
Real Evidence and Misconceptions about Malignant Hyperthermia in Children: A Narrative Review

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Epidemiology
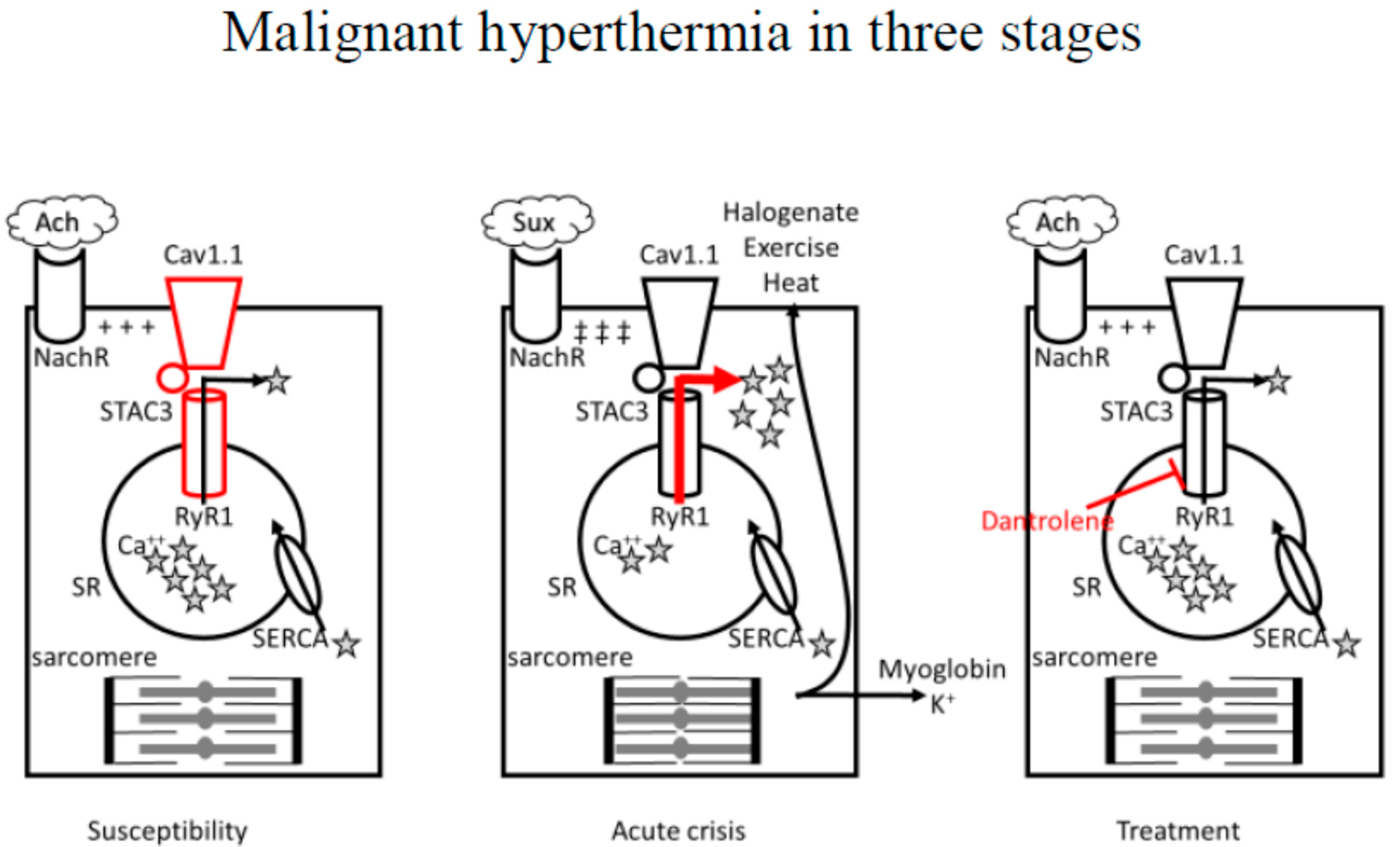
3. Pathophysiology
3.1. Anesthetic-Induced MH
3.2. Non-Anesthetic Induced MH
4. Disorders Associated with Malignant Hyperthermia
5. Clinical Presentation
6. Clinical Diagnostic Pathway
7. Perioperative Recommendations for High-Risk Children
8. Treatment of Malignant Hyperthermia
- -
- Temperature management: despite the syndrome’s name, severe hyperthermia is a sign of a delayed diagnosis or management; accordingly, dantrolene is the most appropriate intervention for body temperature control. However, hyperthermia can lead to coagulopathy, irreversible actin-myosin binding, and worsening of acidosis and electrolyte disturbances; therefore, if the body temperature is >38.5–39 °C, active cooling is indicated in addition to turning off heating devices. The pediatric population can very well benefit from surface cooling due to large body surface area in relation to weight; methods include forced air cooling, ice packs near the great arteries (neck, axillae, groins), wet, cold sheets, cooling blankets or pads set at low temperatures (for example used in targeted temperature management). Caution should be made since direct contact with cold objects with the thin skin of a child can cause frostbite. Ice immersion is the most effective method of external cooling, but not applicable in the operating room setting. Cold intravenous fluids administration (4 °C) is a simple and effective second-line cooling method: while fluid replacement can be beneficial for perspiration and to reduce the risk of acute kidney injury, the risk of fluid overload limits this method of cooling; adult guidelines recommend not to exceed 10–20 mL·kg−1, and it is reasonably adequate in the pediatric population. Invasive cooling methods are usually unnecessary in a