
Research Evidence of the Role of the Glymphatic System and Its Potential Pharmacological Modulation in Neurodegenerative Diseases

Abstract
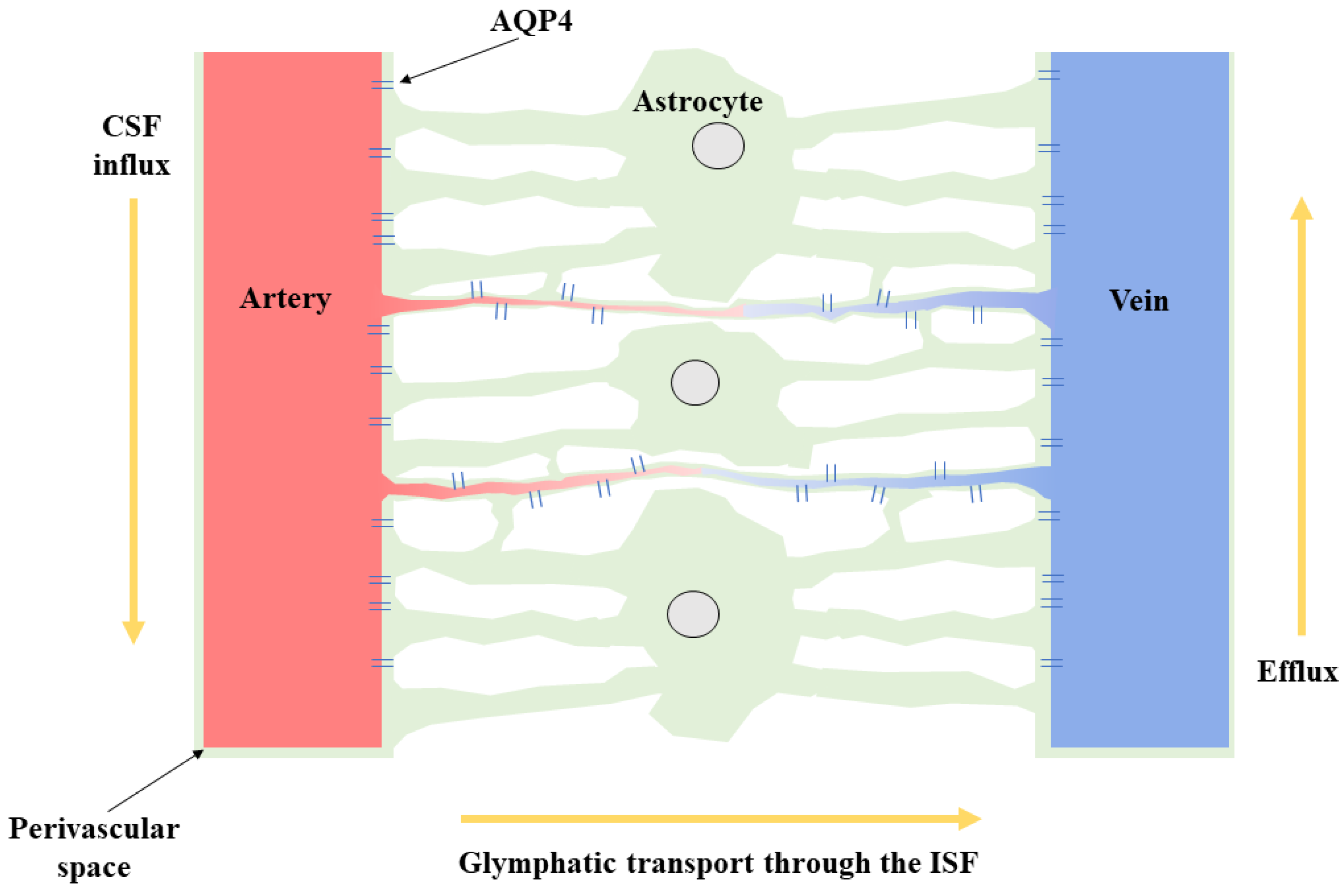
1. Introduction
2. Glymphatic System and Alzheimer’s Disease
3. Glymphatic System and Parkinson’s Disease
4. Glymphatic System and Huntington’s Disease
5. Glymphatic System and Motor Neuron Disease
6. Glymphatic System and Idiopathic Normal Pressure Hydrocephalus
7. Glymphatic System and Multiple Sclerosis:
8. Glymphatic System and Traumatic Brain Injury (TBI)
9. Pharmacological Modulation of the Glymphatic System in Neurodegenerative Diseases
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Nedergaard, M. The Glymphatic System: A Novel Component of Fundamental Neurobiology. J. Neurosci. 2021, 41, 7698–7711. [Google Scholar] [CrossRef]
- Natale, G.; Limanaqi, F.; Busceti, C.L.; Mastroiacovo, F.; Nicoletti, F.; Puglisi-Allegra, S.; Fornai, F. Glymphatic System as a Gateway to Connect Neurodegeneration From Periphery to CNS. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Pizzo, M.E.; Preston, J.E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘glymphatic’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Iliff, J.J.; Nedergaard, M. Is there a cerebral lymphatic system? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Galgani, A.; Puglisi-Allegra, S.; Limanaqi, F.; Busceti, C.L.; Fornai, F. Locus Coeruleus and neurovascular unit: From its role in physiology to its potential role in Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2020, 98, 2406–2434. [Google Scholar] [CrossRef] [PubMed]
- Kiviniemi, V.; Wang, X.; Korhonen, V.; Keinänen, T.; Tuovinen, T.; Autio, J.; LeVan, P.; Keilholz, S.; Zang, Y.F.; Hennig, J.; et al. Ultra-fast magnetic resonance encephalography of physiological brain activity - Glymphatic pulsation mechanisms? J. Cereb. Blood Flow Metab. 2016, 36, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Reddy, O.C.; van der Werf, Y.D. The Sleeping Brain: Harnessing the Power of the Glymphatic System through Lifestyle Choices. Brain Sci. 2020, 10, 868. [Google Scholar] [CrossRef]
- Shokri-Kojori, E.; Wang, G.J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef]
- Gupta, A.; Goyal, R. Amyloid beta plaque: A culprit for neurodegeneration. Acta Neurol. Belg. 2016, 116, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Silva, I.; Silva, J.; Ferreira, R.; Trigo, D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol. Res. Pract. 2021, 3, 5. [Google Scholar] [CrossRef]
- Chen, R.L.; Kassem, N.A.; Redzic, Z.B.; Chen, C.P.; Segal, M.B.; Preston, J.E. Age-related changes in choroid plexus and blood-cerebrospinal fluid barrier function in the sheep. Exp. Gerontol. 2009, 44, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, D.; Berdahl, J.P.; Zaydlarova, J.; Stinnett, S.; Fautsch, M.P.; Allingham, R.R. Cerebrospinal fluid pressure decreases with older age. PLoS ONE 2012, 7, e52664. [Google Scholar] [CrossRef]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef]
- Lv, T.; Zhao, B.; Hu, Q.; Zhang, X. The Glymphatic System: A Novel Therapeutic Target for Stroke Treatment. Front. Aging Neurosci. 2021, 13, 370. [Google Scholar] [CrossRef]
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 91–100. [Google Scholar] [CrossRef]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-X.; Liang, N.; Li, X.-L.; Yang, S.-H.; Wang, Y.-P.; Shi, N.-N. Diagnosis and Treatment for Mild Cognitive Impairment: A Systematic Review of Clinical Practice Guidelines and Consensus Statements. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ismail, Z.; Agüera-Ortiz, L.; Brodaty, H.; Cieslak, A.; Cummings, J.; Fischer, C.E.; Gauthier, S.; Geda, Y.E.; Herrmann, N.; Kanji, J.; et al. The Mild Behavioral Impairment Checklist (MBI-C): A Rating Scale for Neuropsychiatric Symptoms in Pre-Dementia Populations. J. Alzheimers Dis. 2017, 56, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Studart, A.N.; Nitrini, R. Subjective cognitive decline: The first clinical manifestation of Alzheimer’s disease? Dement Neuropsychol. 2016, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, C.C.; Yao, J.; Brinton, R.D. Targeting the prodromal stage of Alzheimer’s disease: Bioenergetic and mitochondrial opportunities. Neurotherapeutics 2015, 12, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.; Lynch, C.A.; Walsh, C.; Coen, R.; Coakley, D.; Lawlor, B.A. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 2005, 6, 347–352. [Google Scholar] [CrossRef]
- Ju, Y.E.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Bitar, R.D.; Torres-Garza, J.L.; Reiter, R.J.; Phillips, W.T. Neural glymphatic system: Clinical implications and potential importance of melatonin. Melatonin Res. 2021, 4, 551–565. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Schubert, J.J.; Veronese, M.; Marchitelli, L.; Bodini, B.; Tonietto, M.; Stankoff, B.; Brooks, D.J.; Bertoldo, A.; Edison, P.; Turkheimer, F.E. Dynamic (11)C-PiB PET Shows Cerebrospinal Fluid Flow Alterations in Alzheimer Disease and Multiple Sclerosis. J. Nucl. Med. 2019, 60, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Reeves, B.C.; Karimy, J.K.; Kundishora, A.J.; Mestre, H.; Cerci, H.M.; Matouk, C.; Alper, S.L.; Lundgaard, I.; Nedergaard, M.; Kahle, K.T. Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol. Med. 2020, 26, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.M.; Kuller, L.H.; Barinas-Mitchell, E.J.; Mackey, R.H.; McDade, E.M.; Klunk, W.E.; Aizenstein, H.J.; Cohen, A.D.; Snitz, B.E.; Mathis, C.A.; et al. Pulse wave velocity is associated with β-amyloid deposition in the brains of very elderly adults. Neurology 2013, 81, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer’s disease mouse model with suppression of glymphatic clearance. Alzheimer’s Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta 2014, 1840, 1492–1506. [Google Scholar] [CrossRef]
