1. Introduction
Acute myeloid leukaemia (AML) is a clonal haematopoietic stem cell disease [
1,
2] characterized by bone marrow infiltration with leukemic myeloid blast cells and low counts of other hematopoietic lineages, producing leucopenia, anemia and thrombocytopenia. AML is frequently associated with a life-threatening haemorrhage. Haemorrhagic complications are a very frequent part of the clinical picture of acute myeloid leukemia. The main causes of this complication are thrombocytopenia and/or defective platelet function, abnormalities of coagulation or fibrinolysis process. It was proven that the high expression of activated GPIIbIIIa, especially p selectin and CD 63, are present in AML patients with a bleeding history. Abnormalities of platelet aggregation were also associated with haemorrhagic complications in the past. Thrombocytopenia is not correlated with a high incidence of bleeding in AML patients [
3]. Abnormalities of coagulation and fibrinolysis are present especially in APL but also, in rare cases, in other subtypes of AML such as AML [
4,
5]. The frequency of disseminated coagulation (DIC) in hematologic malignancies is not low (12.7%), and one half of these patients had an unfavourable evolution, haemorrhage being the main cause of mortality in these patients [
4]. Fibrinolysis is one of the most important factors involved in haemorrhagic complication in AML. Both mechanisms, secondary and primary, of DIC, such as high levels of urokinase-type plasminogen activator (u-PA), annexin-II, tissue-type plasminogen activator (tPA), reduced the levels of plasminogen and α2-antiplasmin, and were present in APL (acute promyelocytic leukemia). Annexin–II, the receptor for plasminogen and tPA, is highly expressed in endothelial cells of cerebral microvasculature, and, likely due to this expression, the incidence of intracerebral haemorrhage is more frequent. Elastase and chymotrypsin from myeloid blasts are involved in the proteolysis of proteins of the coagulation cascade (clotting factors and fibrinogen). Cytokines delivered by leukemic promyelocytes induce APL coagulopathy. These are represented by IL-1β and TNFα and secondary the loss of the thrombomodulin–anticoagulant cofactor [
5].
Mucosal bleeding, petechiae, ecchymosis, fundal or cerebral hemorrhages may occur as a result of thrombocytopenia or thrombocytopathia. Previous research on platelet role in bleeding phenomenona of acute leukaemia patients identified defects in platelet production [
6,
7] and impaired platelet function [
8,
9,
10,
11,
12]. These studies were particularly focused on aggregation defects [
13,
14,
15] and bleeding at diagnosis [
16]. Even if the risk of bleeding was correlated with platelet count in AML, it was shown that platelet aggregation and activation tests were more important in the evaluation of haemorrhagic complication than platelet count alone [
3]. On the other hand, aggregation studies on platelets are very difficult to perform in severe thrombocytopenia, where this method has technical limitations [
17].
From analysing the literature, we found that platelet function analysis using whole-blood flow cytometry was performed in a relative small number of studies [
18,
19,
20,
21] and in a rigorously selected group of patients [
19,
22].
The occurrence of haemorrhages in AML patients was thought to be due to additional risk factors, including fever, sepsis [
20,
23], anticoagulant therapy and drugs [
20,
24,
25], coagulation anomalies, hypoalbuminemia [
26], uraemia [
27,
28], recent bone marrow transplants, low haematocrit [
29,
30], leukopenia or leucocytosis [
20], and vascular integrity [
20]. Data regarding the independent predictive role of these factors in acute leukaemia patients, and for the estimation of severity of the bleeding, are very poor.
Consequently, the goal of our research is to extend the flow cytometry examination of platelet activation (CD63, CD62p,) adhesion (CD42a, CD42b) and aggregation (CD41, CD61) markers to the particular clinical context of the patient with AML.
2. Materials and Methods
The analysed group was composed of 22 patients diagnosed with AML at the Bucharest Emergency University Hospital and Colentina Clinical Hospital; all patients undergoing treatment specific for the stage of the disease. The patients’ diagnosis was established by bone marrow examination in accordance with World Health Organization classification [
31], and the treatment was conducted according to international protocols. The AML classification was conducted using French-American-British (FAB) system [
32,
33] because cytogenetic and molecular analysis was not available during the period in which the patients were enrolled in the study, and usually the therapeutic approach was conducted in relation to FAB classification during the study period. Ten healthy volunteers recruited from the hospital staff members were enrolled as controls.