recognized MH crisis: bladder and gastric lavage are poorly effective, while peritoneal lavage and extracorporeal circulation, albeit effective, require time, equipment, and expertise. Pharmacologic interventions such as acetaminophen or ibuprofen are not effective in this setting. Active cooling should be halted when the core temperature is <38–38.5 °C due to the risk of vasoconstriction and hypothermia in resolving crises.
- -
- Respiratory and metabolic acidosis: minute ventilation should be increased 2–3 times to exhale the excess CO2 production by muscle contraction; a normal EtCO2 (e.g., 40 mmHg) should be targeted. Metabolic acidosis with base excess < −8 mEq· L−1 and pH < 7.2 is treated with sodium bicarbonate 1–2 mEq·kg−1.
- -
- Electrolyte disturbances: first-line treatment for hyperkalemia (K+ > 5.9 mmol/L or QRS widening) is membrane stabilization with calcium (0.1 mmol·kg−1 chloride calcium or 60 mg·kg−1 calcium gluconate) and insulin-glucose system for rapid potassium cell entry (e.g., dextrose: 50%, 50 mL with 50 IU insulin for adults or 0.1 insulin·kg−1 and dextrose 25%, 2 mL·kg−1). Blood glucose should be checked hourly. Other potassium-lowering interventions include sodium bicarbonate, albuterol, furosemide, kayexalate, and hemodialysis.
- -
- Cardiovascular system: arrhythmias should be treated with amiodarone 3 mg·kg−1 up to 300 mg. Persistent tachycardia in the absence of hemodynamic compromise can be treated with beta-blockers, while calcium channel blockers should be avoided due to their relevant interaction.
- -
- Renal system: due to rhabdomyolysis and increased creatine kinase, there is a risk of acute kidney injury; guidelines recommend maintaining a high urine output, at least 2 mL·kg−1, which can be achieved with cold intravenous fluids, furosemide 0.5–1 mg·kg−1, mannitol 1 gr·kg−1 (already contained in dantrolene sodium formulation). Urine alkalinization with sodium bicarbonate 1 mEq·kg−1·h is also an option.