- Benveniste, H.; Liu, X.; Koundal, S.; Sanggaard, S.; Lee, H.; Wardlaw, J. The Glymphatic System and Waste Clearance with Brain Aging: A Review. Gerontology 2019, 65, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Yang, L.; Sun, G.; Qi, S.; Li, B. Mechanism of depression as a risk factor in the development of Alzheimer’s disease: The function of AQP4 and the glymphatic system. Psychopharmacology 2017, 234, 365–379. [Google Scholar] [CrossRef]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, C.; Guo, Q.; Chu, H. Aquaporin-4 and Cognitive Disorders. Aging Dis. 2022, 13, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Huang, X.; Huang, X.; Mai, H.; Li, J.; Jiang, T.; Wang, X.; Lü, T. Aquaporin-4 and Alzheimer’s Disease. J. Alzheimers Dis. 2016, 52, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lunde, L.K.; Nuntagij, P.; Oguchi, T.; Camassa, L.M.; Nilsson, L.N.; Lannfelt, L.; Xu, Y.; Amiry-Moghaddam, M.; Ottersen, O.P.; et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 711–722. [Google Scholar] [CrossRef]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015, 10, 58. [Google Scholar] [CrossRef]
- Chandra, A.; Farrell, C.; Wilson, H.; Dervenoulas, G.; De Natale, E.R.; Politis, M. Aquaporin-4 polymorphisms predict amyloid burden and clinical outcome in the Alzheimer’s disease spectrum. Neurobiol. Aging 2021, 97, 1–9. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Hu, M.T.M. Sleep Disturbance as Potential Risk and Progression Factor for Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Schernhammer, E.; Schwarzschild, M.A.; Ascherio, A. A prospective study of night shift work, sleep duration, and risk of Parkinson’s disease. Am. J. Epidemiol. 2006, 163, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.H.; Chen, Y.T.; Tseng, C.M.; Wu, L.A.; Perng, D.W.; Chen, Y.M.; Chen, T.J.; Chang, S.C.; Chou, K.T. Sleep disorders and an increased risk of Parkinson’s disease in individuals with non-apnea sleep disorders: A population-based cohort study. J. Sleep Res. 2017, 26, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Schenck, C.H.; Mahowald, M.W. REM sleep behavior disorder: Clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep 2002, 25, 120–138. [Google Scholar] [CrossRef] [PubMed]
- St Louis, E.K.; Boeve, A.R.; Boeve, B.F. REM Sleep Behavior Disorder in Parkinson’s Disease and Other Synucleinopathies. Mov. Disord. 2017, 32, 645–658. [Google Scholar] [CrossRef]
- Hawkes, C.H. The prodromal phase of sporadic Parkinson’s disease: Does it exist and if so how long is it? Mov. Disord. 2008, 23, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Jeon, B.S. Clinical implication of REM sleep behavior disorder in Parkinson’s disease. J. Parkinsons Dis. 2014, 4, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef]
- Baumann-Vogel, H.; Hor, H.; Poryazova, R.; Valko, P.; Werth, E.; Baumann, C.R. REM sleep behavior in Parkinson disease: Frequent, particularly with higher age. PLoS ONE 2020, 15, e0243454. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Bertrand, J.A.; Montplaisir, J.; Desjardins, C.; Vendette, M.; Rios Romenets, S.; Panisset, M.; Gagnon, J.F. Rapid eye movement sleep behavior disorder and risk of dementia in Parkinson’s disease: A prospective study. Mov. Disord. 2012, 27, 720–726. [Google Scholar] [CrossRef]
- Diederich, N.J.; Vaillant, M.; Mancuso, G.; Lyen, P.; Tiete, J. Progressive sleep ’destructuring’ in Parkinson’s disease. A polysomnographic study in 46 patients. Sleep Med. 2005, 6, 313–318. [Google Scholar] [CrossRef]
- Chi, Y.; Fan, Y.; He, L.; Liu, W.; Wen, X.; Zhou, S.; Wang, X.; Zhang, C.; Kong, H.; Sonoda, L.; et al. Novel role of aquaporin-4 in CD4+ CD25+ T regulatory cell development and severity of Parkinson’s disease. Aging Cell 2011, 10, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liang, R.; Yang, B.; Zhou, Y.; Liu, M.; Fang, F.; Ding, J.; Fan, Y.; Hu, G. Aquaporin-4 mediates communication between astrocyte and microglia: Implications of neuroinflammation in experimental Parkinson’s disease. Neuroscience 2016, 317, 65–75. [Google Scholar] [CrossRef]
- Xue, X.; Zhang, W.; Zhu, J.; Chen, X.; Zhou, S.; Xu, Z.; Hu, G.; Su, C. Aquaporin-4 deficiency reduces TGF-β1 in mouse midbrains and exacerbates pathology in experimental Parkinson’s disease. J. Cell Mol. Med. 2019, 23, 2568–2582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, B.; Sun, H.; Zhou, Y.; Liu, M.; Ding, J.; Fang, F.; Fan, Y.; Hu, G. Aquaporin-4 deficiency diminishes the differential degeneration of midbrain dopaminergic neurons in experimental Parkinson’s disease. Neurosci. Lett. 2016, 614, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wang, W.; Zheng, X.; Xia, D.; Liu, H.; Qin, C.; Tian, H.; Teng, J. Decreased AQP4 Expression Aggravates ɑ-Synuclein Pathology in Parkinson’s Disease Mice, Possibly via Impaired Glymphatic Clearance. J. Mol. Neurosci. 2021, 71, 2500–2513. [Google Scholar] [CrossRef]
