
Molecular Biomarkers for the Diagnosis, Prognosis, and Pharmacodynamics of Spinal Muscular Atrophy

 , , , ,
, , , , 
Abstract
1. Spinal Muscular Atrophy
2. Spinal Muscular Atrophy Genetics
3. Spinal Muscular Atrophy Pathogenesis
4. Spinal Muscular Atrophy Treatment
5. Spinal Muscular Atrophy Molecular Biomarkers
5.1. Diagnosis and Prognosis of SMA Using Molecular Biomarkers

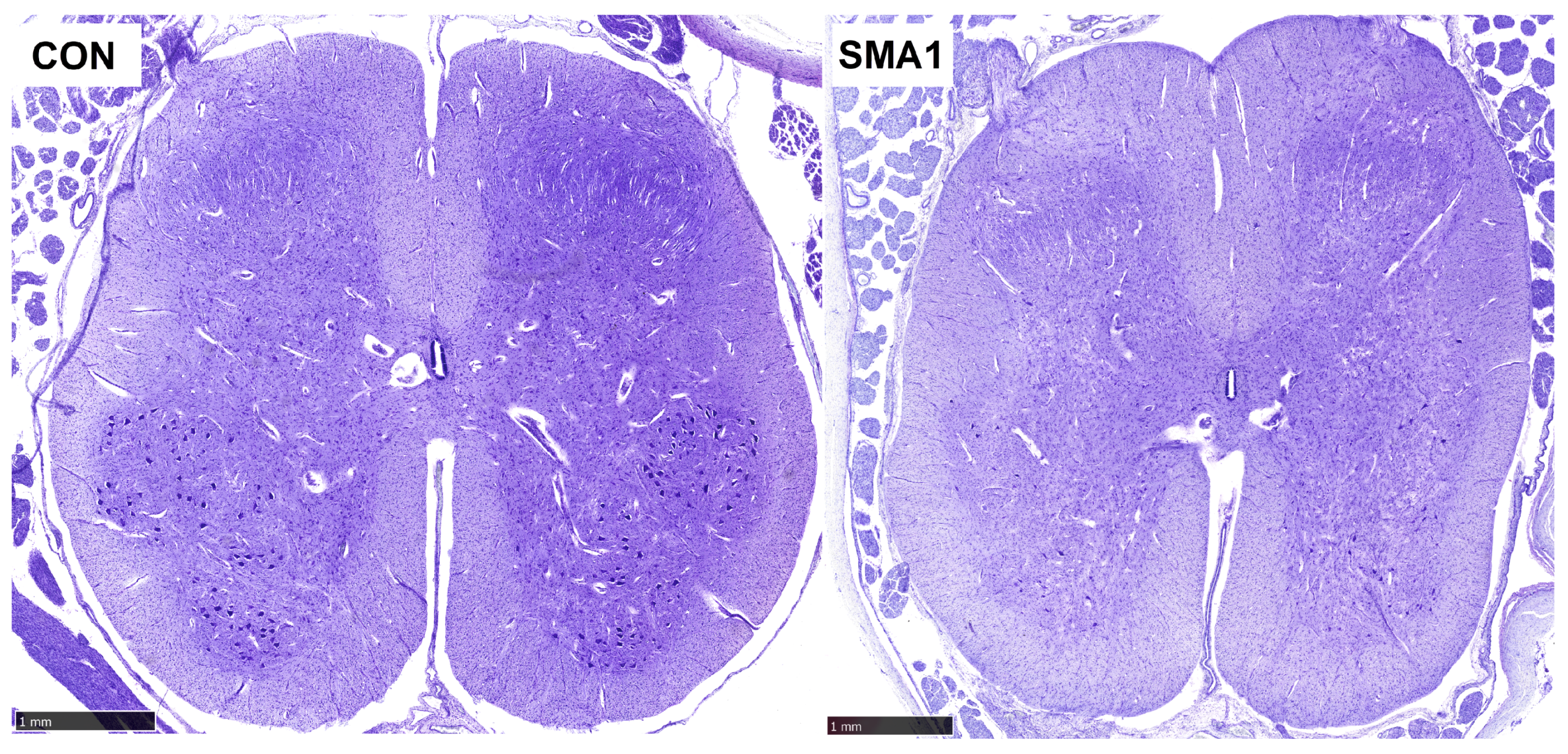{kind=link}
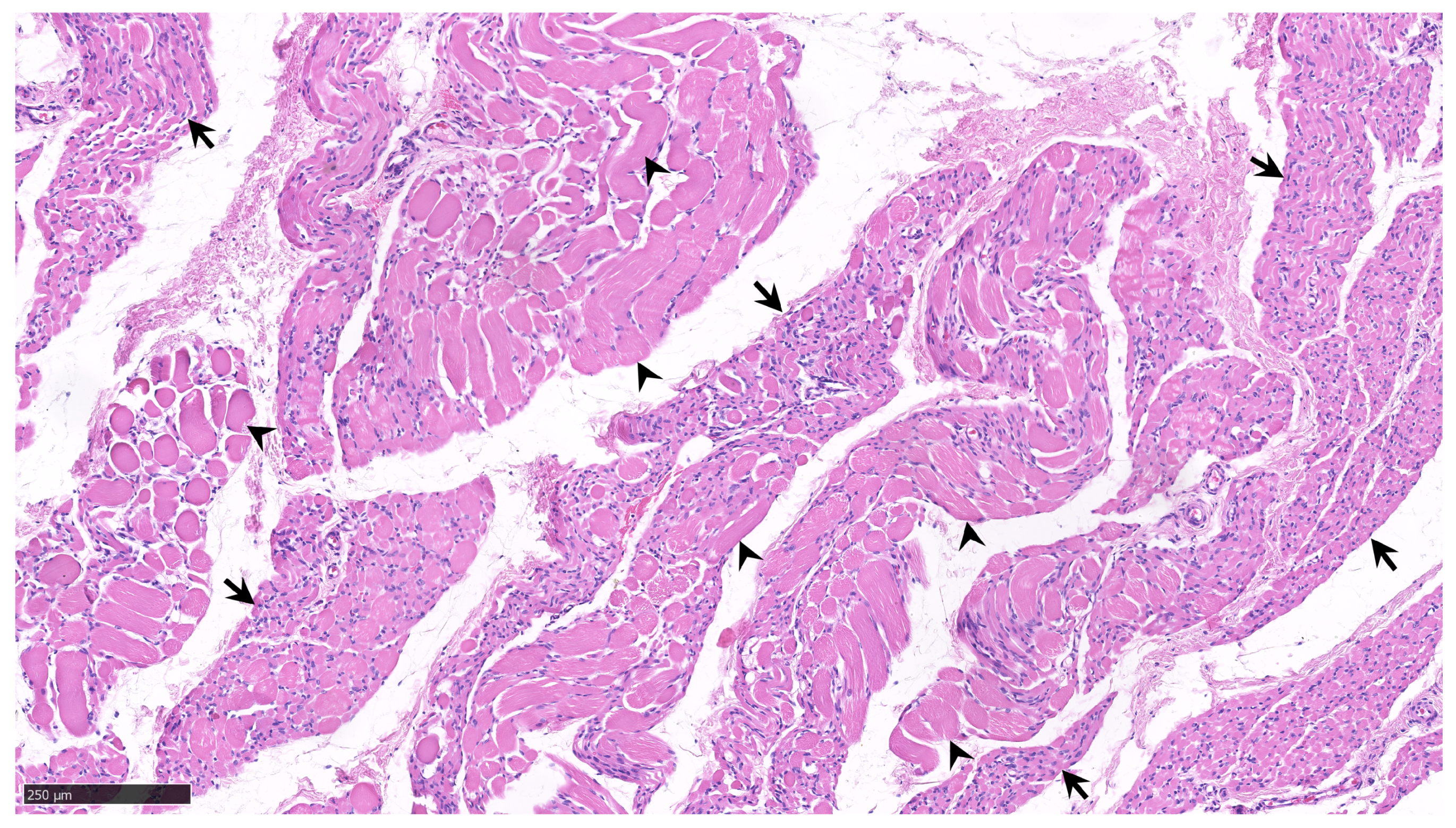{kind=link}
| Reference | Measured Biomarker | Analyzed Bodily Fluid | Number of Participants | Type of Diagnosis (Number of Patients) | Method Used for Measurement of Biomarkers | Biomarkers in SMA Compared to HCs | Correlation of Biomarkers with Scores on Scales for the Assessment of Motor Functions | Type of Biomarker |
|---|---|---|---|---|---|---|---|---|
| [56] | NfL and pNfH | CSF and serum | 39 | Adult SMA patients (33) HCs (6) | Single molecular array | Unchanged | Negative correlation between serum pNfH concentrations and RULM score, upper and lower extremity strength, and total strength | Pharmacodynamic, prognostic |
| [57] | IL-1β, IL-4, IL-6, IL-10, IL-17A, IL-17F, IL-21, IL-22, IL-23, IL-31, TNF-α, and IFN-γ | Serum, CSF | 44 | SMA 1 (4) SMA 2 (13) SMA 3 (16) HCs (11) | Multiplex immunoassay | In serum: ↑IL-1b, ↑IL-4, ↑IL-6, ↑IL-10, ↑IFN-γ, ↑IL-17A, ↑IL-22, ↑IL-23, ↑IL-31, ↑IL-33, ↑TNF-α | No correlation with the HFMSE score | Diagnostic |
| [51] | Whole miRNAome | Serum | 27 | SMA 2 (10) SMA 3 (10) HCs (7) | miRNA next-generation sequencing | 42 miRNAs differentially expressed; 14 miRNAs upregulated and 28 downregulated | Prognostic | |
| [48] | Whole proteome | CSF | 17 | SMA 1 (10) HCs (7) | 2D-PAGE, MS, WB | ↓APOA1 ↑Hemoglobin sub. β ↑Hemoglobin sub. α ↓Transthyretin 2D-PAGE analyses showed 39 protein differences | Not analyzed | Diagnostic |
| [58] | CHIT1 | CSF | 109 | SMA 1 (7) SMA 2 (33) SMA 3 (39) HCs (30) | ELISA | ↑CHIT1 | No association with HFMSE and CHOP-INTEND scores | Diagnostic, pharmacodynamic |
| [55] | Urinary metabolic profiling | Urine | 491 | Pre-symptomatic (5) SMA 1 (9) SMA 2 (8) SMA 3 (7) DMD (18) HCs (444) | 1H-NMR-based metabolic profiling combined with sophisticated algorithms based on machine learning | Urinary metabolic profiling | Not analyzed | Diagnostic, prognostic |
| [49] | Whole proteome | Extracellular vesicles released from fibroblasts | 6 | SMA 1 (1) SMA 2 (2) HCs (3) | MS, WB | 116 differentially expressed proteins (↓94, ↑21) ↑IGFBP3 ↓Plastin 3 ↓PTK7 ↓TCP1 ↑FETUA ↑FXA | Not analyzed | Diagnostic |
| [39] | NfL in CSF and serum | CSF and serum | 115 | SMA 1 (4) SMA 2 (7) SMA 3 (3) SMA 2–3 (4) HCs (97), only serum samples | Single-molecule array (SiMoA) assay | ↑NfL in serum | A negative correlation of serum NfL with CHOP-INTEND score | Pharmacodynamic, prognostic |
| [45] | In CSF: NfL, total tau, p-tau181, Qalb, OCB, CSF white cells In serum: creatinine | CSF, serum | 21 | SMA 2 (3) SMA 3 (6) HCs (12) | ELISA | ↓Creatinine | A positive relationship between creatinine and HFMSE score and RULM | Diagnostic, prognostic |
| [52] | Whole miRNAome | Serum | 88 | SMA 1 (3) SMA 2 (21) SMA 3 (26) SMA 4 (1) HCs (37) | Quantitative real-time PCR | ↑miR-181a-5p ↑miR-324-5p ↑miR-451a | Not analyzed | Diagnostic |
| [59] | HSPA7 mRNA, HSP70B protein, pNfH protein | Blood and serum | 52 | Pre-symptomatic (9) SMA 1 (22) SMA 2 (14) SMA 3 (2) HCs (5) | Enzyme-linked lectin assay (for p-NfH), ELISA (for HSP70B), real-time PCR, whole-blood RNA sequencing | ↑HSPA7 mRNA; Positive association of HSP70B protein with pNfH | Not analyzed | Diagnostic, prognostic |
| [53] | Whole blood transcriptomic screen | Whole blood | 65 | SMA 3 (31) HCs (34) | L1000 profiling technology and RT-PCR | -Seven downregulated and three upregulated KEGG pathways -Most significantly downregulated pathway “Regulation of Actin Cytoskeleton” (including the expression of key genes in this pathway; ROCK1, RHOA, and ACTB) -↓270 genes, ↑287 genes | Not analyzed | Diagnostic |
| [60] | NfL | Serum | 106 | SMA 2 (20) SMA 3 (26) SMA 2/3 (46) HCs (14) | NfL Advantage kit from Quanterix | No difference | Higher levels of NfL were associated with poorer motor performance (as measured by the HFMSE and ALSFRS-R). No correlation between NfL levels and motor scale scores | Pharmacodynamic, prognostic |
| [47] | Creatinine | Serum | 238 | SMA 1 (49) SMA 2 (97) SMA 3 (92) | Creatinine data from Project Cure SMA Longitudinal Population Data Repository | Decrease in creatinine levels with disease severity (SMA 3 > SMA 2 > SMA 1) | A positive association with the HFMS score | Diagnostic, prognostic |
| [61] | pNfH, NfL | CSF | 21 | SMA 3 (12) HCs (9) | ELISA | No difference | No association with the HFMSE score | Pharmacodynamic, prognostic |
| [62] | NfL, total tau, GFAP | CSF | 23 | SMA 1 (12) HCs (11) | ELISA | ↑NfL ↑total tau ↑GFAP | A negative correlation of NfL and total tau levels with the CHOP-INTEND score | Prognostic, diagnostic, pharmacodynamic |
| [63] | pNfH | Plasma | 155 | SMA 1 (121) HCs (34) | Protein Simple platform | ↑pNfH | A negative association between pNfH and the CHOP-INTEND score | Prognostic, diagnostic, pharmacodynamic |
| [64] | SMN protein | Peripheral blood nuclear cells (CD3+, CD19+, and CD33++ cells) | 39 | SMA 1 (4) SMA 2 (17) SMA 3 (4) HCs (14) | Imaging flow cytometry | ↓SMN protein | A positive correlation between SMN-spot+ cell percentage and the HFMS | Prognostic, diagnostic |
| [36] | SMN mRNA and protein | Blood | 53 | Infantile-onset SMA (26) HCs (27) | ECL immunoassay, Droplet Digital PCR | ↓SMN mRNA and protein | A positive association of SMN mRNA with TIMPSI No association with the CHOP-INTEND score | Prognostic, diagnostic |