9. Future Perspectives
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ACFs | Activated charcoal filters |
| Ca2+ | Calcium |
| CCD | Central core disease |
| CHCT | Caffeine/halothane contracture test |
| DHPR | Dihydropyridine receptor |
| EMHG | European Malignant Hyperthermia Group |
| ETCO2 | End-tidal carbon dioxide |
| GA | General anesthesia |
| HypoPP | Hypokalemic periodic paralysis |
| IVCT | In vitro contracture test |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
| MH | Malignant hyperthermia |
| MHANZ | Malignant Hyperthermia Group of Australia and New Zealand |
| MHAUS | Malignant Hyperthermia Association of the United States |
| MHS | Malignant hyperthermia susceptibility |
| MmD | Multi-mini core disease |
| MMR | Masseter muscle rigidity |
| PICU | Pediatric intensive care unit |
| RyR1 | Type 1 ryanodine receptor |
| SERCA | Sarcoendoplasmic reticulum calcium ATPase |
| SR | Sarcoplasmic reticulum |
| TIVA | Total intravenous anesthesia |
References
- Brown, R.C. Hyperpyrexia and Anaesthesia. BMJ 1954, 2, 1526–1527. [Google Scholar] [CrossRef] [PubMed]
- Bigler, J.A. Body temperatures during anesthesia in infants and children. J. Am. Med. Assoc. 1951, 146, 551–556. [Google Scholar] [CrossRef]
- Denborough, M.A.; Forster, J.F.A.; Lovell, R.R.H.; Maplestone, P.A.; Villiers, J.D. Anaesthetic deaths in a family. Br. J. Anaesth. 1962, 34, 395–396. [Google Scholar] [CrossRef]
- Denborough, M.; Ebeling, P.; King, J.; Zapf, P. Myopathy and malignant hyperpyrexia. Lancet 1970, 295, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Denborough, M. Malignant hyperthermia. Lancet 1998, 352, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.; Pollock, N.; Schiemann, A.H.; Bulger, T.; Stowell, K.M. Malignant hyperthermia: A review. Orphanet J. Rare Dis. 2015, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Brady, J.E.; Sun, L.S.; Rosenberg, H.; Li, G. Prevalence of Malignant Hyperthermia Due to Anesthesia in New York State, 2001–2005. Obstet. Anesth. Dig. 2009, 109, 1162–1166. [Google Scholar] [CrossRef]
- Hopkins, P.M.; Girard, T.; Dalay, S.; Jenkins, B.; Thacker, A.; Patteril, M.; McGrady, E. Malignant hyperthermia 2020: Guideline from the Association of Anaesthetists. Anaesthesia 2021, 76, 655–664. [Google Scholar] [CrossRef]
- Rüffert, H.; Bastian, B.; Bendixen, D.; Girard, T.; Heiderich, S.; Hellblom, A.; Hopkins, P.M.; Johannsen, S.; Snoeck, M.M.; Urwyler, A.; et al. Consensus guidelines on perioperative management of malignant hyperthermia suspected or susceptible patients from the European Malignant Hyperthermia Group. Br. J. Anaesth. 2021, 126, 120–130. [Google Scholar] [CrossRef]
- Rosenberg, H.; Sambuughin, N.; Riazi, S.; Dirksen, R. Malignant Hyperthermia Susceptibility. In GeneReviews® [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar]
- Carpenter, D.; Robinson, R.L.; Quinnell, R.J.; Ringrose, C.; Hogg, M.; Casson, F.; Booms, P.; Iles, D.E.; Halsall, P.J.; Steele, D.S.; et al. Genetic variation in RYR1 and malignant hyperthermia phenotypes. Br. J. Anaesth. 2009, 103, 538–548. [Google Scholar] [CrossRef] [Green Version]
- European Malignant Hyperthermia Group (EMHG). Available online: www.emhg.org (accessed on 10 April 2023).
- Watt, S.; McAllister, R.K. Malignant Hyperthermia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Parness, J.; Bandschapp, O.; Girard, T. The Myotonias and Susceptibility to Malignant Hyperthermia. Obstet. Anesth. Dig. 2009, 109, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Larach, M.G. A Primer for Diagnosing and Managing Malignant Hyperthermia Susceptibility. Anesthesiology 2018, 128, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Brislin, R.P.; Theroux, M.C. Core myopathies and malignant hyperthermia susceptibility: A review. Pediatr. Anesth. 2013, 23, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Bin, X.; Wang, B.; Tang, Z. Malignant Hyperthermia: A Killer If Ignored. J. PeriAnesthesia Nurs. 2022, 37, 435–444. [Google Scholar] [CrossRef]
