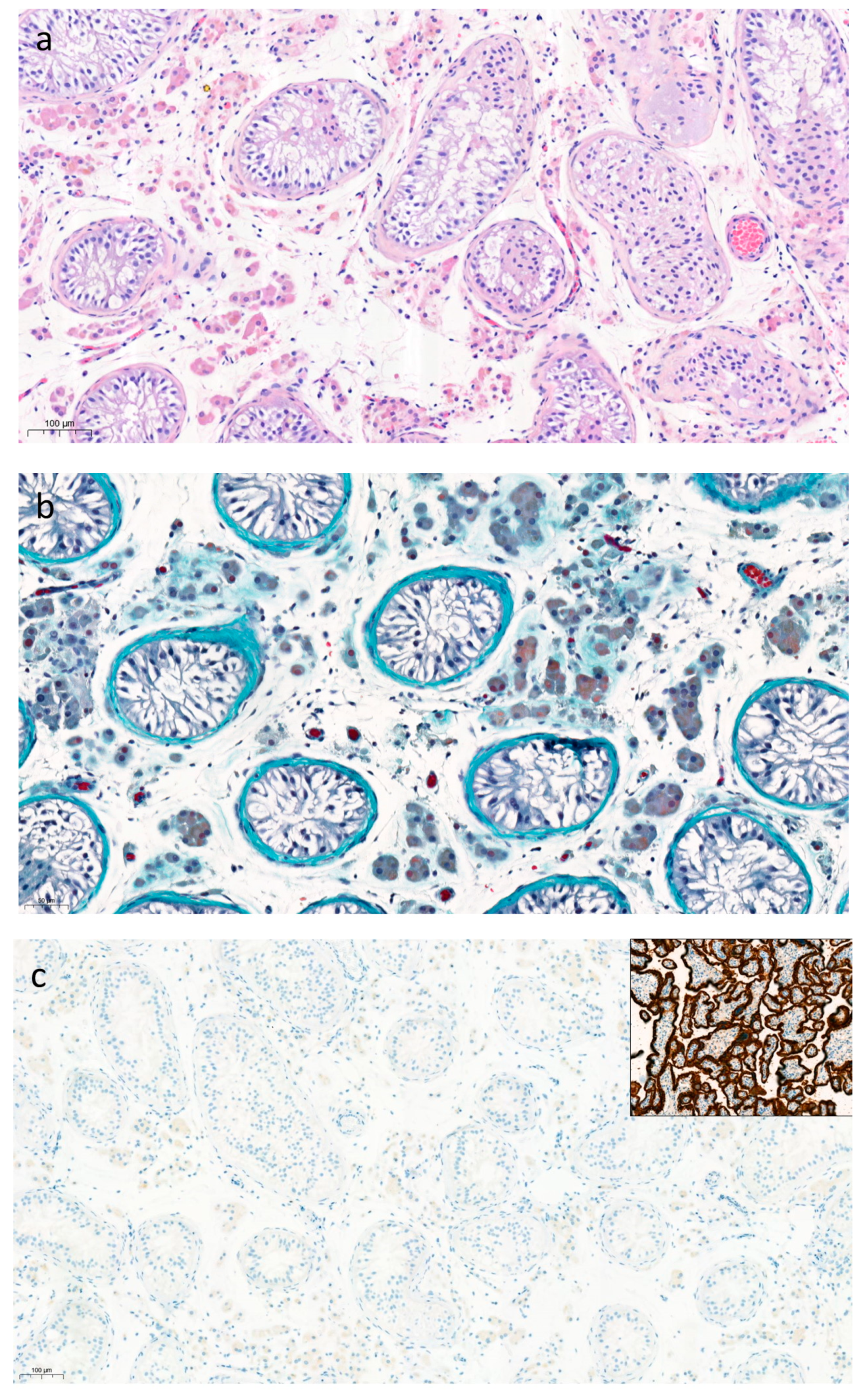
3.3. Male Characteristics and Testicular Histopathology
3.4. Bone Phenotype and Investigations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boivin, J.; Bunting, L.; Collins, J.A.; Nygren, K.G. International Estimates of Infertility Prevalence and Treatment-Seeking: Potential Need and Demand for Infertility Medical Care. Hum. Reprod. 2007, 22, 1506–1512. [Google Scholar] [CrossRef] [Green Version]
- Volozonoka, L.; Miskova, A.; Kornejeva, L.; Kempa, I.; Bargatina, V.; Gailite, L. A Systematic Review and Standardized Clinical Validity Assessment of Genes Involved in Female Reproductive Failure. Reproduction 2022, 163, 351–363. [Google Scholar] [CrossRef]
- Patel, D.P.; Jenkins, T.G.; Aston, K.I.; Guo, J.; Pastuszak, A.W.; Hanson, H.A.; Hotaling, J.M. Harnessing the Full Potential of Reproductive Genetics and Epigenetics for Male Infertility in the Era of “Big Data”. Fertil. Steril. 2020, 113, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Vogt, E.C.; Real, F.G.; Husebye, E.S.; Björnsdottir, S.; Benediktsdottir, B.; Bertelsen, R.J.; Demoly, P.; Franklin, K.A.; Gallastegui, L.S.d.A.; González, F.J.C.; et al. Premature Menopause and Autoimmune Primary Ovarian Insufficiency in Two International Multi-Center Cohorts. Endocr. Connect. 2022, 11, e220024. [Google Scholar] [CrossRef]
- Webber, L.; Davies, M.; Anderson, R.; Bartlett, J.; Braat, D.; Cartwright, B.; Cifkova, R.; De Muinck Keizer-Schrama, S.; Hogervorst, E.; Janse, F.; et al. ESHRE Guideline: Management of Women with Premature Ovarian Insufficiency. Hum. Reprod. 2016, 31, 926–937. [Google Scholar] [CrossRef]
- Yang, Q.; Mumusoglu, S.; Qin, Y.; Sun, Y.; Hsueh, A.J. A Kaleidoscopic View of Ovarian Genes Associated with Premature Ovarian Insufficiency and Senescence. FASEB J. 2021, 35, e21753. [Google Scholar] [CrossRef]
- Agarwal, A.; Mulgund, A.; Hamada, A.; Chyatte, M.R. A Unique View on Male Infertility around the Globe. Reprod. Biol. Endocrinol. 2015, 13, 37. [Google Scholar] [CrossRef] [Green Version]
- Krausz, C.; Cioppi, F. Genetic Factors of Non-Obstructive Azoospermia: Consequences on Patients’ and Offspring Health. J. Clin. Med. 2021, 10, 4009. [Google Scholar] [CrossRef]
- Chen, S.; Wang, G.; Zheng, X.; Ge, S.; Dai, Y.; Ping, P.; Chen, X.; Liu, G.; Zhang, J.; Yang, Y.; et al. Whole-Exome Sequencing of a Large Chinese Azoospermia and Severe Oligospermia Cohort Identifies Novel Infertility Causative Variants and Genes. Hum. Mol. Genet. 2020, 29, 2451–2459. [Google Scholar] [CrossRef]
- Wyrwoll, M.J.; Köckerling, N.; Vockel, M.; Dicke, A.K.; Rotte, N.; Pohl, E.; Emich, J.; Wöste, M.; Ruckert, C.; Wabschke, R.; et al. Genetic Architecture of Azoospermia—Time to Advance the Standard of Care. Eur. Urol. 2022. [Google Scholar] [CrossRef]