2.1. Monoclonal Antibodies
The following six monoclonal antibodies were used for the identification of platelet defects: CD61 clone SZ21 IM1758 (FITC conjugated) [
34], CD41 clone P2IM1416 (PE conjugated) [
33], CD42a clone SZ1IM1757 (FITC conjugated), CD42b clone SZ2IM1417 (PE conjugated), CD62P clone CLBThromb/6IM1164 (FITC conjugated) and CD63 clone CLBGran/12IM1914 (PE conjugated)—all were produced by Coulter-Immunotech [
31].
Anti CD61 antibody is directed against the Glycoprotein [Gp] IIIa (β3), sub-unit of the Gp IIb-IIIa, the most frequent platelet surface integrin. The CD41 antigen (αIIb chain from integrin) is always non-covalently associated with the CD61. CD41 is expressed by platelets, megakaryocytes and a small subset of CD34+ cells, suggesting that CD41/CD61 is one of the earliest markers of the megakaryocytic lineage. The P2 antibody reacts with CD41a (GpIIbα) in the intact complex with GpIIIa but not with the GpIIb or GpIIIa separately.
Anti CD42b antibody is targeted against Gp Ib-α chain, the moiety of the von Willebrand receptor, GpIb. GpIbα, GpIbβ, GpIX (CD42a), and GpV represent a multifunctional receptor (the glycoprotein Ib-V-IX). The CD42a/CD42b complex is the receptor for von Willebrand’s factor and is known as von Willebrand factor-dependent adhesion receptor. CD42a expression is restricted to platelets and megakaryocytes. The SZ1 monoclonal antibody reacts with the complex CD42a/CD42b but does not recognize GpIb or GpIX individually [
35].
Anti CD 62P antibody is specific for p selectin, a protein present in megakaryocytes, in Weibel–Palade bodies of endothelial cells and in alpha granules of platelets. It is translocated to the platelet surface membrane upon platelet activation [
36]. The p-selectin role in vivo is still under study but there is strong evidence that it interferes in leukocyte recruitment and potentially supports platelet rolling [
37,
38,
39].
Anti CD63 antibody is directed against Gp53 (granulophysin), a four transmembrane protein member of the tetraspanin (TM4SF) family present on the membranes of dense granules and lysosomes b. It relocates to the plasma membrane following platelet activation and degranulation. On the membrane surface it associates with the platelet integrin αIIbβ3-CD9 complex and with the actin cytoskeleton [
40].
2.2. Preparation of Whole Blood Samples for Flow Cytometry
Blood samples (3 mL peripheral blood) from patients (n = 22) and volunteers (n = 10) were used for analyses. The samples were collected by venous punction from the antecubital vein with a minimum of stasis in Beckton Dickinson vacutainers with anticoagulant sodium citrate. The analysis was performed within 4–6 h from the venous punction with a FACS Calibur BD four channels flowcytometer, using the CellQuest Software.
The protocol for the platelet immunophenotyping is described below:
The blood samples were centrifuged at 20 °C and 200× g for 10 min, and the platelet-rich plasma obtained was removed and centrifuged at 20 °C and 800× g for 10 min to prepare the platelet pellet. The platelet pellet was washed three times with phosphate-buffered saline (PBS) containing EDTA (0.009 mol/L Na2 EDTA; 0.01 mol/L Na2 HPO4; 0.0018 mol/L KH2PO4; 0.17 mol/L NaCl and 0.0033 mol/L KCl), and then fixed by incubation for 10 min at room temperature with 2% paraformaldehyde in PBS-EDTA. The fixed platelets were washed twice with PBS-EDTA and adjusted to a concentration of 5 × 109/L. Aliquots (200 µL) of the platelet suspension were added to 12 mm × 75 mm polystyrene tubes previously coated with 30 µL of 5% bovine albumin.