- Denborough, M.; Hopkinson, K.C.; O’Brien, R.O.; Foster, P.S. Overheating Alone Can Trigger Malignant Hyperthermia in Piglets. Anaesth. Intensiv. Care 1996, 24, 348–354. [Google Scholar] [CrossRef]
- Sumitani, M.; Uchida, K.; Yasunaga, H.; Horiguchi, H.; Kusakabe, Y.; Matsuda, S.; Yamada, Y. Prevalence of Malignant Hyperthermia and Relationship with Anesthetics in Japan: Data from the diagnosis procedure combination database. Anesthesiology 2011, 114, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Riazi, S.; Larach, M.G.; Hu, C.; Wijeysundera, D.; Massey, C.; Kraeva, N. Malignant hyperthermia in Canada: Characteristics of index anesthetics in 129 malignant hyperthermia susceptible probands. Anesth. Analg. 2014, 118, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Tatsukawa, H.; Okuda, J.; Kondoh, M.; Inoue, M.; Terashima, S.; Katoh, S.; Ida, K. Malignant Hyperthermia Caused by Intravenous Lidocaine for Ventricular Arrhythmia. Intern. Med. 1992, 31, 1069–1072. [Google Scholar] [CrossRef] [Green Version]
- Schieren, M.; Defosse, J.; Böhmer, A.; Wappler, F.; Gerbershagen, M.U. Anaesthetic management of patients with myopathies. Eur. J. Anaesthesiol. 2017, 34, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.; Ellis, F.; Halsall, P. Evidence for related myopathies in exertional heat stroke and malignant hyperthermia. Lancet 1991, 338, 1491–1492. [Google Scholar] [CrossRef]
- Lehmann-Horn, F.; Klingler, W.; Jurkat-Rott, K. Nonanesthetic Malignant Hyperthermia. Anesthesiology 2011, 115, 915–917. [Google Scholar] [CrossRef]
- Larach, M.G.; Allen, G.C.; Rosenberg, H. Confirmation of Nonanesthetic-induced Malignant Hyperthermia. Anesthesiology 2012, 116, 1398–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, J.R. Malignant Hyperthermia and Apparent Heat Stroke. JAMA 2001, 286, 168–169. [Google Scholar] [CrossRef] [PubMed]
- Carsana, A. Exercise-Induced Rhabdomyolysis and Stress-Induced Malignant Hyperthermia Events, Association with Malignant Hyperthermia Susceptibility, and RYR1 Gene Sequence Variations. Sci. World J. 2013, 2013, 531465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, M.E.; Kenney, K.; Deuster, P.; Campbell, W. Exertional Rhabdomyolysis. J. Clin. Neuromuscul. Dis. 2012, 13, 122–136. [Google Scholar] [CrossRef]
- Laitano, O.; Murray, K.; Leon, L.R. Overlapping Mechanisms of Exertional Heat Stroke and Malignant Hyperthermia: Evidence vs. Conjecture. Sports Med. 2020, 50, 1581–1592. [Google Scholar] [CrossRef]
- E Nelson, T.; Jones, E.W.; Venable, J.H.; Kerr, D.D. Malignant Hyperthermia of Poland China Swine. Anesthesiology 1972, 36, 52–56. [Google Scholar] [CrossRef]
- Corona, B.T.; Rouviere, C.; Hamilton, S.L.; Ingalls, C.P. FKBP12 deficiency reduces strength deficits after eccentric contraction-induced muscle injury. J. Appl. Physiol. 2008, 105, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Maryansky, A.; Rose, J.C.; Rosenblatt, M.A.; Lai, Y.H. Postoperative Hyperthermia and Hemodynamic Instability in a Suspected Malignant Hyperthermia–Susceptible Patient: A Case Report. A A Pract. 2021, 15, e01314. [Google Scholar] [CrossRef]
- Gronert, G.A.; Tobin, J.R.; Muldoon, S. Malignant hyperthermia—Human stress triggering. Biochim. Biophys. Acta 2011, 1813, 2191–2192. [Google Scholar] [CrossRef] [Green Version]
- Sbaraglia, F.; Racca, F.; Maiellare, F.; Longhitano, Y.; Zanza, C.; Caputo, C.T. Prevenzione, Gestione e Trattamento Dell’ipertermia Maligna. 2020. Available online: www.siaarti.it (accessed on 10 April 2023). (In Italian).