- Hoshi, A.; Tsunoda, A.; Tada, M.; Nishizawa, M.; Ugawa, Y.; Kakita, A. Expression of Aquaporin 1 and Aquaporin 4 in the Temporal Neocortex of Patients with Parkinson’s Disease. Brain Pathol. 2017, 27, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Garon, M.; Weis, L.; Fiorenzato, E.; Pistonesi, F.; Cagnin, A.; Bertoldo, A.; Anglani, M.; Cecchin, D.; Antonini, A.; Biundo, R. Quantification of Brain β-Amyloid Load in Parkinson’s Disease With Mild Cognitive Impairment: A PET/MRI Study. Front. Neurol. 2021, 12, 760518. [Google Scholar] [CrossRef]
- Chen, H.; Wan, H.; Zhang, M.; Wardlaw, J.M.; Feng, T.; Wang, Y. Perivascular space in Parkinson’s disease: Association with CSF amyloid/tau and cognitive decline. Parkinsonism. Relat. Disord. 2022, 95, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Mashima, K.; Ito, D.; Kameyama, M.; Osada, T.; Tabuchi, H.; Nihei, Y.; Yoshizaki, T.; Noguchi, E.; Tanikawa, M.; Iizuka, T.; et al. Extremely Low Prevalence of Amyloid Positron Emission Tomography Positivity in Parkinson’s Disease without Dementia. Eur. Neurol. 2017, 77, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Melzer, T.R.; Stark, M.R.; Keenan, R.J.; Myall, D.J.; MacAskill, M.R.; Pitcher, T.L.; Livingston, L.; Grenfell, S.; Horne, K.L.; Young, B.N.; et al. Beta Amyloid Deposition Is Not Associated With Cognitive Impairment in Parkinson’s Disease. Front. Neurol. 2019, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, E.; Roe, R.M.; Mailman, R.B.; Chambers, J.E. (Eds.) Dictionary of Toxicology, 3rd ed.; Academic Press: Boston, MA, USA, 2015; pp. 1–46. [Google Scholar] [CrossRef]
- Huang, M.; Wang, Y.; Wang, L.; Chen, B.; Wang, X.; Hu, Y. APOE rs405509 polymorphism and Parkinson’s disease risk in the Chinese population. Neurosci. Lett. 2020, 736, 135256. [Google Scholar] [CrossRef]
- Kim, H.; Nanavaty, N.; Ahmed, H.; Mathur, V.A.; Anderson, B.A. Motivational Salience Guides Attention to Valuable and Threatening Stimuli: Evidence from Behavior and Functional Magnetic Resonance Imaging. J. Cogn. Neurosci. 2021. [Google Scholar] [CrossRef]
- Tipton, P.; Bülbül, N.; Crook, J.; Quicksall, Z.; Ross, O.; Uitti, R.; Wszolek, Z.; Ertekin-Taner, N. Effects of sex and APOE on Parkinson’s Disease-related cognitive decline. Neurol. I Neurochir. Pol. 2021, 55, 559–566. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Bol, J.G.; Van Haastert, E.S.; Rozemuller, A.J.; Bu, G.; Drukarch, B.; Hoozemans, J.J. Apolipoprotein E and LRP1 Increase Early in Parkinson’s Disease Pathogenesis. Am. J. Pathol. 2011, 179, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lin, L.; Yin, L.; Hao, X.; Tian, J.; Zhang, X.; Ren, Y.; Li, C.; Yang, Y. Acutely Inhibiting AQP4 With TGN-020 Improves Functional Outcome by Attenuating Edema and Peri-Infarct Astrogliosis After Cerebral Ischemia. Front. Immunol. 2022, 13, 870029. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2022, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, J.; Hwang, J. On the hunt for a cure: A guide to Huntington disease. JAAPA 2021, 34. [Google Scholar] [CrossRef] [PubMed]
- Potkin, K.T.; Potkin, S.G. New directions in therapeutics for Huntington disease. Future Neurol. 2018, 13, 101–121. [Google Scholar] [CrossRef]
- Herzog–Krzywoszanska, R.; Krzywoszanski, L. Sleep Disorders in Huntington’s Disease. Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef]
- Caron, N.S.; Banos, R.; Yanick, C.; Aly, A.E.; Byrne, L.M.; Smith, E.D.; Xie, Y.; Smith, S.E.P.; Potluri, N.; Findlay Black, H.; et al. Mutant Huntingtin Is Cleared from the Brain via Active Mechanisms in Huntington Disease. J. Neurosci. 2021, 41, 780–796. [Google Scholar] [CrossRef]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J. Clin. Invest. 2015, 125, 1979–1986. [Google Scholar] [CrossRef]
- Foster, L.A.; Salajegheh, M.K. Motor Neuron Disease: Pathophysiology, Diagnosis, and Management. Am. J. Med. 2019, 132, 32–37. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Nicaise, C.; Soyfoo, M.S.; Authelet, M.; De Decker, R.; Bataveljic, D.; Delporte, C.; Pochet, R. Aquaporin-4 overexpression in rat ALS model. Anat. Rec. 2009, 292, 207–213. [Google Scholar] [CrossRef]
- Bataveljić, D.; Nikolić, L.; Milosević, M.; Todorović, N.; Andjus, P.R. Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia 2012, 60, 1991–2003. [Google Scholar] [CrossRef]
- Dai, J.; Lin, W.; Zheng, M.; Liu, Q.; He, B.; Luo, C.; Lu, X.; Pei, Z.; Su, H.; Yao, X. Alterations in AQP4 expression and polarization in the course of motor neuron degeneration in SOD1G93A mice. Mol. Med. Rep. 2017, 16, 1739–1746. [Google Scholar] [CrossRef]
- Cui, Y.; Masaki, K.; Yamasaki, R.; Imamura, S.; Suzuki, S.O.; Hayashi, S.; Sato, S.; Nagara, Y.; Kawamura, M.F.; Kira, J. Extensive dysregulations of oligodendrocytic and astrocytic connexins are associated with disease progression in an amyotrophic lateral sclerosis mouse model. J. Neuroinflamm. 2014, 11, 42. [Google Scholar] [CrossRef]