- Bouali, N.; Francou, B.; Bouligand, J.; Imanci, D.; Dimassi, S.; Tosca, L.; Zaouali, M.; Mougou, S.; Young, J.; Saad, A.; et al. New MCM8 Mutation Associated with Premature Ovarian Insufficiency and Chromosomal Instability in a Highly Consanguineous Tunisian Family. Fertil. Steril. 2017, 108, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Kherraf, Z.E.; Cazin, C.; Bouker, A.; Fourati Ben Mustapha, S.; Hennebicq, S.; Septier, A.; Coutton, C.; Raymond, L.; Nouchy, M.; Thierry-Mieg, N.; et al. Whole-Exome Sequencing Improves the Diagnosis and Care of Men with Non-Obstructive Azoospermia. Am. J. Hum. Genet. 2022, 109, 508–517. [Google Scholar] [CrossRef]
- Park, J.; Long, D.T.; Lee, K.Y.; Abbas, T.; Shibata, E.; Negishi, M.; Luo, Y.; Schimenti, J.C.; Gambus, A.; Walter, J.C.; et al. The MCM8-MCM9 Complex Promotes RAD51 Recruitment at DNA Damage Sites To Facilitate Homologous Recombination. Mol. Cell. Biol. 2013, 33, 1632–1644. [Google Scholar] [CrossRef] [Green Version]
- Lutzmann, M.; Bernex, F.; da Costa de Jesus, C.; Hodroj, D.; Marty, C.; Plo, I.; Vainchenker, W.; Tosolini, M.; Forichon, L.; Bret, C.; et al. MCM8- and MCM9 Deficiencies Cause Lifelong Increased Hematopoietic DNA Damage Driving P53-Dependent Myeloid Tumors. Cell Rep. 2019, 28, 2851–2865.e4. [Google Scholar] [CrossRef]
- Lutzmann, M.; Grey, C.; Traver, S.; Ganier, O.; Maya-Mendoza, A.; Ranisavljevic, N.; Bernex, F.; Nishiyama, A.; Montel, N.; Gavois, E.; et al. MCM8- and MCM9-Deficient Mice Reveal Gametogenesis Defects and Genome Instability Due to Impaired Homologous Recombination. Mol. Cell 2012, 47, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, Y.; Aleme, O.; Peled-Perets, L.; Castellvi-Bel, S.; Nielsen, M.; Shalev, S.A. MCM9 Is Associated with Germline Predisposition to Early-Onset Cancer—Clinical Evidence. Npj Genomic Med. 2021, 6, 78. [Google Scholar] [CrossRef]
- Wood-Trageser, M.A.; Gurbuz, F.; Yatsenko, S.A.; Jeffries, E.P.; Kotan, L.D.; Surti, U.; Ketterer, D.M.; Matic, J.; Chipkin, J.; Jiang, H.; et al. MCM9 Mutations Are Associated with Ovarian Failure, Short Stature, and Chromosomal Instability. Am. J. Hum. Genet. 2014, 95, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Rossetti, R.; Moleri, S.; Guizzardi, F.; Gentilini, D.; Libera, L.; Marozzi, A.; Moretti, C.; Brancati, F.; Bonomi, M.; Persani, L. Targeted Next-Generation Sequencing Indicates a Frequent Oligogenic Involvement in Primary Ovarian Insufficiency Onset. Front. Endocrinol. 2021, 12, 664645. [Google Scholar] [CrossRef]
- Desai, S.; Rajkovic, A. Genetics of Reproductive Aging from Gonadal Dysgenesis through Menopause. Semin. Reprod. Med. 2017, 35, 147–159. [Google Scholar] [CrossRef]
- McLachlan, R.I.; Rajpert-De Meyts, E.; Hoei-Hansen, C.E.; de Kretser, D.M.; Skakkebaek, N.E. Histological Evaluation of the Human Testis—Approaches to Optimizing the Clinical Value of the Assessment: Mini Review. Hum. Reprod. 2007, 22, 2–16. [Google Scholar] [CrossRef]