| [65] | SMN protein | Exosomes released from fibroblasts and serum | 2 | SMA 3 (1) HCs (1) | WB | ↓SMN protein | Not analyzed | Diagnostic |
| [66] | SMN1, SMN2-FL, SMN2-Δ7, GAPDH and 18S mRNA, SMN protein | PBMCs, skin-derived fibroblasts | 443 | SMA 1 (18) SMA 2 (60) SMA 3 (52) SMA 4 (5) HCs (293) | ELISA, real-time PCR | ↓SMN protein | Correlation between SMN protein concentration in fibroblasts and HFMSE | Prognostic, diagnostic |
| [67] | SMN protein | Whole blood | 52 | SMA 1 (5) SMA 2 (22) SMA 3 (22) HCs (3) | ECL immunoassay | ↓SMN protein | Not analyzed | Diagnostic |
| [68] | SMN mRNA, SMN protein | Blood | 131 | SMA 1 (7) SMA 2 (14) SMA 3 (15) HCs (95) | In-house ELISA, multiplex qRT-PCR | A positive association between SMN protein and SMN mRNA levels in patients with SMA 1 and SMA 2 | Not analyzed | Diagnostic |
| [69] | SMN protein | Fibroblasts | SMA 1 (1) HC | Imaging flow cytometry technique | ↓SMN protein | Not analyzed | Diagnostic | |
| [50] | Analysis of nearly 1000 plasma proteins | Plasma | 288 | BforSMA samples: SMA 1 (17) SMA 2 (49) SMA 3 (42) HCs (22) PNCRN samples: SMA 1 (35) SMA 2 (66) SMA 3 (57) | Multiplex immunoassay | Development of commercial SMA-MAP biomarker panel (APOB, APCS, ASHG, AXL, CCL2, CD93, CFH, CDH13, CHI3L1, CLEC3B, COMP, CRP, CTSD, DPP4, ENG, ERBB2, FBLN1, IGF1, IGFBP6, LEP, LUM, MB, PEPD, PGF, SPP1, THBS4, TNXB) | A positive association of AXL, CD93, CDH13, COMP, DPP4, LUM, MB, PEPD, SPP1, and THBS4, and negative association of CHI3L1, APCS, and LEP with the MHFMS | Prognostic, diagnostic |
| [70] | GRP75/Mortalin, Calreticulin | Biopsy of skeletal muscle (m. quadratus femoris) | 6 | SMA 2/3 (3) HCs (3) | Quantitative fluorescent WB | ↑Calreticulin | Not analyzed | Prognostic |
| [71] | SMN2-FL transcripts, SMN-∆D7, SMN protein | Blood | 45 | SMA 3 | ELISA, real-time PCR | Not analyzed | A positive correlation between SMN2-FL transcripts and lower limb Medical Research Council (MRC) score and total MRC score | Prognostic |
| [34] | Proteome, transcriptome, metabolome | Plasma (proteome, metabolome, amino acids, free fatty acids), urine (metabolome), whole blood (transcriptome) | 130 | SMA 1 (17) SMA 2 (49) SMA 3 (42) HCs (22) | LC-MALDI-MS/MS Proteomics, Lipid LC/MS Metabolomics, GC/MS Metabolomics, Affymetrix Exon Array | Top 20 markers: (in plasma) TNXB, CILP2, COMP, Glu, CLEC3B, ADAMTSL4, THBS4, OMD, LUM, DPP4, PEPD, C10:0-fatty-acid, CDH13, Asp, Hyp, (in urine) Inositol, Uric acid, Pantothenic acid | 97 plasma proteins, 59 plasma metabolites, and 44 urine metabolites correlated with the MHFMS | Prognostic |
| [72] | SMN protein, SMN transcripts (SMN2-FL, SMN1-FL, SMN-∆D7), GAPDH transcript | Blood | 130 | SMA 1 (17) SMA 2 (49) SMA 3 (42) HCs (22) | ELISA, real-time PCR | ↓SMN protein SMN2-FL (SMA 3 > SMA 2 > SMA 1) ↑SMN-∆7 (in SMA 2 and 3) ↓GAPDH (in SMA 2 and 3) | No association with the MHFMS | Prognostic, diagnostic |
| [73] | SMN-FL, SMN-Δ7 mRNA | Blood | 61 | SMA 2 (42) SMA 3 (19) | Real-time PCR | ↓SMN-FL/SMNΔ7 ratio | Not analyzed | Prognostic |
| [54] | Transcriptome (Gene Expression Plate “Neurodegeneration”), α-synuclein (SNCA) protein | Fibroblasts, spinal cord mRNA | 6 | SMA 1 (3) HCs (3) | Real-time PCR, WB | ↓SMN1 mRNA, ↓SNCA mRNA, ↓SNCA protein, ↓SV2A mRNA and ↓SYN2 mRNA | Not analyzed | Diagnostic |
| [74] | SMN protein | PBMCs | 7 | SMA 1 (1) HCs (6) | ELISA | ↓SMN protein | Not analyzed | Diagnostic |
| [75] | SMN mRNA and SMN protein | Blood | 86 | SMA 1 (6) SMA 2 (9) SMA 3 (14) Carriers (29) HCs (28) | Cell immunoassay, quantitative reverse transcription PCR | ↑SMN-Δ7 mRNA in SMA 2 and 3 patients ↓SMN-7+ mRNA in SMA 1 patients ↓SMN protein in SMA 1 patients | Diagnostic |
5.2. Monitoring of Therapeutic Response in SMA Using Molecular Biomarkers
| Reference | Measured Biomarker | Analyzed Bodily Fluid | Number of Subjects | Type of Diagnosis (Number of Patients) | The Highest Number of Nusinersen Doses Administered or Treatment Duration | Changes in Biomarker Levels Following Nusinersen Therapy | Correlation between Biomarkers and Motor Function Assessment Scale Values |
|---|---|---|---|---|---|---|---|
| [93] | IL-1β, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p70), IL-13, IL-15, IL-17A, IP10, eotaxin, G-CSF, GM-CSF, IFNγ, MCAF/MCP-1, MIP-1α, MIP-1β, RANTES, TNF-α, PDGF-BB, VEGF, FGF-basic | CSF | 48 | SMA 1 (18) SMA 2 (19) SMA 3 (11) HCs (4) | 6 doses (302 days) | In SMA 1 patients: ↓IL-2, ↓IL-4, ↓IL-7, ↓IL-9, ↓IL-12, ↓IL-17, ↓VEGF, ↓eotaxin, ↓TNF-α In SMA 2 patients: ↑G-CSF, ↑IL-8, ↑MCP-1, ↑MIP-1α, ↑MIP-1β In SMA 3 patients: ↓IL-1ra | No association with CHOP-INTEND and HFMSE scores |
| [89] | CHIT1, IFN-γ, TNF-α | CSF | 6 | SMA 1 (2) SMA 2 (4) | 24 months | ↓CHIT1 | No association |
| [94] | IL-1β, IL-3, IL-6, IL-10, IL-12, IL-13, IL-17, IL-18, IFN-γ, BDNF, FAS, VEGF, TNF-α, ANG1, C5a, MCP-1/CCL2 | CSF | 38 | SMA 1 (4) SMA 2 (22) SMA 3 (12) | 10 months | ↑IL-10, ↑leukocyte counts, ↑MCP-1/CCL2 | No association |
| [76] | In serum extracellular vesicles: SMN transcript In serum and CSF: pNfH | Serum, CSF, serum extracellular vesicles | SMA 2 | 14 months | ↑flSMN transcript, ↓pNfH | ||
| [82] | Total tau, NfL, S100B | CSF | 26 | SMA 1 (13) SMA 2 (4) SMA 3 (9) | 5–15 doses | ↓Total tau | Not analyzed |
| [91] | NfL, MCP-1, fractalkine/CXCL3, IP-10/CXCL-10, IL-8/CXCL-8, sAPPα, sAPPβ | CSF | 13 | Older children with SMA (4) Younger children with SMA (9) | 36 months | ↓NfL, ↓sAPPα, ↓sAPPβ, ↓IL-8 (only in younger patients), ↓IP-10 (only in younger patients), ↑fractalkine | A negative correlation of IL-8, MCP-1, and sAPPα with CHOP-INTEND score |