- Zou, S.; Lan, Y.L.; Wang, H.; Zhang, B.; Sun, Y.G. The potential roles of aquaporin 4 in amyotrophic lateral sclerosis. Neurol. Sci. 2019, 40, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Papadopoulos, M.C.; Manley, G.T.; Verkman, A.S. Aquaporin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J. Neurochem. 2005, 95, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Auguste, K.I.; Manley, G.T.; Verkman, A.S. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J. Cereb. Blood Flow Metab. 2006, 26, 1527–1537. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, H.; Verkman, A.S. Greatly attenuated experimental autoimmune encephalomyelitis in aquaporin-4 knockout mice. BMC Neurosci. 2009, 10, 94. [Google Scholar] [CrossRef]
- Akdemir, G.; Ratelade, J.; Asavapanumas, N.; Verkman, A.S. Neuroprotective effect of aquaporin-4 deficiency in a mouse model of severe global cerebral ischemia produced by transient 4-vessel occlusion. Neurosci. Lett. 2014, 574, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Matsumoto, S.; Moriwaki, Y.; Okuda, T.; Ohara, S.; Yamanaka, K.; Abe, Y.; Yasui, M.; Misawa, H. Dissociation of blood-brain barrier disruption and disease manifestation in an aquaporin-4-deficient mouse model of amyotrophic lateral sclerosis. Neurosci. Res. 2018, 133, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Nassar, B.R.; Lippa, C.F. Idiopathic Normal Pressure Hydrocephalus: A Review for General Practitioners. Gerontol. Geriatr. Med. 2016, 2, 2333721416643702. [Google Scholar] [CrossRef]
- Gallia, G.L.; Rigamonti, D.; Williams, M.A. The diagnosis and treatment of idiopathic normal pressure hydrocephalus. Nat. Clin. Pract. Neurol. 2006, 2, 375–381. [Google Scholar] [CrossRef]
- Martín-Láez, R.; Caballero-Arzapalo, H.; Valle-San Román, N.; López-Menéndez, L.; Arango-Lasprilla, J.C.; Vázquez-Barquero, A. Incidence of Idiopathic Normal-Pressure Hydrocephalus in Northern Spain. World Neurosurg. 2016, 87, 298–310. [Google Scholar] [CrossRef]
- Tan, C.; Wang, X.; Wang, Y.; Wang, C.; Tang, Z.; Zhang, Z.; Liu, J.; Xiao, G. The Pathogenesis Based on the Glymphatic System, Diagnosis, and Treatment of Idiopathic Normal Pressure Hydrocephalus. Clin. Interv. Aging 2021, 16, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-m.; Yao, Y.; Xu, S.; Li, X.; Hu, F.; Wang, H.; Ding, J.; Wang, X. White Matter Microstructural Damage Associated With Gait Abnormalities in Idiopathic Normal Pressure Hydrocephalus. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Ringstad, G.; Vatnehol, S.A.S.; Eide, P.K. Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain 2017, 140, 2691–2705. [Google Scholar] [CrossRef] [PubMed]
- Eide, P.K.; Ringstad, G. Delayed clearance of cerebrospinal fluid tracer from entorhinal cortex in idiopathic normal pressure hydrocephalus: A glymphatic magnetic resonance imaging study. J. Cereb. Blood Flow Metab. 2019, 39, 1355–1368. [Google Scholar] [CrossRef]
- Eide, P.K.; Hansson, H.A. Astrogliosis and impaired aquaporin-4 and dystrophin systems in idiopathic normal pressure hydrocephalus. Neuropathol. Appl. Neurobiol. 2018, 44, 474–490. [Google Scholar] [CrossRef] [PubMed]
- Hasan-Olive, M.M.; Enger, R.; Hansson, H.A.; Nagelhus, E.A.; Eide, P.K. Loss of perivascular aquaporin-4 in idiopathic normal pressure hydrocephalus. Glia 2019, 67, 91–100. [Google Scholar] [CrossRef]
- Ishikawa, M.; Oowaki, H.; Takezawa, M.; Takenaka, T.; Yamada, S.; Yamamoto, K.; Okamoto, S. Disproportionately Enlarged Subarachnoid Space Hydrocephalus in Idiopathic Normal-Pressure Hydrocephalus and Its Implication in Pathogenesis. Acta Neurochir. Suppl. 2016, 122, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, K.; Vakis, A.; Tsitsipanis, C. Pathogenesis of idiopathic Normal Pressure Hydrocephalus: A review of knowledge. J. Clin. Neurosci. 2019, 61, 10–13. [Google Scholar] [CrossRef]
- Jingami, N.; Uemura, K.; Asada-Utsugi, M.; Kuzuya, A.; Yamada, S.; Ishikawa, M.; Kawahara, T.; Iwasaki, T.; Atsuchi, M.; Takahashi, R.; et al. Two-Point Dynamic Observation of Alzheimer’s Disease Cerebrospinal Fluid Biomarkers in Idiopathic Normal Pressure Hydrocephalus. J. Alzheimers Dis. 2019, 72, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Trevick, S.; Younger, D.S. Epidemiology of Multiple Sclerosis. Neurol. Clin. 2016, 34, 919–939. [Google Scholar] [CrossRef] [PubMed]
- Faguy, K. Multiple Sclerosis: An Update. Radiol. Technol. 2016, 87, 529–550. [Google Scholar]
- Rohr, S.O.; Greiner, T.; Joost, S.; Amor, S.; Valk, P.V.; Schmitz, C.; Kipp, M. Aquaporin-4 Expression during Toxic and Autoimmune Demyelination. Cells 2020, 9, 2187. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Ikeda, K.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003, 106, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Aoki-Yoshino, K.; Uchihara, T.; Duyckaerts, C.; Nakamura, A.; Hauw, J.J.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with inflammatory diseases. Acta Neuropathol. 2005, 110, 281–288. [Google Scholar] [CrossRef]
- Vos, C.M.; Geurts, J.J.; Montagne, L.; van Haastert, E.S.; Bö, L.; van der Valk, P.; Barkhof, F.; de Vries, H.E. Blood-brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol. Dis. 2005, 20, 953–960. [Google Scholar] [CrossRef]
- Carotenuto, A.; Cacciaguerra, L.; Pagani, E.; Preziosa, P.; Filippi, M.; Rocca, M.A. Glymphatic system impairment in multiple sclerosis: Relation with brain damage and disability. Brain 1093. [Google Scholar] [CrossRef]
- Cristofori, I.; Levin, H.S. Chapter 37 - Traumatic brain injury and cognition. In Handbook of Clinical Neurology; Grafman, J., Salazar, A.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 128, pp. 579–611. [Google Scholar]