- Radkowski, P.; Suren, L.; Podhorodecka, K.; Harikumar, S.; Jamrozik, N. A Review on the Anesthetic Management of Patients with Neuromuscular Diseases. Anesthesiol. Pain Med. 2023, 13, e132088. [Google Scholar] [CrossRef]
- Adams, D.C.; Heyer, E.J. Problems of anesthesia in patients with neuromuscular disease. Anesthesiol. Clin. N. Am. 1997, 15, 673–689. [Google Scholar] [CrossRef]
- Montagnese, F.; Schoser, B. Dystrophische und nicht-dystrophische Myotonien [Dystrophic and non-dystrophic myotonias]. Fortschr. Neurol. Psychiatr. 2018, 86, 575–583. [Google Scholar] [CrossRef]
- Bandschapp, O.; Iaizzo, P.A. Pathophysiologic and anesthetic considerations for patients with myotonia congenita or periodic paralyses. Pediatr. Anesth. 2013, 23, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Gurnaney, H.; Brown, A.; Litman, R.S. Malignant Hyperthermia and Muscular Dystrophies. Obstet. Anesthesia Dig. 2009, 109, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Cruz, B. Mitochondrial diseases and anesthesia: A literature review of current opinions. AANA J. 2013, 81, 237–243. [Google Scholar]
- Morgan, P.G.; Hoppel, C.L.; Sedensky, M.M. Mitochondrial Defects and Anesthetic Sensitivity. Anesthesiology 2002, 96, 1268–1270. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Anesthesia Analg. 2015, 17, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Sakamizu, A.; Yaguchi, E.; Hamaguchi, S. Anesthetic Management for an Adult With Glycogen Storage Disease Type 0. Anesthesia Prog. 2020, 67, 233–234. [Google Scholar] [CrossRef]
- Bollig, G. McArdle’s disease (glycogen storage disease type V) and anesthesia—A case report and review of the literature. Pediatr. Anesthesia 2013, 23, 817–823. [Google Scholar] [CrossRef]
- Hunter, A.; Pinsky, L. An evaluation of the possible association of malignant hyperpyrexia with the Noonan syndrome using serum creatine phosphokinase levels. J. Pediatr. 1975, 86, 412–415. [Google Scholar] [CrossRef]
- Benca, J.; Hogan, K. Malignant Hyperthermia, Coexisting Disorders, and Enzymopathies: Risks and Management Options. Obstet. Anesth. Dig. 2009, 109, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.M.; Rüffert, H.; Snoeck, M.M.; Girard, T.; Glahn, K.P.; Ellis, F.R.; Müller, C.R.; Urwyler, A.; European Malignant Hyperthermia Group. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br. J. Anaesth. 2015, 115, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, L.; Rockoff, M.A.; Koka, B.V. Masseter Spasm with Anesthesia: Incidence and implications. Anesthesiology 1984, 61, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Lazzell, V.A.; Carr, A.S.; Lerman, J.; Burrows, F.A.; Creighton, R.E. The incidence of masseter muscle rigidity after succinylcholine in infants and children. Can. J. Anaesth. 1994, 41, 475–479. [Google Scholar] [CrossRef] [Green Version]
- O’Flynn, R.P.; Shutack, J.G.; Rosenberg, H.; Fletcher, J.E. Masseter Muscle Rigidity and Malignant Hyperthermia Susceptibility in Pediatric Patients An Update on Management and Diagnosis. Anesthesiology 1994, 80, 1228–1233. [Google Scholar] [CrossRef]
- Vukcevic, M.; Broman, M.; Islander, G.; Bodelsson, M.; Ranklev-Twetman, E.; Müller, C.R.; Treves, S. Functional Properties of RYR1 Mutations Identified in Swedish Patients with Malignant Hyperthermia and Central Core Disease. Obstet. Anesth. Dig. 2010, 111, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Jungbluth, H.; Sewry, C.A.; Muntoni, F. Core Myopathies. Semin. Pediatr. Neurol. 2011, 18, 239–249. [Google Scholar] [CrossRef]
- Quinlivan, R.M.; Muller, C.R.; A Davis, M.; Laing, N.; A Evans, G.; Dwyer, J.; Dove, J.B.; Roberts, A.P.; A Sewry, C. Central core disease: Clinical, pathological, and genetic features. Arch. Dis. Child. 2003, 88, 1051–1055. [Google Scholar] [CrossRef] [Green Version]
- Ríos, E. Calcium-induced release of calcium in muscle: 50 years of work and the emerging consensus. J. Gen. Physiol. 2018, 150, 521–537. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Koop, A.; Liu, Y.; Guo, W.; Wei, J.; Wang, R.; MacLennan, D.H.; Dirksen, R.T.; Chen, S.R.W. Reduced threshold for store overload-induced Ca2+ release is a common defect of RyR1 mutations associated with malignant hyperthermia and central core disease. Biochem. J. 2017, 474, 2749–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Ibarra, M.C.A.; Malicdan, M.C.V.; Murayama, K.; Ichihara, Y.; Kikuchi, H.; Nonaka, I.; Noguchi, S.; Hayashi, Y.K.; Nishino, I. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain 2006, 129, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungbluth, H. Central core disease. Orphanet J. Rare Dis. 2007, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Dirksen, R.T. Ryanodinopathies: RyR-Linked Muscle Diseases. Curr. Top Membr. 2010, 66, 139–167. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, S.; Zorzato, F.; Ferreiro, A.; Jungbluth, H.; Muntoni, F.; Monnier, N.; Müller, C.R.; Treves, S. Functional properties of ryanodine receptors carrying three amino acid substitutions identified in patients affected by multi-minicore disease and central core disease, expressed in immortalized lymphocytes. Biochem. J. 2006, 395, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.; Litman, R.S. Malignant Hyperthermia in Children: An analysis of the North American malignant hyperthermia registry. Obstet. Anesth. Dig. 2014, 118, 369–374. [Google Scholar] [CrossRef]