The platelets were incubated for 15 min with the above-described monoclonal antibodies conjugated with fluorocromes for surface receptors
2.3. Flow Cytometry Analysis of Platelets in Whole Blood
The flow cytometry settings were optimized for the acquisition of platelets by logarithmic signal amplification in all detectors. Electronic compensation was used to control for spectral overlap and the flow cytometer was aligned daily with CaliBriteTM beads (BDIS). A threshold on FL-1 was set to ensure that only platelet CD61-positive events were collected. A minimum of 5000 CD61-positive platelets was collected from each sample.
For analysis, an electronic gate enclosing platelets was set, as defined by forward scatter (FSC) and 90° side scatter (SSC) characteristics and CD61/CD41 platelet positivity markers, excluding platelet-leukocytes aggregates as well as platelet-erythrocyte and platelet-leukocytes doublets [
18]. Further analysis of platelet activation markers CD62P, CD63, CD42b, and CD42a was performed on non-aggregated platelets as defined in
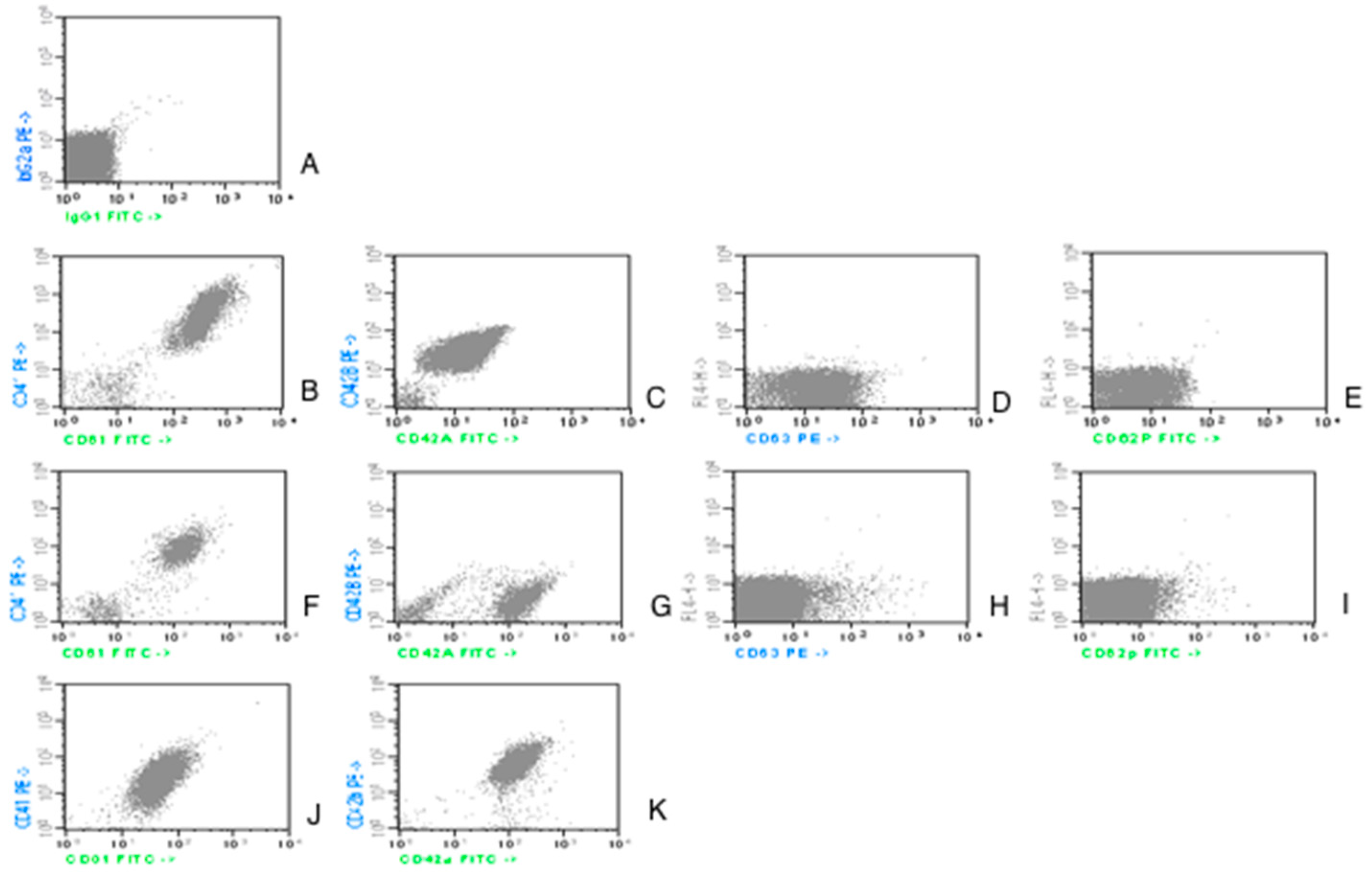
Figure 1. Cells stained with an isotype control antibody were used as a reference when defining the threshold in order to distinguish negative from positive cells.