- Ng, S.Y.; Lee, A.Y.W. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Visser, K.; Koggel, M.; Blaauw, J.; van der Horn, H.J.; Jacobs, B.; van der Naalt, J. Blood-based biomarkers of inflammation in mild traumatic brain injury: A systematic review. Neurosci. Biobehav. Rev. 2022, 132, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.A.; Serpa, R.; Moattari, C.R.; Brown, A.; Greco, T.; Prins, M.L.; Teng, E. Repeat Mild Traumatic Brain Injury in Adolescent Rats Increases Subsequent β-Amyloid Pathogenesis. J. Neurotrauma 2017, 35, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Wright, D.K.; Yamakawa, G.R.; Shultz, S.R.; Mychasiuk, R. Repetitive Mild Traumatic Brain Injury Alters Glymphatic Clearance Rates in Limbic Structures of Adolescent Female Rats. Sci. Rep. 2020, 10, 6254. [Google Scholar] [CrossRef]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chopp, M.; Ding, G.; Davoodi-Bojd, E.; Zhang, L.; Li, Q.; Zhang, Y.; Xiong, Y.; Jiang, Q. MRI detection of impairment of glymphatic function in rat after mild traumatic brain injury. Brain Res. 2020, 1747, 147062. [Google Scholar] [CrossRef] [PubMed]
- Sullan, M.J.; Asken, B.M.; Jaffee, M.S.; DeKosky, S.T.; Bauer, R.M. Glymphatic system disruption as a mediator of brain trauma and chronic traumatic encephalopathy. Neurosci. Biobehav. Rev. 2018, 84, 316–324. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef]
- Doustar, J.; Danan, I.J. Glymphatic System Dysfunction in Mild Traumatic Brain Injury. Neurology 2022, 98, S24–S25. [Google Scholar] [CrossRef]
- Lohela, T.J.; Lilius, T.O.; Nedergaard, M. The glymphatic system: Implications for drugs for central nervous system diseases. Nat. Rev. Drug Discov. 2022, 21, 763–779. [Google Scholar] [CrossRef]
- Cao, X.; Xu, H.; Feng, W.; Su, D.; Xiao, M. Deletion of aquaporin-4 aggravates brain pathology after blocking of the meningeal lymphatic drainage. Brain Res. Bull. 2018, 143, 83–96. [Google Scholar] [CrossRef]
- Lan, Y.L.; Chen, J.J.; Hu, G.; Xu, J.; Xiao, M.; Li, S. Aquaporin 4 in Astrocytes is a Target for Therapy in Alzheimer’s Disease. Curr. Pharm. Des. 2017, 23, 4948–4957. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Yu, X.; Xu, W.; Lenahan, C.; Tu, S.; Shao, A. The role of glymphatic system in the cerebral edema formation after ischemic stroke. Exp. Neurol. 2021, 340, 113685. [Google Scholar] [CrossRef]
- Huber, V.J.; Igarashi, H.; Ueki, S.; Kwee, I.L.; Nakada, T. Aquaporin-4 facilitator TGN-073 promotes interstitial fluid circulation within the blood-brain barrier: [17O]H2O JJVCPE MRI study. Neuroreport 2018, 29, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Nakada, T. Identification of Aquaporin 4 inhibitors using in vitro and in silico methods. Bioorganic Med. Chem. 2009, 17, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Pirici, I.; Balsanu, T.A.; Bogdan, C.; Margaritescu, C.; Divan, T.; Vitalie, V.; Mogoanta, L.; Pirici, D.; Carare, R.O.; Muresanu, D.F. Inhibition of Aquaporin-4 Improves the Outcome of Ischaemic Stroke and Modulates Brain Paravascular Drainage Pathways. Int. J. Mol. Sci. 2017, 19, 46. [Google Scholar] [CrossRef]
- Migliati, E.; Meurice, N.; DuBois, P.; Fang, J.S.; Somasekharan, S.; Beckett, E.; Flynn, G.; Yool, A.J. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol. Pharmacol. 2009, 76, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Kwee, I.L.; Nakada, T. Inhibition of aquaporin 4 by antiepileptic drugs. Bioorg. Med. Chem. 2009, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, H.; Verkman, A.S. Lack of aquaporin-4 water transport inhibition by antiepileptics and arylsulfonamides. Bioorg. Med. Chem. 2008, 16, 7489–7493. [Google Scholar] [CrossRef] [PubMed]
- Detmers, F.J.; de Groot, B.L.; Müller, E.M.; Hinton, A.; Konings, I.B.; Sze, M.; Flitsch, S.L.; Grubmüller, H.; Deen, P.M. Quaternary ammonium compounds as water channel blockers. Specificity, potency, and site of action. J. Biol. Chem. 2006, 281, 14207–14214. [Google Scholar] [CrossRef]
- Jha, R.M.; Raikwar, S.P.; Mihaljevic, S.; Casabella, A.M.; Catapano, J.S.; Rani, A.; Desai, S.; Gerzanich, V.; Simard, J.M. Emerging therapeutic targets for cerebral edema. Expert Opin. Ther. Targets 2021, 25, 917–938. [Google Scholar] [CrossRef]
- Cui, D.; Jia, S.; Yu, J.; Li, D.; Li, T.; Liu, Y.; Chang, J.; Wang, X.; Liu, X.; Wang, Y.-F. Alleviation of Cerebral Infarction of Rats With Middle Cerebral Artery Occlusion by Inhibition of Aquaporin 4 in the Supraoptic Nucleus. ASN Neuro. 2020, 12, 1759091420960550. [Google Scholar] [CrossRef]
- Rauen, K.; Pop, V.; Trabold, R.; Badaut, J.; Plesnila, N. Vasopressin V(1a) Receptors Regulate Cerebral Aquaporin 1 after Traumatic Brain Injury. J. Neurotrauma 2020, 37, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Taya, K.; Gulsen, S.; Okuno, K.; Prieto, R.; Marmarou, C.R.; Marmarou, A. Modulation of AQP4 expression by the selective V1a receptor antagonist, SR49059, decreases trauma-induced brain edema. In Acta Neurochirurgica Supplements; Springer: Vienna, Austria, 2009; pp. 425–429. [Google Scholar]