| [100] | CSF miRNA signature | CSF | 34 | SMA 2 (16) SMA 3 (18) | 6 months | Lower baseline levels of miR-206 and miR-133 predict clinical response to nusinersen ↑miR-103b and ↓miR-1-3p, miR-133a/b and miR-206 in nusinersen responders | Inverse correlation of miR-206 with the difference in HFMSE score post and pre-treatment with nusinersen |
| [56] | NfL and pNfH | CSF and serum | 39 | Adult SMA patients (33) HCs (6) | Unchanged | A negative correlation of serum pNfH levels with RULM score, upper and lower extremity strength, and total strength | |
| [77] | Whole proteome | CSF | 44 | SMA 1 (12) SMA 2 (9) SMA 3 (6) Asymptomatic individuals (4) HCs (13) | 300 days | ↓Cathepsin D, ↓pNfH, ↓pNfL | Cathepsin D alters in response to treatment based on CHOP-INTEND-, HFMSE- and HINE2- scores |
| [51] | 41 miRNAs | Serum | 22 | SMA 1 | 6 months | ↑miR-335-5p, ↑miR-328-3p, ↑miR-423-3p, ↑miR-142- 5p and ↓miR-26b-5p | Several miRNA levels correlated positively with the functional improvement induced by nusinersen treatment (as measured by the CHOP-INTEND and HINE scales) |
| [101] | Extracellular RNAs and miRNAs | CSF | 12 | SMA 1 (4) SMA 2 (5) SMA 3 (3) | 4–13 doses | ↓expression of 48 genes ↑expression of 53 genes ↑miR-132, miR-218, miR-9, miR-23a, miR-146a | Not analyzed |
| [83] | pNfH | CSF | 11 | SMA 2 | Up to 34 months | No difference | Not analyzed |
| [103] | Metabolome | CSF | 27 | SMA 1 (12) SMA 2 (7) SMA 3 (8) | 302 days | -Altered amino acids and glucose metabolism in the CSF of SMA 1 patients -Altered amino acids and ketone body metabolism in the CSF of SMA 2 patients | A negative correlation between 3-hydroxybutyrate levels and CHOP-INTEND scores; a positive correlation between 3-hydroxybutyrate, alanine, and valine and HFMSE scores |
| [78] | pNfH | Plasma | 93 | Infantile-onset SMA | Up to 757 days | ↓pNfH | Not analyzed |
| [95] | Total protein, glucose, cell count | CSF | 50 | SMA 1 (22) SMA 2 (17) SMA 3 (11) | 22 months | ↑total protein (for SMA 2 and 3) | No association |
| [48] | Whole proteome | CSF | 17 | SMA 1 (10) HCs (7) | 4 doses (6 months) | ↑APOA1 ↑Transthyretin ↓Haptoglobin ↓Hemoglobin sub. β 2D-PAGE analyses showed differential expression of 30 proteins | Not analyzed |
| [104] | Creatinine kinase, Cystatin C | Serum | 16 | SMA 3 SMA 4 | 14 months | No difference | Not analyzed |
| [45] | In CSF: NfL, total tau, p-tau181, Qalb, OCB, CSF white cells In serum: creatinine | CSF, serum | 21 | SMA 2 (3) SMA 3 (6) HCs (12) | 5 (540 days) | ↑Qalb, development of systemic and intrathecal OCBs (in some SMA patients) | A positive relationship between creatinine and HFMSE score and RULM |
| [96] | Leukocyte count, lactate, total protein, Qalb, OCB | CSF | 28 | SMA 2 (10)SMA 3 (17) SMA 4 (1) | 22 months | ↑total proteins ↑Qalb | No association with HFMSE, 6MWT, and RULM scores |
| [58] | CHIT1, total proteins, Qalb | CSF | 109 | SMA 1 (7) SMA 2 (33) SMA 3 (39) HCs (30) | 6 doses (14 months) | ↑CHIT1 ↑Qalb ↑total proteins | No association with HFMSE and CHOP-INTEND scores |
| [90] | Amyloid β1–40 and amyloid β1–42 | CSF | 8 | SMA 2 (3) SMA 3 (5) | 420 days | ↑amyloid β1–42 | Not analyzed |
| [79] | pNfH, NfL, total tau, neurogranin, BACE-1, α-synuclein | CSF | 44 | SMA 1 (16) SMA 2 (16) SMA 3 (12) | 300 days | ↓pNfH (in SMA 1 and 2) ↓NfL (in SMA 1) ↓total tau (in SMA 1 and 2) ↑neurogranin (in SMA 3) ↑α-synuclein (in SMA 1) | Inverse correlation between total tau and pNfH levels and the CHOP-INTEND score in SMA type 1 patients; a positive association between neurogranin and RULM score in SMA type 2 and 3 patients; an inverse correlation of NfL with HFMSE score and pNfH with HFMSE and RULM score (in SMA 2 and 3 patients). |
| [97] | In CSF: WBC, glucose, lactate, total protein In blood: WBC, platelets, INR, aPTT, Crea, Urea, AST, ALT, GGT | CSF and blood | 50 | SMA 2 (14) SMA 3 (36) | Up to 12 doses (34 months) | In CSF: ↓Glucose, ↑total protein In blood: ↓aPTT, ↓Crea | Not analyzed |
| [39] | NfL in CSF and serum | CSF and serum | 115 | SMA 1 (4) SMA 2 (7) SMA 3 (3) SMA 2–3 (4) HCs (97) | Up to 12 doses (34 months) | ↓NfL in CSF and serum | Negative correlation between serum NfL and CHOP-INTEND score |
| [80] | NfL | CSF | 1 | SMA 1 | 12 months | ↓NfL | Not analyzed |
| [60] | NfL | Serum | 106 | SMA 2 (20) SMA 3 (26) SMA 2/3 (46) HCs (14) | 7 doses (over 14 months of treatment) | No difference | Higher levels of NfL are associated with poorer motor performance (as measured using the HFMSE and ALSFRS-R); no correlation between NfL levels and motor scale scores |
| [98] | Whole proteome, total proteins, Qalb, QIgA, QIgG, QIgM, OCB, glucose, lactate, cell count, reactive mononuclear transformations | CSF | 20 | SMA 2 (1) SMA 3 (9) HCs (10) | 10 months | ↑total proteins, ↑Qalb; Two protein clusters were identified after nusinersen treatment that differed from protein clusters at the baseline | After treatment with nusinersen, two protein clusters with substantially different HFMSE scores were identified |
| [81] | NfL, GFAP | CSF | 17 | SMA 2 (6) SMA 3 (11) | 14.33 months | ↓NfL, ↓GFAP (only in SMA 3 patients) | No association with the RULM scores |
| [102] | Muscle-specific miRNAs (myomiRs); miR-133a, miR-133b, miR-206, miR-1 | Plasma | 21 | SMA 2 (16) SMA 3 (5) | 4 doses (6 months) | ↓miR-133a ↓miR-133b ↓miR-1 | A negative correlation of miR-133a with the HFMSE score |
| [61] | pNfH, NfL | CSF | 21 | SMA 3 (12) HCs (9) | 6 months | ↓pNfH, ↓NfL | No association with the HFMSE score |
| [62] | NfL, total tau, GFAP | CSF | 23 | SMA 1 (12) HCs (11) | 8 doses | ↓NfL, ↓total tau, ↓GFAP | A negative relationship between NfL and total tau levels and the CHOP-INTEND score |