- Otsuki, S.; Miyoshi, H.; Mukaida, K.; Yasuda, T.; Nakamura, R.; Tsutsumi, Y.M. Age-Specific Clinical Features of Pediatric Malignant Hyperthermia: A Review of 187 Cases Over 60 Years in Japan. Obstet. Anesth. Dig. 2021, 135, 128–135. [Google Scholar] [CrossRef]
- Larach, M.G.; Brandom, B.W.; Allen, G.C.; Gronert, G.A.; Lehman, E.B. Malignant Hyperthermia Deaths Related to Inadequate Temperature Monitoring, 2007–2012: A report from the North American malignant hyperthermia registry of the malignant hyperthermia association of the United States. Obstet. Anesth. Dig. 2014, 119, 1359–1366. [Google Scholar] [CrossRef]
- Litman, R.S.; Flood, C.D.; Kaplan, R.F.; Kim, Y.L.; Tobin, J.R. Postoperative malignant hyperthermia: An analysis of cases from the North American Malignant Hyperthermia Registry. Anesthesiology 2008, 109, 825–829. [Google Scholar] [CrossRef] [Green Version]
- Sessler, M.D.I.; Warner, D.S.; Warner, M.A. Temperature Monitoring and Perioperative Thermoregulation. Anesthesiology 2008, 109, 318–338. [Google Scholar] [CrossRef] [Green Version]
- Karan, S.M.; Crowl, F.; Muldoon, S.M. Malignant Hyperthermia Masked by Capnographic Monitoring. Obstet. Anesth. Dig. 1994, 78, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Klincová, M.; Štěpánková, D.; Schröderová, I.; Klabusayová, E.; Štourač, P. Malignant Hyperthermia in PICU—From Diagnosis to Treatment in the Light of Up-to-Date Knowledge. Children 2022, 9, 1692. [Google Scholar] [CrossRef] [PubMed]
- Uitenbosch, G.; Sng, D.; Carvalho, H.N.; Cata, J.P.; De Boer, H.D.; Erdoes, G.; Heytens, L.; Lois, F.J.; Rousseau, A.-F.; Pelosi, P.; et al. Expert Multinational Consensus Statement for Total Intravenous Anaesthesia (TIVA) Using the Delphi Method. J. Clin. Med. 2022, 11, 3486. [Google Scholar] [CrossRef]
- Yassen, K.A.; Jabaudon, M.; Alsultan, H.A.; Almousa, H.; I Shahwar, D.; Alhejji, F.Y.; Aljaziri, Z.Y. Inhaled Sedation with Volatile Anesthetics for Mechanically Ventilated Patients in Intensive Care Units: A Narrative Review. J. Clin. Med. 2023, 12, 1069. [Google Scholar] [CrossRef]
- Johannsen, S.; Mögele, S.; Roewer, N.; Schuster, F. Malignant hyperthermia on ICU—Sudden attack of the “snake”. BMC Anesthesiol. 2014, 14, A11. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.; Daly, C.; Aboelsaod, E.; Gardner, L.; Hobson, S.; Riasat, K.; Shepherd, S.; Robinson, R.; Bilmen, J.; Gupta, P.; et al. Genetic epidemiology of malignant hyperthermia in the UK. Br. J. Anaesth. 2018, 121, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Litman, R.S.; Rosenberg, H. Malignant Hyperthermia: Update on susceptibility testing. JAMA 2005, 293, 2918–2924. [Google Scholar] [CrossRef]
- Bersselaar, L.R.v.D.; Hellblom, A.; Gashi, B.M.; Kamsteeg, E.-J.; Voermans, N.C.; Jungbluth, H.; de Puydt, J.; Heytens, L.; Riazi, S.; Snoeck, M.M.J. Referral Indications for Malignant Hyperthermia Susceptibility Diagnostics in Patients without Adverse Anesthetic Events in the Era of Next-generation Sequencing. Anesthesiology 2022, 136, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Larach, M.G. Standardization of the caffeine halothane muscle contracture test. North American Malignant Hyperthermia Group. Anesth. Analg. 1989, 69, 511–515. [Google Scholar] [CrossRef]
- Riazi, M.S.; Kraeva, N.; Hopkins, M.P.M. Malignant Hyperthermia in the Post-Genomics Era: New Perspectives on an Old Concept. Anesthesiology 2018, 128, 168–180. [Google Scholar] [CrossRef]
- Larach, M.G. Should We Use Muscle Biopsy to Diagnose Malignant Hyperthermia Susceptibility? Anesthesiology 1993, 79, 1–4. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Schiemann, A.H.; Burling, S.M.; Bjorksten, A.; Bulger, T.; Gillies, R.; Hopkins, P.M.; Kamsteeg, E.-J.; Machon, R.G.; Massey, S.; et al. Functional analysis of RYR1 variants in patients with confirmed susceptibility to malignant hyperthermia. Br. J. Anaesth. 2022, 129, 879–888. [Google Scholar] [CrossRef]
- Britt, B.A.; Gordon, R.A. Three cases of malignant hyperthermia with special consideration of management. Can. J. Anaesth. 1969, 16, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissonnette, B. Temperature monitoring in pediatric anesthesia. Int. Anesthesiol. Clin. 1992, 30, 63–76. [Google Scholar] [PubMed]
- Available online: www.accessdata.fda.gov/drugsatfda_docs/label/2008/018264s025lbl.pdf (accessed on 10 April 2023).