- Oliveira, G.B.; Fontes Ede, A., Jr.; de Carvalho, S.; da Silva, J.B.; Fernandes, L.M.; Oliveira, M.C.; Prediger, R.D.; Gomes-Leal, W.; Lima, R.R.; Maia, C.S. Minocycline mitigates motor impairments and cortical neuronal loss induced by focal ischemia in rats chronically exposed to ethanol during adolescence. Brain Res. 2014, 1561, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.H.; Gilbert, M.E. Modest thyroid hormone insufficiency during development induces a cellular malformation in the corpus callosum: A model of cortical dysplasia. Endocrinology 2007, 148, 2593–2597. [Google Scholar] [CrossRef]
- Costa, L.E.S.; Clementino-Neto, J.; Mendes, C.B.; Franzon, N.H.; Costa, E.O.; Moura-Neto, V.; Ximenes-da-Silva, A. Evidence of Aquaporin 4 Regulation by Thyroid Hormone During Mouse Brain Development and in Cultured Human Glioblastoma Multiforme Cells. Front. Neurosci. 2019, 13, 317. [Google Scholar] [CrossRef] [PubMed]
- Sadana, P.; Coughlin, L.; Burke, J.; Woods, R.; Mdzinarishvili, A. Anti-edema action of thyroid hormone in MCAO model of ischemic brain stroke: Possible association with AQP4 modulation. J. Neurol. Sci. 2015, 354, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Vandebroek, A.; Yasui, M. Regulation of AQP4 in the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 1603. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical Perspectives in Neurodegeneration. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin regulates Aβ production/clearance balance and Aβ neurotoxicity: A potential therapeutic molecule for Alzheimer’s disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef]
- Tordjman, S.; Chokron, S.; Delorme, R.; Charrier, A.; Bellissant, E.; Jaafari, N.; Fougerou, C. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr. Neuropharmacol. 2017, 15, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Matsubara, E.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Zagorski, M.; Sambamurti, K. Melatonin Treatment Enhances Aβ Lymphatic Clearance in a Transgenic Mouse Model of Amyloidosis. Curr. Alzheimer. Res. 2018, 15, 637–642. [Google Scholar] [CrossRef]
- Ren, X.; Liu, S.; Lian, C.; Li, H.; Li, K.; Li, L.; Zhao, G. Dysfunction of the Glymphatic System as a Potential Mechanism of Perioperative Neurocognitive Disorders. Front. Aging Neurosci. 2021, 13, 659457. [Google Scholar] [CrossRef] [PubMed]
- Hablitz, L.M.; Vinitsky, H.S.; Sun, Q.; Stæger, F.F.; Sigurdsson, B.; Mortensen, K.N.; Lilius, T.O.; Nedergaard, M. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci. Adv. 2019, 5, eaav5447. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Chung, H.W.; Lee, E.J.; Jung, K.H.; Paik, J.Y.; Lee, K.H. α2-Adrenergic agonists including xylazine and dexmedetomidine inhibit norepinephrine transporter function in SK-N-SH cells. Neurosci. Lett. 2013, 541, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018, 13, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Han, H.; Yang, J.; Sun, M.; Cui, D.; Li, Y.; Gao, Y.; Zou, J. Brain interstitial fluid drainage and extracellular space affected by inhalational isoflurane: In comparison with intravenous sedative dexmedetomidine and pentobarbital sodium. Sci. China Life Sci. 2020, 63, 1363–1379. [Google Scholar] [CrossRef]
- Bah, T.M.; Goodman, J.; Iliff, J.J. Sleep as a Therapeutic Target in the Aging Brain. Neurotherapeutics 2019, 16, 554–568. [Google Scholar] [CrossRef] [PubMed]
- Lilius, T.O.; Blomqvist, K.; Hauglund, N.L.; Liu, G.; Stæger, F.F.; Bærentzen, S.; Du, T.; Ahlström, F.; Backman, J.T.; Kalso, E.A.; et al. Dexmedetomidine enhances glymphatic brain delivery of intrathecally administered drugs. J. Control. Release 2019, 304, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Singh, P.M. Current role of dexmedetomidine in clinical anesthesia and intensive care. Anesth. Essays Res. 2011, 5, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Persson, N.; Uusalo, P.; Nedergaard, M.; Lohela, T.J.; Lilius, T.O. Could dexmedetomidine be repurposed as a glymphatic enhancer? Trends Pharmacol. Sci. 2022, 43, 1030–1040. [Google Scholar] [CrossRef]
- Semyachkina-Glushkovskaya, O.; Postnov, D.; Penzel, T.; Kurths, J. Sleep as a Novel Biomarker and a Promising Therapeutic Target for Cerebral Small Vessel Disease: A Review Focusing on Alzheimer’s Disease and the Blood-Brain Barrier. Int. J. Mol. Sci. 2020, 21, 6293. [Google Scholar] [CrossRef] [PubMed]
- Wafford, K.A. Aberrant waste disposal in neurodegeneration: Why improved sleep could be the solution. Cerebral. Circ. Cogn. Behav. 2021, 2, 100025. [Google Scholar] [CrossRef] [PubMed]
- Lucey, B.P.; McCullough, A.; Landsness, E.C.; Toedebusch, C.D.; McLeland, J.S.; Zaza, A.M.; Fagan, A.M.; McCue, L.; Xiong, C.; Morris, J.C.; et al. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaau6550. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.K.; Hall-Porter, J.M.; Griffin, K.S.; Dodson, E.R.; Forst, E.H.; Curry, D.T.; Eisenstein, R.D.; Schweitzer, P.K. Enhancing Slow Wave Sleep with Sodium Oxybate Reduces the Behavioral and Physiological Impact of Sleep Loss. Sleep 2010, 33, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Gerashchenko, D.; Timofeev, I.; Bacskai, B.J.; Kastanenka, K.V. Slow Wave Sleep Is a Promising Intervention Target for Alzheimer’s Disease. Front. Neurosci. 2020, 14, 705. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.; Oishi, Y.; Bjorness, T.E.; Greene, R.W. Gating and the Need for Sleep: Dissociable Effects of Adenosine A1 and A2A Receptors. Front. Neurosci. 2019, 13, 740. [Google Scholar] [CrossRef] [PubMed]