| [86] | NfH, total tau, S100B and NSE | CSF and serum | 11 | SMA 3 | 4 doses | Unchanged | Not analyzed |
| [99] | White cell count, total protein, Qalb, lactate, and OCB | CSF | 60 | SMA 1 (2) SMA 2 (28) SMA 3 (30) | 540 days | ↑total protein, ↑Qalb | No association with HFMSE score |
| [84] | pNfH, NSE, amyloid β1–40, amyloid β1–42, p-tau, tau protein, proteins, creatine kinase | CSF | 19 | SMA 3 | 300 days | ↓NSE, ↓p-tau | Not analyzed |
| [63] | pNfH | Plasma | 155 | SMA 1 (121, of whom 48 completed treatment) HCs (34) | 5 doses (302 days) | ↓pNfH | A negative association between pNfH and CHOP-INTEND score |
| [105] | SMN protein | CSF | 28 | SMA 2 (15) SMA 3 (13) | 9–14 months | ↑SMN protein | Correlation between SMN protein and enhancement in motor function (as measured using HFMSE score) |
5.3. Limitations and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Vill, K.; Kölbel, H.; Schwartz, O.; Blaschek, A.; Olgemöller, B.; Harms, E.; Burggraf, S.; Röschinger, W.; Durner, J.; Gläser, D.; et al. One Year of Newborn Screening for SMA—Results of a German Pilot Project. J. Neuromuscul. Dis. 2019, 6, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Li, D.K.; Tisdale, S.; Lotti, F.; Pellizzoni, L. SMN control of RNP assembly: From post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev. Biol. 2014, 32, 22–29. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.T.; Gillingwater, T.H.; Faller, K.M.E. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [PubMed]
- Pierzchlewicz, K.; Kępa, I.; Podogrodzki, J.; Kotulska, K. Spinal Muscular Atrophy: The Use of Functional Motor Scales in the Era of Disease-Modifying Treatment. Child Neurol. Open 2021, 8, 2329048X2110087. [Google Scholar] [CrossRef]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Glanzman, A.M.; Mazzone, E.; Main, M.; Pelliccioni, M.; Wood, J.; Swoboda, K.J.; Scott, C.; Pane, M.; Messina, S.; Bertini, E.; et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscul. Disord. 2010, 20, 155–161. [Google Scholar] [CrossRef]
- Main, M.; Kairon, H.; Mercuri, E.; Muntoni, F. The Hammersmith functional motor scale for children with spinal muscular atrophy: A scale to test ability and monitor progress in children with limited ambulation. Eur. J. Paediatr. Neurol. 2003, 7, 155–159. [Google Scholar] [CrossRef]
- Glanzman, A.M.; O’Hagen, J.M.; McDermott, M.P.; Martens, W.B.; Flickinger, J.; Riley, S.; Quigley, J.; Montes, J.; Dunaway, S.; Deng, L.; et al. Validation of the Expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J. Child Neurol. 2011, 26, 1499–1507. [Google Scholar] [CrossRef]
- Lunn, M.R.; Wang, C.H. Spinal muscular atrophy. Lancet 2008, 371, 2120–2133. [Google Scholar] [CrossRef]
- Bürglen, L.; Lefebvre, S.; Clermont, O.; Burlet, P.; Viollet, L.; Cruaud, C.; Munnich, A.; Melki, J. Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996, 32, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Wilson, R.B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 2002, 111, 477–500. [Google Scholar] [CrossRef]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.J.; Yum, M.S.; Kim, E.H.; Yoo, H.W.; Lee, B.H.; Kim, G.H.; Ko, T.S. Genotype-Phenotype Correlation of SMN1 and NAIP Deletions in Korean Patients with Spinal Muscular Atrophy. J. Clin. Neurol. 2017, 13, 27–31. [Google Scholar] [CrossRef]
- Hassan, H.A.; Zaki, M.S.; Issa, M.Y.; El-Bagoury, N.M.; Essawi, M.L. Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt. J. Med. Hum. Genet. 2020, 21, 4. [Google Scholar] [CrossRef]
- Bernal, S.; Also-Rallo, E.; Martínez-Hernández, R.; Alías, L.; Rodríguez-Alvarez, F.J.; Millán, J.M.; Hernández-Chico, C.; Baiget, M.; Tizzano, E.F. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul. Disord. 2011, 21, 413–419. [Google Scholar] [CrossRef]
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossoll, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Hosseinibarkooie, S.; Peters, M.; Torres-Benito, L.; Rastetter, R.H.H.; Hupperich, K.; Hoffmann, A.; Mendoza-Ferreira, N.; Kaczmarek, A.; Janzen, E.; Milbradt, J.; et al. The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype. Am. J. Hum. Genet. 2016, 99, 647–665. [Google Scholar] [CrossRef]
- Werdnig, G. Zwei frühinfantile hereditäre Fälle von progressiver Muskelatrophie unter dem Bilde der Dystrophie, aber anf neurotischer Grundlage. Arch. für Psychiatr. und Nervenkrankh. 1891, 22, 437–480. [Google Scholar] [CrossRef]
- Šimić, G. Pathogenesis of proximal autosomal recessive spinal muscular atrophy. Acta Neuropathol. 2008, 116, 223–234. [Google Scholar] [CrossRef]
- Šimić, G.; Mladinov, M.; Šimić, Đ.Š.; Milošević, N.J.; Islam, A.; Pajtak, A.; Barišić, N.; Sertić, J.; Lucassen, P.J.; Hof, P.R.; et al. Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol. 2008, 115, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Wadman, R.I.; Vrancken, A.F.J.E.; Van Den Berg, L.H.; Van Der Pol, W.L. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology 2012, 79, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W. ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Hensel, N.; Kubinski, S.; Claus, P. The Need for SMN-Independent Treatments of Spinal Muscular Atrophy (SMA) to Complement SMN-Enhancing Drugs. Front. Neurol. 2020, 11, 45. [Google Scholar] [CrossRef]
- Sturm, S.; Günther, A.; Jaber, B.; Jordan, P.; Al Kotbi, N.; Parkar, N.; Cleary, Y.; Frances, N.; Bergauer, T.; Heinig, K.; et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br. J. Clin. Pharmacol. 2019, 85, 181–193. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Fagan, A.M.; Holtzman, D.M. Biomarkers of Alzheimer’s disease. Neurobiol. Dis. 2009, 35, 128–140. [Google Scholar] [CrossRef]