- Ooba, T.; Ishikawa, T.; Yamaguchi, K.; Kondo, Y.; Sakamoto, Y.; Fujisawa, M. Expression and Distribution of Laminin Chains in the Testis for Patients with Azoospermia. J. Androl. 2008, 29, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Fauchereau, F.; Shalev, S.; Chervinsky, E.; Beck-Fruchter, R.; Legois, B.; Fellous, M.; Caburet, S.; Veitia, R.A. A Non-Sense MCM9 Mutation in a Familial Case of Primary Ovarian Insufficiency. Clin. Genet. 2016, 89, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Touraine, P.; Desai, S.; Humphreys, G.; Jiang, H.; Yatsenko, A.; Rajkovic, A. Gene Variants Identified by Whole-Exome Sequencing in 33 French Women with Premature Ovarian Insufficiency. J. Assist. Reprod. Genet. 2019, 36, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Jolly, A.; Bayram, Y.; Turan, S.; Aycan, Z.; Tos, T.; Abali, Z.Y.; Hacihamdioglu, B.; Coban Akdemir, Z.H.; Hijazi, H.; Bas, S.; et al. Exome Sequencing of a Primary Ovarian Insufficiency Cohort Reveals Common Molecular Etiologies for a Spectrum of Disease. J. Clin. Endocrinol. Metab. 2019, 104, 3049–3067. [Google Scholar] [CrossRef] [PubMed]
- Traver, S.; Coulombe, P.; Peiffer, I.; Hutchins, J.R.A.; Kitzmann, M.; Latreille, D.; Méchali, M. MCM9 Is Required for Mammalian DNA Mismatch Repair. Mol. Cell 2015, 59, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Hartford, S.A.; Luo, Y.; Southard, T.L.; Min, I.M.; Lis, J.T.; Schimenti, J.C. Minichromosome Maintenance Helicase Paralog MCM9 Is Dispensible for DNA Replication but Functions in Germ-Line Stem Cells and Tumor Suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 17702–17707. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Schimenti, J.C. MCM9 Deficiency Delays Primordial Germ Cell Proliferation Independent of the ATM Pathway. Genesis 2015, 53, 678–684. [Google Scholar] [CrossRef]
- Golubicki, M.; Bonjoch, L.; Acuña-Ochoa, J.G.; Díaz-Gay, M.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline Biallelic Mcm8 Variants Are Associated with Early-Onset Lynch-like Syndrome. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Collet, C.; Ostertag, A.; Ricquebourg, M.; Delecourt, M.; Tueur, G.; Isidor, B.; Guillot, P.; Schaefer, E.; Javier, R.M.; Funck-Brentano, T.; et al. Primary Osteoporosis in Young Adults: Genetic Basis and Identification of Novel Variants in Causal Genes. JBMR Plus 2018, 2, 12–21. [Google Scholar] [CrossRef]
- Rocha-Braz, M.G.M.; França, M.M.; Fernandes, A.M.; Lerario, A.M.; Zanardo, E.A.; de Santana, L.S.; Kulikowski, L.D.; Martin, R.M.; Mendonca, B.B.; Ferraz-De-Souza, B. Comprehensive Genetic Analysis of 128 Candidate Genes in a Cohort with Idiopathic, Severe, or Familial Osteoporosis. J. Endocr. Soc. 2020, 4, bvaa148. [Google Scholar] [CrossRef]