- Leenaars, C.H.C.; Savelyev, S.A.; Van der Mierden, S.; Joosten, R.; Dematteis, M.; Porkka-Heiskanen, T.; Feenstra, M.G.P. Intracerebral Adenosine During Sleep Deprivation: A Meta-Analysis and New Experimental Data. J. Circadian Rhythm. 2018, 16, 11. [Google Scholar] [CrossRef]
- Zhao, Z.-A.; Li, P.; Ye, S.-Y.; Ning, Y.-L.; Wang, H.; Peng, Y.; Yang, N.; Zhao, Y.; Zhang, Z.-H.; Chen, J.-F.; et al. Perivascular AQP4 dysregulation in the hippocampal CA1 area after traumatic brain injury is alleviated by adenosine A2A receptor inactivation. Sci. Rep. 2017, 7, 2254. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.; Shen, H.Y.; Cherasse, Y.; Qu, W.M.; Huang, Z.L.; Bass, C.E.; Winsky-Sommerer, R.; Semba, K.; Fredholm, B.B.; Boison, D.; et al. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J. Neurosci. 2011, 31, 10067–10075. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.R. Mechanisms and Mitochondrial Redox Signaling in Photobiomodulation. Photochem. Photobiol. 2018, 94, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, E.; Navolokin, N.; Shirokov, A.; Khlebtsov, B.; Dubrovsky, A.; Saranceva, E.; Abdurashitov, A.; Khorovodov, A.; Terskov, A.; Mamedova, A.; et al. Pilot study of transcranial photobiomodulation of lymphatic clearance of beta-amyloid from the mouse brain: Breakthrough strategies for non-pharmacologic therapy of Alzheimer’s disease. Biomed. Opt. Express 2019, 10, 4003–4017. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina-Glushkovskaya, O.; Abdurashitov, A.; Dubrovsky, A.; Klimova, M.; Agranovich, I.; Terskov, A.; Shirokov, A.; Vinnik, V.; Kuzmina, A.; Lezhnev, N.; et al. Photobiomodulation of lymphatic drainage and clearance: Perspective strategy for augmentation of meningeal lymphatic functions. Biomed. Opt. Express 2020, 11, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Khademi, M.; Bragin, D.E.; DiDuro, J.O. Photobiomodulation Therapy and the Glymphatic System: Promising Applications for Augmenting the Brain Lymphatic Drainage System. Int. J. Mol. Sci. 2022, 23, 2975. [Google Scholar] [CrossRef]
- Kylkilahti, T.M.; Berends, E.; Ramos, M.; Shanbhag, N.C.; Töger, J.; Markenroth Bloch, K.; Lundgaard, I. Achieving brain clearance and preventing neurodegenerative diseases—A glymphatic perspective. J. Cereb. Blood Flow Metab. 2021, 41, 2137–2149. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cohort | Main Findings | References |
|---|---|---|
| 100 LMCI or mild AD (Aβ +ve), 469 MCI (168 Aβ +ve, 145 Aβ -ve), 244 LMCI, 97 Aβ -ve CN controls | AQP4 SNP rs72878794 = ↓ uptake of Aβ AQP4 SNP rs151244 = ↑ uptake of Aβ | (Chandra et al. 2021) [48] |
| APP695/PS1-dE9 transgenic (APP/PS1), AQP4−/−/APP/PS1, WT, APQ4 KO (AQP4−/−) rodents (3 mnth) | Relative to APP/PS1 groups, AQP4-/-APP/PS1 rodents = ↑ cerebral cortex microglial Aβ activation and phagocytosis | (Feng et al. 2020) [37] |
| C57BL/6 rodents (8–12 wks M) | ↓ CSF influx in AQP4- rodents | (Iliff et al. 2012) [5] |
| Rodents [M + F, APPswe/PS1dE9 (APP/PS1), C57BL/6J, Tg (Cspg4-Ds Red.T1)1Akik/J (NG2-DsRed reporter mice), LM controls | APP/PS1 = Aβ accumulation, ↓ glymphatic clearance | (Peng et al. 2016) [36] |
| 11 AD 12 MCI 20 MS Matched controls | [11C]-PiB PET = ↓ signal clearance (lat ventricles) AD vs. HC. | (Schubert et al. 2019) [33] |
| APP/PS1 mice (12-mnths) | AQP4- APP/PS1 = ↑ astrocyte atrophy, CAA, Aβ build-up, ↓ cognition | (Xu et al. 2015) [47] |
| 79 total PM (cog intact 33–57yrs, Cog intact 61–96, AD 60+) | Assoc. between AD and localization of AQP4 (PV) Assoc. with AQP4 (PV) localization and ↑ Aβ | (Zeppenfeld et al. 2017) [45] |
| Cohort | Main Findings | References | |
|---|---|---|---|
| PD: Mouse models | Inflammatory changes | AQP4−/− mice models showed reduced anti-inflammatory activity levels (reduced transforming growth factor-β1 levels along with reduced CD4+ and CD25+ regulatory T-Cells levels) compared to AQP4+/+ mice models. | (Chi et al. 2011; Sun et al. 2016; Xue et al. 2019) [64,65,66] |
| AQP4−/− mice models showed increased pro-inflammatory pathway activity and cytokines levels (increased TNF-α, IL-1 and IL-1β levels with increased NF-κB activity) compared to AQP4+/+ mice models. | (Sun et al. 2016; Zhang et al. 2016) [65,67] | ||
| Inflammatory changes after MPTP changes | AQP4−/− mice models were at significantly greater risk of MPTP neurotoxicity compared to mice with AQP4+/+ mice models. | (Zhang et al. 2016) [67] | |
| AQP4−/− mice models had significantly greater pro-inflammatory markers after MPTP delivery than AQP4+/+ mice models. | (Zhang et al. 2016) [74] | ||
| AQP4−/− mice models had significantly lower transforming growth factor- β1 (a suppressive cytokine) after MPTP delivery than AQP4+/+ mice models. | (Xue et al. 2019) [66] | ||
| α-synuclein | Reduced AQP4 expression was associated with greater α-synuclein deposition and progressive dopaminergic neurone loss within the SN (when comparing AQP4−/− and AQP4+/+ mice models) | [68] | |
| PD: Human | α-synuclein | Greater expression of AQP4 and AQP1 positive astrocytes were seen in the temporal lobe of the neocortical PD group compared to HC as well limbic and brain stem PD subgroups. | (Hoshi et al. 2017) [69] |
| PD cohort revealing a negative correlation between AQP1/AQP4 to α-synuclein deposition in neuronal layers II-III and V-VI respectively. | (Hoshi et al. 2017) [74] | ||