- Available online: www.ema.europa.eu/en/medicines/human/orphan-designations/eu3141379 (accessed on 10 April 2023).
- Kim, H.J.; Koh, W.U.; Choi, J.M.; Ro, Y.J.; Yang, H.S. Malignant hyperthermia and dantrolene sodium. Korean J. Anesthesiol. 2019, 72, 78–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyota, Y.; Kondo, T.; Shorin, D.; Sumii, A.; Kido, K.; Watanabe, T.; Otsuki, S.; Kanzaki, R.; Miyoshi, H.; Yasuda, T.; et al. Rapid Dantrolene Administration with Body Temperature Monitoring Is Associated with Decreased Mortality in Japanese Malignant Hyperthermia Events. BioMed Res. Int. 2023, 2023, 8340209. [Google Scholar] [CrossRef]
- Chan, C.H. Dantrolene sodium and hepatic injury. Neurology 1990, 40, 1427. [Google Scholar] [CrossRef]
- Glahn, K.P.; Bendixen, D.; Girard, T.; Hopkins, P.M.; Johannsen, S.; Rüffert, H.; Snoeck, M.M.; Urwyler, A.; European Malignant Hyperthermia Group. Availability of dantrolene for the management of malignant hyperthermia crises: European Malignant Hyperthermia Group guidelines. Br. J. Anaesth. 2020, 125, 133–140. [Google Scholar] [CrossRef]
- Miller, C.; Bräuer, A.; Wieditz, J.; Klose, K.; Pancaro, C.; Nemeth, M. Modeling iatrogenic intraoperative hyperthermia from external warming in children: A pooled analysis from two prospective observational studies. Pediatr. Anesth. 2022, 33, 114–122. [Google Scholar] [CrossRef]
- Safety Committee of Japanese Society of Anesthesiologists JSA guideline for the management of malignant hyperthermia crisis 2016. J. Anesthesia 2017, 31, 307–317. [CrossRef]
- Glahn, K.P.E.; Ellis, F.R.; Halsall, P.J.; Müller, C.R.; Snoeck, M.M.J.; Urwyler, A.; Wappler, F.; European Malignant Hyperthermia Group. Recognizing and managing a malignant hyperthermia crisis: Guidelines from the European Malignant Hyperthermia Group. Br. J. Anaesth. 2010, 105, 417–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkman, J.M.; Posner, K.L.; Domino, K.B. Analysis of the Clinical Variables Associated with Recrudescence after Malignant Hyperthermia Reactions. Anesthesiology 2007, 106, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Krause, T.; Gerbershagen, M.U.; Fiege, M.; Weißhorn, R.; Wappler, F. Dantrolene–A review of its pharmacology, therapeutic use and new developments. Anaesthesia 2004, 59, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Chaffman, M.O.; Sorkin, E.M. Dantrolene. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in malignant hyperthermia, the neuroleptic malignant syndrome and an update of its use in muscle spasticity. Drugs 1986, 32, 130–168. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, M.; van Engelen, B.G.M.; Küsters, B.; Lammens, M.; Meijer, R.; Molenaar, J.P.F.; Raaphorst, J.; Verschuuren-Bemelmans, C.C.; Straathof, C.S.M.; Sie, L.T.L.; et al. RYR1-related myopathies: A wide spectrum of phenotypes throughout life. Eur. J. Neurol. 2015, 22, 1094–1112. [Google Scholar] [CrossRef]
- Johnston, J.J.; Dirksen, R.T.; Girard, T.; Gonsalves, S.G.; Hopkins, P.M.; Riazi, S.; Saddic, L.A.; Sambuughin, N.; Saxena, R.; Stowell, K.; et al. Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genet. Med. 2021, 23, 1288–1295. [Google Scholar] [CrossRef]