- Aluise, C.D.; Sowell, R.A.; Butterfield, D.A. Peptides and proteins in plasma and cerebrospinal fluid as biomarkers for the prediction, diagnosis, and monitoring of therapeutic efficacy of Alzheimer’s disease. Biochim. Biophys. Acta 2008, 1782, 549–558. [Google Scholar] [CrossRef]
- Perrin, R.J.; Fagan, A.M.; Holtzman, D.M. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 2009, 461, 916–922. [Google Scholar] [CrossRef]
- The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer’s Disease”. Neurobiol. Aging 1998, 19, 109–116. [CrossRef]
- Hampel, H.; Frank, R.; Broich, K.; Teipel, S.J.; Katz, R.G.; Hardy, J.; Herholz, K.; Bokde, A.L.W.; Jessen, F.; Hoessler, Y.C.; et al. Biomarkers for Alzheimer’s disease: Academic, industry and regulatory perspectives. Nat. Rev. Drug Discov. 2010, 9, 560–574. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer’s disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Crawford, T.O.; Swoboda, K.J.; Kaufmann, P.; Juhasz, P.; Li, X.; Guo, Y.; Li, R.H.; Trachtenberg, F.; Forrest, S.J.; et al. Candidate proteins, metabolites and transcripts in the Biomarkers for Spinal Muscular Atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e35462. [Google Scholar] [CrossRef]
- Bonati, U.; Holiga, Š.; Hellbach, N.; Risterucci, C.; Bergauer, T.; Tang, W.; Hafner, P.; Thoeni, A.; Bieri, O.; Gerlach, I.; et al. Longitudinal characterization of biomarkers for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2017, 4, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Natural history of infantile-onset spinal muscular atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef]
- Pino, M.G.; Rich, K.A.; Kolb, S.J. Update on Biomarkers in Spinal Muscular Atrophy. Biomark. Insights 2021, 16, 11772719211035644. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments at a glance. J. Cell Sci. 2012, 125, 3257–3263. [Google Scholar] [CrossRef]
- Nitz, E.; Smitka, M.; Schallner, J.; Akgün, K.; Ziemssen, T.; von der Hagen, M.; Tüngler, V. Serum neurofilament light chain in pediatric spinal muscular atrophy patients and healthy children. Ann. Clin. Transl. Neurol. 2021, 8, 2013–2024. [Google Scholar] [CrossRef]
- Schwartz, O.; Kölbel, H.; Blaschek, A.; Gläser, D.; Burggraf, S.; Röschinger, W.; Schara, U.; Müller-Felber, W.; Vill, K. Spinal Muscular Atrophy—Is Newborn Screening Too Late for Children with Two SMN2 Copies? J. Neuromuscul. Dis. 2022, 9, 389–396. [Google Scholar] [CrossRef]
- Martin, S.J.; McGlasson, S.; Hunt, D.; Overell, J. Cerebrospinal fluid neurofilament light chain in multiple sclerosis and its subtypes: A meta-analysis of case-control studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1059–1067. [Google Scholar] [CrossRef]
- Dhiman, K.; Gupta, V.B.; Villemagne, V.L.; Eratne, D.; Graham, P.L.; Fowler, C.; Bourgeat, P.; Li, Q.X.; Collins, S.; Bush, A.I.; et al. Cerebrospinal fluid neurofilament light concentration predicts brain atrophy and cognition in Alzheimer’s disease. Alzheimer Dement. 2020, 12, e12005. [Google Scholar] [CrossRef]
- Dreger, M.; Steinbach, R.; Gaur, N.; Metzner, K.; Stubendorff, B.; Witte, O.W.; Grosskreutz, J. Cerebrospinal Fluid Neurofilament Light Chain (NfL) Predicts Disease Aggressiveness in Amyotrophic Lateral Sclerosis: An Application of the D50 Disease Progression Model. Front. Neurosci. 2021, 15, 651651. [Google Scholar] [CrossRef]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef]
- Milella, G.; Introna, A.; D’Errico, E.; Fraddosio, A.; Scaglione, G.; Morea, A.; Ucci, M.; Ruggieri, M.; Mastrapasqua, M.; Megna, M.; et al. Cerebrospinal Fluid and Clinical Profiles in Adult Type 2-3 Spinal Muscular Atrophy Patients Treated with Nusinersen: An 18-Month Single-Centre Experience. Clin. Drug Investig. 2021, 41, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Freigang, M.; Wurster, C.D.; Hagenacker, T.; Stolte, B.; Weiler, M.; Kamm, C.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Kowski, A.; et al. Serum creatine kinase and creatinine in adult spinal muscular atrophy under nusinersen treatment. Ann. Clin. Transl. Neurol. 2021, 8, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.R.R.; Zhang, R.; Johnstone, A.J.; Garner, R.; Nwe, P.H.; Siranosian, J.J.; Swoboda, K.J. Serum creatinine is a biomarker of progressive denervation in spinal muscular atrophy. Neurology 2020, 94, e921–e931. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Sframeli, M.; Vantaggiato, L.; Vita, G.L.; Ciranni, A.; Polito, F.; Oteri, R.; Gitto, E.; Di Giuseppe, F.; Angelucci, S.; et al. Nusinersen Modulates Proteomics Profiles of Cerebrospinal Fluid in Spinal Muscular Atrophy Type 1 Patients. Int. J. Mol. Sci. 2021, 22, 4329. [Google Scholar] [CrossRef]
- Roberto, J.; Poulin, K.L.; Parks, R.J.; Vacratsis, P.O. Label-free quantitative proteomic analysis of extracellular vesicles released from fibroblasts derived from patients with spinal muscular atrophy. Proteomics 2021, 21, e2000301. [Google Scholar] [CrossRef]
- Kobayashi, D.T.; Shi, J.; Stephen, L.; Ballard, K.L.; Dewey, R.; Mapes, J.; Chung, B.; McCarthy, K.; Swoboda, K.J.; Crawford, T.O.; et al. SMA-MAP: A plasma protein panel for spinal muscular atrophy. PLoS ONE 2013, 8, e60113. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, I.T.; Scoto, M.; Aragon-Gawinska, K.; Ridout, D.; Doreste, B.; Servais, L.; Muntoni, F.; Zhou, H. Response of plasma microRNAs to nusinersen treatment in patients with SMA. Ann. Clin. Transl. Neurol. 2022, 9, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Abiusi, E.; Infante, P.; Cagnoli, C.; Severini, L.L.; Pane, M.; Coratti, G.; Pera, M.C.; D’amico, A.; Diano, F.; Novelli, A.; et al. SMA-miRs (MiR-181a- 5p, -324-5p, and -451a) are overexpressed in spinal muscular atrophy skeletal muscle and serum samples. Elife 2021, 10, e68054. [Google Scholar] [CrossRef] [PubMed]