- Foresta, C.; Strapazzon, G.; De Toni, L.; Perilli, L.; Di Mambro, A.; Muciaccia, B.; Sartori, L.; Selice, R. Bone Mineral Density and Testicular Failure: Evidence for a Role of Vitamin D 25-Hydroxylase in Human Testis. J. Clin. Endocrinol. Metab. 2011, 96, E646–E652. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| IV.1 | IV.5 | IV.2 | IV.3 | IV.6 | III.3 | III.4 | |
|---|---|---|---|---|---|---|---|
| Gender | female | female | female | male | male | male | female |
| MCM9 status | c.394C>T, p.Arg132* homozyg. | c.394C>T, p.Arg132* homozyg. | c.394C>T, p.Arg132* homozyg. | c.394C>T, p.Arg132* homozyg. | c.394C>T, p.Arg132* heterozyg. | NA | NA |
| Pubertal status | Absent | Absent | Absent | Normal | Normal | Normal; fertile | Normal; fertile |
| FSH (U/L) | 117 | 112 | 106 | 5 | 4.5 | NA | NA |
| LH (U/L) | 41 | 24 | 56 | 4.8 | 3.8 | NA | NA |
| Estradiol (ng/L) | <24 | <24 | <24 | 31 | NA | NA | NA |
| Testosterone (nmol/L) | 1.36 | 1.2 | 0.5 | 23.7 | 28.8 | NA | NA |
| L spine Z-score | B/L 18 mo. −4.2 −3.2 | B/L 18 mo −4.8 −3.0 | −3.4 | −3.1 | −1.1 | −3.0 | −1.2 |
| Fem neck Z-score | B/L 18 mo. −3.7 −2.9 | B/L 18 mo −2.9 −1.7 | −2.6 | −1.7 | −2.2 | −2.3 | −0.7 |
| Sperm analysis | - | - | - | Non-obstructive azoospermia | ND | ND | - |
| COL1A2 status | c.106G/G | c.106G/G | c.106G/A | c.106G/A | c.106G/A | c.106G/G | c.106G/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potorac, I.; Laterre, M.; Malaise, O.; Nechifor, V.; Fasquelle, C.; Colleye, O.; Detrembleur, N.; Verdin, H.; Symoens, S.; De Baere, E.; et al. The Role of MCM9 in the Etiology of Sertoli Cell-Only Syndrome and Premature Ovarian Insufficiency. J. Clin. Med. 2023, 12, 990. https://doi.org/10.3390/jcm12030990
Potorac I, Laterre M, Malaise O, Nechifor V, Fasquelle C, Colleye O, Detrembleur N, Verdin H, Symoens S, De Baere E, et al. The Role of MCM9 in the Etiology of Sertoli Cell-Only Syndrome and Premature Ovarian Insufficiency. Journal of Clinical Medicine. 2023; 12(3):990. https://doi.org/10.3390/jcm12030990
Chicago/Turabian StylePotorac, Iulia, Marie Laterre, Olivier Malaise, Vlad Nechifor, Corinne Fasquelle, Orphal Colleye, Nancy Detrembleur, Hannah Verdin, Sofie Symoens, Elfride De Baere, and et al. 2023. "The Role of MCM9 in the Etiology of Sertoli Cell-Only Syndrome and Premature Ovarian Insufficiency" Journal of Clinical Medicine 12, no. 3: 990. https://doi.org/10.3390/jcm12030990
APA StylePotorac, I., Laterre, M., Malaise, O., Nechifor, V., Fasquelle, C., Colleye, O., Detrembleur, N., Verdin, H., Symoens, S., De Baere, E., Daly, A. F., Bours, V., Pétrossians, P., & Pintiaux, A. (2023). The Role of MCM9 in the Etiology of Sertoli Cell-Only Syndrome and Premature Ovarian Insufficiency. Journal of Clinical Medicine, 12(3), 990. https://doi.org/10.3390/jcm12030990