| ApoE | APOE rs405509 T allele is correlated with increased susceptibility of PD in a Chinese population. | (Huang et al. 2020) [75] | |
| Early PD patients with ApoE ε4 allele mutations have shown a more rate rapid cognitive decline than early PD patients without ApoE ε4 allele mutations. | (Kim et al. 2021) [76] | ||
| Non-demented PD patients with ApoE ε4 allele mutations have shown worse cognitive performance scores than non-demented PD patients without ApoE ε4 allele mutations. | (Tipton et al. 2021) [77] | ||
| Increased LRP1 and ApoE in LB and melanised neurons of the SN in PD patients and other LB diseases. | (Wilhelmus et al. 2011) [78] | ||
| LRP1 | Increased LRP1 and ApoE in LB and melanised neurons of the SN in PD patients and other LB diseases. | [78] | |
| Aβ | Aβ-positivity is still relatively small in non-demented PD and MCI- PD (though prevalence may rise in increasing cognitive impairment and PDD). | (Mashima et al. 2017; Garon et al. 2021) [70,72] | |
| PD patients with enlarged Basal Ganglia PVS (BG-PVS) at baseline had reduced CSF Aβ42 and had lower Montreal cognitive assessment (MoCA) scores at 3-year follow-up compared to PD patients without enlarged BG-PVS | (Chen et al. 2022) [79] | ||
| Increasing age was associated with increased cortical Florbetaben (an amyloid PET tracer) within PD patients. | (Melzer et al. 2019) [73] | ||
| When age was adjusted, no correlation was found between cortical FBB uptake and global cognitive ability within the PD cohort. | (Melzer et al. 2019) [73] | ||
| Aβ synergises with the other pathological processes, which can accelerate primary cognitive impairment seen in PD. | (Melzer et al. 2019; Garon et al. 2021) [70,73] | ||
| Cohort | Main Findings | References | |
|---|---|---|---|
| ALS: Mouse models | AQP4 changes in the spinal cord | AQP4 expression increased in the spinal cord of SOD1G93A mice as the disease progressed | (Dai et al. 2017) [91] |
| AQP4 polarization decreased as the disease progressed, and AQP4 polarized localization at the endfeet of astrocytes was decreased in the spinal ventral horn of SOD1G93A mice at the disease onset and end stages. | (Dai et al. 2017) [91] | ||
| AQP4 changes in the BBB | Alternations to AQP4 in ALS may cause reduced BBB integrity, as a result, AQP4 changes lead to impaired potassium and connexin regulation, resulting in increased BBB permeability | (Cui et al. 2014; Zou et al. 2019) [92,93] | |
| SOD1G93A AQP4-/- ALS mouse models had improved BBB permeability compared to AQP4+/+ SOD1G93A mouse models, with reduced hemosiderin deposition and immunoglobulin leakage. | (Watanabe-Matsumoto et al. 2018) [99] | ||
| Changes in the glymphatic system and disease outcomes due to AQP4 changes | Disease onset and lifespan were decreased significantly in AQP4−/− SOD1G93A mouse models compared to AQP4+/+ SOD1G93A models | [99] | |
| Cohort | Main Findings | References | |
|---|---|---|---|
| MS: Mouse models | AQP4 changes in demyelinated areas | In immunohistological analysis, cuprizone + EAE mice showed increased AQP4 expression at the centre of the inflammatory lesions with reduced AQP4 expression and polarity at the edge of the lesion | (Rohr et al. 2020) [114] |
| In immunohistological analysis, Cuprizone toxin-induced demyelination mice models exhibited loss of AQP4 polarity within the endfeet of astrocytes surrounding perivascular structures at the perivascular endfeet of astrocytes and diffuse increase in AQP4 expression. | (Rohr et al. 2020) [114] | ||
| MS: Humans | Inflammatory changes in demyelinated areas | In immunohistological analysis, the post-mortem advanced MS cohort showed diffuse AQP4 expression increases in chronic-active lesions in advanced MS. | [114] |
| MRI: DTI-ALPS | ALPS index was lower in both RRMS and progressive multiple sclerosis patients compared to HC, with progressive multiple sclerosis patients exhibiting lower ALPS values than RRMS patients | (Carotenuto et al. 2021) [118] | |
| Lower ALPS index score in the MS groups was associated with more severe clinical disability, more significant lesion load, grey matter (GM) atrophy, reduced mean diffusivity, and fractional anisotropy in normal-appearing white matter | (Carotenuto et al. 2021) [118] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verghese, J.P.; Terry, A.; de Natale, E.R.; Politis, M. Research Evidence of the Role of the Glymphatic System and Its Potential Pharmacological Modulation in Neurodegenerative Diseases. J. Clin. Med. 2022, 11, 6964. https://doi.org/10.3390/jcm11236964
Verghese JP, Terry A, de Natale ER, Politis M. Research Evidence of the Role of the Glymphatic System and Its Potential Pharmacological Modulation in Neurodegenerative Diseases. Journal of Clinical Medicine. 2022; 11(23):6964. https://doi.org/10.3390/jcm11236964
Chicago/Turabian StyleVerghese, Joji Philip, Alana Terry, Edoardo Rosario de Natale, and Marios Politis. 2022. "Research Evidence of the Role of the Glymphatic System and Its Potential Pharmacological Modulation in Neurodegenerative Diseases" Journal of Clinical Medicine 11, no. 23: 6964. https://doi.org/10.3390/jcm11236964
APA StyleVerghese, J. P., Terry, A., de Natale, E. R., & Politis, M. (2022). Research Evidence of the Role of the Glymphatic System and Its Potential Pharmacological Modulation in Neurodegenerative Diseases. Journal of Clinical Medicine, 11(23), 6964. https://doi.org/10.3390/jcm11236964