- Siranosian, J.J.; Nery, F.C.; Alves, C.R.R.; Siranosian, B.A.; Lyons, N.J.; Eichelberger, E.J.; Garner, R.; Da Silva Duarte Lepez, S.; Johnstone, A.J.; Subramanian, A.; et al. Whole-blood dysregulation of actin-cytoskeleton pathway in adult spinal muscular atrophy patients. Ann. Clin. Transl. Neurol. 2020, 7, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Acsadi, G.; Li, X.; Murphy, K.J.; Swoboda, K.J.; Parker, G.C. Alpha-Synuclein Loss in Spinal Muscular Atrophy. J. Mol. Neurosci. 2011, 43, 275–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saffari, A.; Cannet, C.; Blaschek, A.; Hahn, A.; Hoffmann, G.F.; Johannsen, J.; Kirsten, R.; Kockaya, M.; Kölker, S.; Müller-Felber, W.; et al. 1H-NMR-based metabolic profiling identifies non-invasive diagnostic and predictive urinary fingerprints in 5q spinal muscular atrophy. Orphanet J. Rare Dis. 2021, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Rich, K.A.; Fox, A.; Yalvac, M.; Heintzman, S.; Tellez, M.; Bartlett, A.; Severyn, S.; Linsenmayer, M.; Kelly, K.; Reynolds, J.; et al. Neurofilament Levels in CSF and Serum in an Adult SMA Cohort Treated with Nusinersen. J. Neuromuscul. Dis. 2022, 9, 111–119. [Google Scholar] [CrossRef]
- Bonanno, S.; Cavalcante, P.; Salvi, E.; Giagnorio, E.; Malacarne, C.; Cattaneo, M.; Andreetta, F.; Venerando, A.; Pensato, V.; Gellera, C.; et al. Identification of a cytokine profile in serum and cerebrospinal fluid of pediatric and adult spinal muscular atrophy patients and its modulation upon nusinersen treatment. Front. Cell. Neurosci. 2022, 16, 982760. [Google Scholar] [CrossRef]
- Freigang, M.; Steinacker, P.; Wurster, C.D.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Koch, J.C.; Rostásy, K.; Falkenburger, B.; Ludolph, A.C.; et al. Increased chitotriosidase 1 concentration following nusinersen treatment in spinal muscular atrophy. Orphanet J. Rare Dis. 2021, 16, 1–11. [Google Scholar] [CrossRef]
- Eichelberger, E.J.; Alves, C.R.R.; Zhang, R.; Petrillo, M.; Cullen, P.; Farwell, W.; Hurt, J.A.; Staropoli, J.F.; Swoboda, K.J. Increased systemic HSP70B levels in spinal muscular atrophy infants. Ann. Clin. Transl. Neurol. 2021, 8, 1495–1501. [Google Scholar] [CrossRef]
- Wurster, C.D.; Steinacker, P.; Günther, R.; Koch, J.C.; Lingor, P.; Uzelac, Z.; Witzel, S.; Wollinsky, K.; Winter, B.; Osmanovic, A.; et al. Neurofilament light chain in serum of adolescent and adult SMA patients under treatment with nusinersen. J. Neurol. 2020, 267, 36–44. [Google Scholar] [CrossRef]
- Faravelli, I.; Meneri, M.; Saccomanno, D.; Velardo, D.; Abati, E.; Gagliardi, D.; Parente, V.; Petrozzi, L.; Ronchi, D.; Stocchetti, N.; et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J. Cell. Mol. Med. 2020, 24, 3034–3039. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T.; Crawford, T.O.; Finkel, R.S.; Mercuri, E.; De Vivo, D.C.; Oskoui, M.; Tizzano, E.F.; Ryan, M.M.; Muntoni, F.; Zhao, G.; et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, N.; Arakawa, R.; Kaneko, K.; Aoki, R.; Arakawa, M.; Saito, K. A new biomarker candidate for spinal muscular atrophy: Identification of a peripheral blood cell population capable of monitoring the level of survival motor neuron protein. PLoS ONE 2018, 13, e0201764. [Google Scholar] [CrossRef]
- Nash, L.A.; McFall, E.R.; Perozzo, A.M.; Turner, M.; Poulin, K.L.; De Repentigny, Y.; Burns, J.K.; McMillan, H.J.; Warman Chardon, J.; Burger, D.; et al. Survival Motor Neuron Protein is Released from Cells in Exosomes: A Potential Biomarker for Spinal Muscular Atrophy. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Wadman, R.I.; Stam, M.; Jansen, M.D.; Van Der Weegen, Y.; Wijngaarde, C.A.; Harschnitz, O.; Sodaar, P.; Braun, K.P.J.; Dooijes, D.; Lemmink, H.H.; et al. A Comparative Study of SMN Protein and mRNA in Blood and Fibroblasts in Patients with Spinal Muscular Atrophy and Healthy Controls. PLoS ONE 2016, 11, e0167087. [Google Scholar] [CrossRef]
- Zaworski, P.; Von Herrmann, K.M.; Taylor, S.; Sunshine, S.S.; Mccarthy, K.; Risher, N.; Newcomb, T.; Weetall, M.; Prior, T.W.; Swoboda, K.J.; et al. SMN Protein Can Be Reliably Measured in Whole Blood with an Electrochemiluminescence (ECL) Immunoassay: Implications for Clinical Trials. PLoS ONE 2016, 11, e0150640. [Google Scholar] [CrossRef]
- Czech, C.; Tang, W.; Bugawan, T.; Mano, C.; Horn, C.; Iglesias, V.A.; Fröhner, S.; Zaworski, P.G.; Paushkin, S.; Chen, K.; et al. Biomarker for Spinal Muscular Atrophy: Expression of SMN in Peripheral Blood of SMA Patients and Healthy Controls. PLoS ONE 2015, 10, e0139950. [Google Scholar] [CrossRef]
- Arakawa, M.; Arakawa, R.; Tatsumi, S.; Aoki, R.; Saito, K.; Nomoto, A. A novel evaluation method of survival motor neuron protein as a biomarker of spinal muscular atrophy by imaging flow cytometry. Biochem. Biophys. Res. Commun. 2014, 453, 368–374. [Google Scholar] [CrossRef]
- Mutsaers, C.A.; Lamont, D.J.; Hunter, G.; Wishart, T.M.; Gillingwater, T.H. Label-free proteomics identifies Calreticulin and GRP75/Mortalin as peripherally accessible protein biomarkers for spinal muscular atrophy. Genome Med. 2013, 5, 1–11. [Google Scholar] [CrossRef]
- Tiziano, F.D.; Lomastro, R.; Di Pietro, L.; Pasanisi, M.B.; Fiori, S.; Angelozzi, C.; Abiusi, E.; Angelini, C.; Sorarù, G.; Gaiani, A.; et al. Clinical and molecular cross-sectional study of a cohort of adult type III spinal muscular atrophy patients: Clues from a biomarker study. Eur. J. Hum. Genet. 2013, 21, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Paushkin, S.V.; Kobayashi, D.T.; Forrest, S.J.; Joyce, C.L.; Finkel, R.S.; Kaufmann, P.; Swoboda, K.J.; Tiziano, D.; Lomastro, R.; et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE 2012, 7, e33572. [Google Scholar] [CrossRef]