- Yang, L.; Dedkova, E.N.; Allen, P.D.; Jafri, M.S.; Fomina, A.F. T lymphocytes from malignant hyperthermia-susceptible mice display aberrations in intracellular calcium signaling and mitochondrial function. Cell Calcium 2021, 93, 102325. [Google Scholar] [CrossRef]
- Zitnik, M.; Nguyen, F.; Wang, B.; Leskovec, J.; Goldenberg, A.; Hoffman, M.M. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf. Fusion 2019, 50, 71–91. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Disease | Evidence in Adults | Evidence in Children | Suggested Perioperative Pathway |
|---|---|---|---|
| Upper motor neurons disease | |||
| Amyotrophic lateral sclerosis | None | None | standard |
| Myelin sheath disease | |||
| Multiple sclerosis | None | None | Standard |
| Guillain–Barré syndrome | None | None | Standard |
| Chronic inflammatory demyelinating polyneuropathy | None | None | Standard |
| Alexander disease | None | None | Standard |
| Krabbe disease | None | None | Standard |
| Adrenoleukodystrophy | None | None | Standard |
| Neuromyelitis optica spectrum disorders | None | None | Standard |
| Neuromuscular junction disease | |||
| Miastenia gravis | None | None | Standard |
| Muscular Dystrophy | |||
| Duchenne muscular dystrophy | Mild | Mild | Trigger-Free |
| Congenital muscular dystrophy | Mild | Mild | Trigger-Free |
| Facioscapulohumeral muscular dystrophy | Mild | Mild | Trigger-Free |
| Emery–Dreifuss muscular dystrophy | Mild | Mild | Trigger-Free |
| Becker muscular dystrophy | Mild | Mild | Trigger-Free |
| Channel disease | |||
| Myotonia congenita | None | None | Standard |
| Hypokalemic periodic paralysis | Strong | Strong | Trigger-Free |
| Central core disease | Strong | Strong | Trigger-Free |
| Cellular Metabolism disease | |||
| Mitochondrial disease | None | None | Standard |
| Kearns–Sayre syndrome | None | None | Standard |
| Glycogen storage disease | None | None | Standard |
| Lipid storage disorder | None | None | Standard |
| Other | |||
| Neonatal Palsy | None | None | Standard |
| Traumatic Damage | None | None | Standard |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frassanito, L.; Sbaraglia, F.; Piersanti, A.; Vassalli, F.; Lucente, M.; Filetici, N.; Zanfini, B.A.; Catarci, S.; Draisci, G. Real Evidence and Misconceptions about Malignant Hyperthermia in Children: A Narrative Review. J. Clin. Med. 2023, 12, 3869. https://doi.org/10.3390/jcm12123869
Frassanito L, Sbaraglia F, Piersanti A, Vassalli F, Lucente M, Filetici N, Zanfini BA, Catarci S, Draisci G. Real Evidence and Misconceptions about Malignant Hyperthermia in Children: A Narrative Review. Journal of Clinical Medicine. 2023; 12(12):3869. https://doi.org/10.3390/jcm12123869
Chicago/Turabian StyleFrassanito, Luciano, Fabio Sbaraglia, Alessandra Piersanti, Francesco Vassalli, Monica Lucente, Nicoletta Filetici, Bruno Antonio Zanfini, Stefano Catarci, and Gaetano Draisci. 2023. "Real Evidence and Misconceptions about Malignant Hyperthermia in Children: A Narrative Review" Journal of Clinical Medicine 12, no. 12: 3869. https://doi.org/10.3390/jcm12123869
APA StyleFrassanito, L., Sbaraglia, F., Piersanti, A., Vassalli, F., Lucente, M., Filetici, N., Zanfini, B. A., Catarci, S., & Draisci, G. (2023). Real Evidence and Misconceptions about Malignant Hyperthermia in Children: A Narrative Review. Journal of Clinical Medicine, 12(12), 3869. https://doi.org/10.3390/jcm12123869