- Zheleznyakova, G.Y.; Kiselev, A.V.; Vakharlovsky, V.G.; Rask-Andersen, M.; Chavan, R.; Egorova, A.A.; Schiöth, H.B.; Baranov, V.S. Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med. Genet. 2011, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.T.; Olson, R.J.; Sly, L.; Swanson, C.J.; Chung, B.; Naryshkin, N.; Narasimhan, J.; Bhattacharyya, A.; Mullenix, M.; Chen, K.S. Utility of survival motor neuron ELISA for spinal muscular atrophy clinical and preclinical analyses. PLoS ONE 2011, 6, e24269. [Google Scholar] [CrossRef] [PubMed]
- Sumner, C.J.; Kolb, S.J.; Harmison, G.G.; Jeffries, N.O.; Schadt, K.; Finkel, R.S.; Dreyfuss, G.; Fischbeck, K.H. SMN mRNA and protein levels in peripheral blood: Biomarkers for SMA clinical trials. Neurology 2006, 66, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Trifunov, S.; Natera-de Benito, D.; Carrera-García, L.; Codina, A.; Expósito-Escudero, J.; Ortez, C.; Medina, J.; Torres Alcala, S.; Bernal, S.; Alias, L.; et al. Full-Length SMN Transcript in Extracellular Vesicles as Biomarker in Individuals with Spinal Muscular Atrophy Type 2 Treated with Nusinersen. J. Neuromuscul. Dis. 2023, 10, 653–665. [Google Scholar] [CrossRef]
- Schorling, D.C.; Kölbel, H.; Hentschel, A.; Pechmann, A.; Meyer, N.; Wirth, B.; Rombo, R.; Sickmann, A.; Kirschner, J.; Schara-Schmidt, U.; et al. Cathepsin D as biomarker in cerebrospinal fluid of nusinersen-treated patients with spinal muscular atrophy. Eur. J. Neurol. 2022, 29, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Ryan, M.M.; Pascual Pascual, S.I.; Day, J.W.; Mercuri, E.; De Vivo, D.C.; Foster, R.; Montes, J.; Gurgel-Giannetti, J.; MacCannell, D.; et al. Scientific rationale for a higher dose of nusinersen. Ann. Clin. Transl. Neurol. 2022, 9, 819–829. [Google Scholar] [CrossRef]
- Johannsen, J.; Weiss, D.; Daubmann, A.; Schmitz, L.; Denecke, J. Evaluation of putative CSF biomarkers in paediatric spinal muscular atrophy (SMA) patients before and during treatment with nusinersen. J. Cell. Mol. Med. 2021, 25, 8419–8431. [Google Scholar] [CrossRef]
- Tozawa, T.; Kasai, T.; Tatebe, H.; Shiomi, K.; Nishio, H.; Tokuda, T.; Chiyonobu, T. Intrathecal nusinersen treatment after ventriculo-peritoneal shunt placement: A case report focusing on the neurofilament light chain in cerebrospinal fluid. Brain Dev. 2020, 42, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.; Fadda, G.; McMillan, C.; Michon, S.-C.; Kichula, E.; Zhao, L.; Banwell, B.; Bar-Or, A.; Elman, L. Effect of Nusinsersen in an Adult SMA Cohort: CSF Biomarkers and RULM (4725). Neurology 2020, 94, 4725. [Google Scholar]
- Šimić, G.; Vukić, V.; Babić, M.; Banović, M.; Berečić, I.; Španić, E.; Zubčić, K.; Golubić, A.T.; Barišić Kutija, M.; Merkler Šorgić, A.; et al. Total tau in cerebrospinal fluid detects treatment responders among spinal muscular atrophy types 1-3 patients treated with nusinersen. CNS Neurosci. Ther. 2022; Early View. [Google Scholar] [CrossRef]
- Carson, V.J.; Young, M.; Brigatti, K.W.; Robinson, D.L.; Reed, R.M.; Sohn, J.; Petrillo, M.; Farwell, W.; Miller, F.; Strauss, K.A. Nusinersen by subcutaneous intrathecal catheter for symptomatic spinal muscular atrophy patients with complex spine anatomy. Muscle Nerve 2022, 65, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Wenninger, S.; Thiele, S.; Stauber, J.; Hiebeler, M.; Greckl, E.; Stahl, K.; Pechmann, A.; Lochmüller, H.; Kirschner, J.; et al. Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3—A Prospective Observational Study. J. Neuromuscul. Dis. 2019, 6, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef]
- Totzeck, A.; Stolte, B.; Kizina, K.; Bolz, S.; Schlag, M.; Thimm, A.; Kleinschnitz, C.; Hagenacker, T. Neurofilament Heavy Chain and Tau Protein Are Not Elevated in Cerebrospinal Fluid of Adult Patients with Spinal Muscular Atrophy during Loading with Nusinersen. Int. J. Mol. Sci. 2019, 20, 5397. [Google Scholar] [CrossRef]
- Santamaria-Kisiel, L.; Rintala-Dempsey, A.C.; Shaw, G.S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 2006, 396, 201–214. [Google Scholar] [CrossRef]
- Kanneganti, M.; Kamba, A.; Mizoguchi, E. Role of chitotriosidase (chitinase 1) under normal and disease conditions. J. Epithel. Biol. Pharmacol. 2012, 5, 1–9. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ishikawa, N.; Tateishi, Y.; Izumo, H.; Eto, S.; Eguchi, Y.; Okada, S. Evaluation of cerebrospinal fluid biomarkers in pediatric patients with spinal muscular atrophy. Brain Dev. 2023, 45, 2–7. [Google Scholar] [CrossRef]
- Introna, A.; Milella, G.; D’Errico, E.; Fraddosio, A.; Scaglione, G.; Ucci, M.; Ruggieri, M.; Simone, I.L. Is cerebrospinal fluid amyloid-β42 a promising biomarker of response to nusinersen in adult spinal muscular atrophy patients? Muscle Nerve 2021, 63, 905–909. [Google Scholar] [CrossRef]
- Verma, S.; Perry, K.; Razdan, R.; Howell, J.C.; Dawson, A.L.; Hu, W.T. CSF IL-8 Associated with Response to Gene Therapy in a Case Series of Spinal Muscular Atrophy. Neurotherapeutics 2023, 20, 245–253. [Google Scholar] [CrossRef]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef]
- Nuzzo, T.; Russo, R.; Errico, F.; D’Amico, A.; Tewelde, A.G.; Valletta, M.; Hassan, A.; Tosi, M.; Panicucci, C.; Bruno, C.; et al. Nusinersen mitigates neuroinflammation in severe spinal muscular atrophy patients. Commun. Med. 2023, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Scheijmans, F.E.V.; Cuppen, I.; Zwartkruis, M.M.; Signoria, I.; van Ekris, C.; Asselman, F.; Wadman, R.I.; Knol, E.F.; van der Pol, W.L.; Groen, E.J.N. Inflammatory markers in cerebrospinal fluid of paediatric spinal muscular atrophy patients receiving nusinersen treatment. Eur. J. Paediatr. Neurol. 2023, 42, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Orbach, R.; Sagi, L.; Sadot, E.; Tokatly Latzer, I.; Shtamler, A.; Zisberg, T.; Fattal-Valevski, A. Cerebrospinal fluid characteristics of patients treated with intrathecal nusinersen for spinal muscular atrophy. Muscle Nerve 2022, 66, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Müschen, L.H.; Osmanovic, A.; Binz, C.; Jendretzky, K.F.; Ranxha, G.; Bronzlik, P.; Abu-fares, O.; Wiehler, F.; Möhn, N.; Hümmert, M.W.; et al. Cerebrospinal Fluid Parameters in Antisense Oligonucleotide-Treated Adult 5q-Spinal Muscular Atrophy Patients. Brain Sci. 2021, 11, 296. [Google Scholar] [CrossRef]
- Stolte, B.; Nonnemacher, M.; Kizina, K.; Bolz, S.; Totzeck, A.; Thimm, A.; Wagner, B.; Deuschl, C.; Kleinschnitz, C.; Hagenacker, T. Nusinersen treatment in adult patients with spinal muscular atrophy: A safety analysis of laboratory parameters. J. Neurol. 2021, 268, 4667–4679. [Google Scholar] [CrossRef]
- Kessler, T.; Latzer, P.; Schmid, D.; Warnken, U.; Saffari, A.; Ziegler, A.; Kollmer, J.; Möhlenbruch, M.; Ulfert, C.; Herweh, C.; et al. Cerebrospinal fluid proteomic profiling in nusinersen-treated patients with spinal muscular atrophy. J. Neurochem. 2020, 153, 650–661. [Google Scholar] [CrossRef]
- Wurster, C.D.; Koch, J.C.; Cordts, I.; Dreyhaupt, J.; Otto, M.; Uzelac, Z.; Witzel, S.; Winter, B.; Kocak, T.; Schocke, M.; et al. Routine Cerebrospinal Fluid (CSF) Parameters in Patients With Spinal Muscular Atrophy (SMA) Treated With Nusinersen. Front. Neurol. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Magen, I.; Aharoni, S.; Yacovzada, N.S.; Tokatly Latzer, I.; Alves, C.R.R.; Sagi, L.; Fattal-Valevski, A.; Swoboda, K.J.; Katz, J.; Bruckheimer, E.; et al. Muscle microRNAs in the cerebrospinal fluid predict clinical response to nusinersen therapy in type II and type III spinal muscular atrophy patients. Eur. J. Neurol. 2022, 29, 2420–2430. [Google Scholar] [CrossRef]
- Welby, E.; Rehborg, R.J.; Harmelink, M.; Ebert, A.D. Assessment of cerebral spinal fluid biomarkers and microRNA-mediated disease mechanisms in spinal muscular atrophy patient samples. Hum. Mol. Genet. 2022, 31, 1830–1843. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, S.; Marcuzzo, S.; Malacarne, C.; Giagnorio, E.; Masson, R.; Zanin, R.; Arnoldi, M.T.; Andreetta, F.; Simoncini, O.; Venerando, A.; et al. Circulating MyomiRs as Potential Biomarkers to Monitor Response to Nusinersen in Pediatric SMA Patients. Biomedicines 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Errico, F.; Marino, C.; Grimaldi, M.; Nuzzo, T.; Bassareo, V.; Valsecchi, V.; Panicucci, C.; Di Schiavi, E.; Mazza, T.; Bruno, C.; et al. Nusinersen Induces Disease-Severity-Specific Neurometabolic Effects in Spinal Muscular Atrophy. Biomolecules 2022, 12, 1431. [Google Scholar] [CrossRef]
- De Wel, B.; Goosens, V.; Sobota, A.; Van Camp, E.; Geukens, E.; Van Kerschaver, G.; Jagut, M.; Claes, K.; Claeys, K.G. Nusinersen treatment significantly improves hand grip strength, hand motor function and MRC sum scores in adult patients with spinal muscular atrophy types 3 and 4. J. Neurol. 2021, 268, 923–935. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 897. [Google Scholar] [CrossRef]
- Glascock, J.; Darras, B.T.; Crawford, T.O.; Sumner, C.J.; Kolb, S.J.; DiDonato, C.; Elsheikh, B.; Howell, K.; Farwell, W.; Valente, M.; et al. Identifying Biomarkers of Spinal Muscular Atrophy for Further Development. J. Neuromuscul. Dis. 2023, 1–18. [Google Scholar] [CrossRef]
- Ogbonmide, T.; Rathore, R.; Rangrej, S.B.; Hutchinson, S.; Lewis, M.; Ojilere, S.; Carvalho, V.; Kelly, I. Gene Therapy for Spinal Muscular Atrophy (SMA): A Review of Current Challenges and Safety Considerations for Onasemnogene Abeparvovec (Zolgensma). Cureus 2023, 15, e36197. [Google Scholar] [CrossRef]
- Navarrete-Opazo, A.; Garrison, S.; Waite, M. Molecular Biomarkers for Spinal Muscular Atrophy: A Systematic Review. Neurol. Clin. Pract. 2021, 11, e524–e536. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babić, M.; Banović, M.; Berečić, I.; Banić, T.; Babić Leko, M.; Ulamec, M.; Junaković, A.; Kopić, J.; Sertić, J.; Barišić, N.; et al. Molecular Biomarkers for the Diagnosis, Prognosis, and Pharmacodynamics of Spinal Muscular Atrophy. J. Clin. Med. 2023, 12, 5060. https://doi.org/10.3390/jcm12155060
Babić M, Banović M, Berečić I, Banić T, Babić Leko M, Ulamec M, Junaković A, Kopić J, Sertić J, Barišić N, et al. Molecular Biomarkers for the Diagnosis, Prognosis, and Pharmacodynamics of Spinal Muscular Atrophy. Journal of Clinical Medicine. 2023; 12(15):5060. https://doi.org/10.3390/jcm12155060
Chicago/Turabian StyleBabić, Marija, Maria Banović, Ivana Berečić, Tea Banić, Mirjana Babić Leko, Monika Ulamec, Alisa Junaković, Janja Kopić, Jadranka Sertić, Nina Barišić, and et al. 2023. "Molecular Biomarkers for the Diagnosis, Prognosis, and Pharmacodynamics of Spinal Muscular Atrophy" Journal of Clinical Medicine 12, no. 15: 5060. https://doi.org/10.3390/jcm12155060
APA StyleBabić, M., Banović, M., Berečić, I., Banić, T., Babić Leko, M., Ulamec, M., Junaković, A., Kopić, J., Sertić, J., Barišić, N., & Šimić, G. (2023). Molecular Biomarkers for the Diagnosis, Prognosis, and Pharmacodynamics of Spinal Muscular Atrophy. Journal of Clinical Medicine, 12(15), 5060. https://doi.org/10.3390/jcm12155060