The platelet fluorescence of activation markers was measured as percentage of platelets expressing fluorescence above a threshold set to include only 1% of platelets incubated with the respective control.
Counting from the upper part of the picture and from the left side:
- -
the first line, image A, is an isotype control;
- -
the second line, a case of control group; image B for CD61/CD41; C for CD42a/CD42b; D for CD63; E for CD62P;
- -
the third and fourth lines represent AML patients. The images are illustrative for the variability in expression:
Images F and G from line 3 illustrate the expression of the aggregation (CD41/CD61) and adhesion (CD42a/CD42b) markers. In these examples, CD61, CD41 and CD42b have a lower fluorescence level in patients vs. controls.
Images J and K from line 4 show a different expression pattern with CD42b expression, which is as high as the expression at the controls.
Images H and I from line 3 present the fluorescence level for the activation markers (CD63, CD62P): reduced in patients vs. controls.
The acquisition was made with BD FACS Calibur flowcytometer and the analysis performed with Cellquest Software, from Becton Dickinson and Company, 1997, Becton Dickinson Immunocytometry Systems, San Jose, CA 95131, United States of America. Reagents were produced by IMMUNOTECH SAS, a Beckman Coulter Company, Marseille, France.
2.4. Statistical Analysis
For the medians, the 25th and 75th quartiles of the data were used to describe the observed values. Antigen expressions were considerate, continuous variables and were compared using non-parametric Wilcoxon rank sum test.
Spearman’s rank correlation was used for correlation analyses between platelets’ surface antigens and values of haematocrit, leukocytes, platelets, the percentage of peripheral blasts, level of uric acid, creatinine, BUN, and body temperature, all considered continuous variables. A general two-tailed significance level of 5% was applied.
Other clinical determinants, such as the presence of infection (localized or generalized) or cutaneous haemorrhagic syndrome, were analysed using crosstabs procedure.
All analyses were performed using SPSS statistical software, version 12.0 for Windows.
3. Results
The present study examines a group of 22 patients (13 females and 9 males) with ages ranging between 28 and 83 years. At the time of the analysis, they were already diagnosed with acute myeloid leukaemia and in different stages of disease.
Each patient was investigated by clinical examination, full blood count and peripheral blood smear with a detailed lymphocyte count. In every case, biochemistry samples (uric acid, creatinine, BUN) were taken and analysed.
As the majority of the patients were hospitalised at the time of the study, information about the treatment received a day before collecting the probes was available: chemotherapy, antibiotic therapy (beta-lactam antibiotics) and blood products transfusion therapy (
Table 1).
The diagnosis of acute myeloid leukaemia was established prior to the beginning of the study, and the patients were assessed against AML subtypes according to FAB classification and EGIL criteria for AML0 [
33,
41]. Almost half of the group had AML type 4 (40.9%) and one-third (27.27%) had AML type 2. The AML type 3 FAB (promyelocytic) patients were excluded from the study design, due to frequently associated coagulation disorders. The number of cases with AML type 1 and AML type 0 were comparable (18.18% versus 13.64%).
The presence or absence of infection (localized or generalized), body temperature and cutaneous haemorrhagic syndrome were clinically assessed. Fever was present in almost half of patients with infection. Skin haemorrhage was present in 59% of AML patients with no significant difference according to AML subtype.
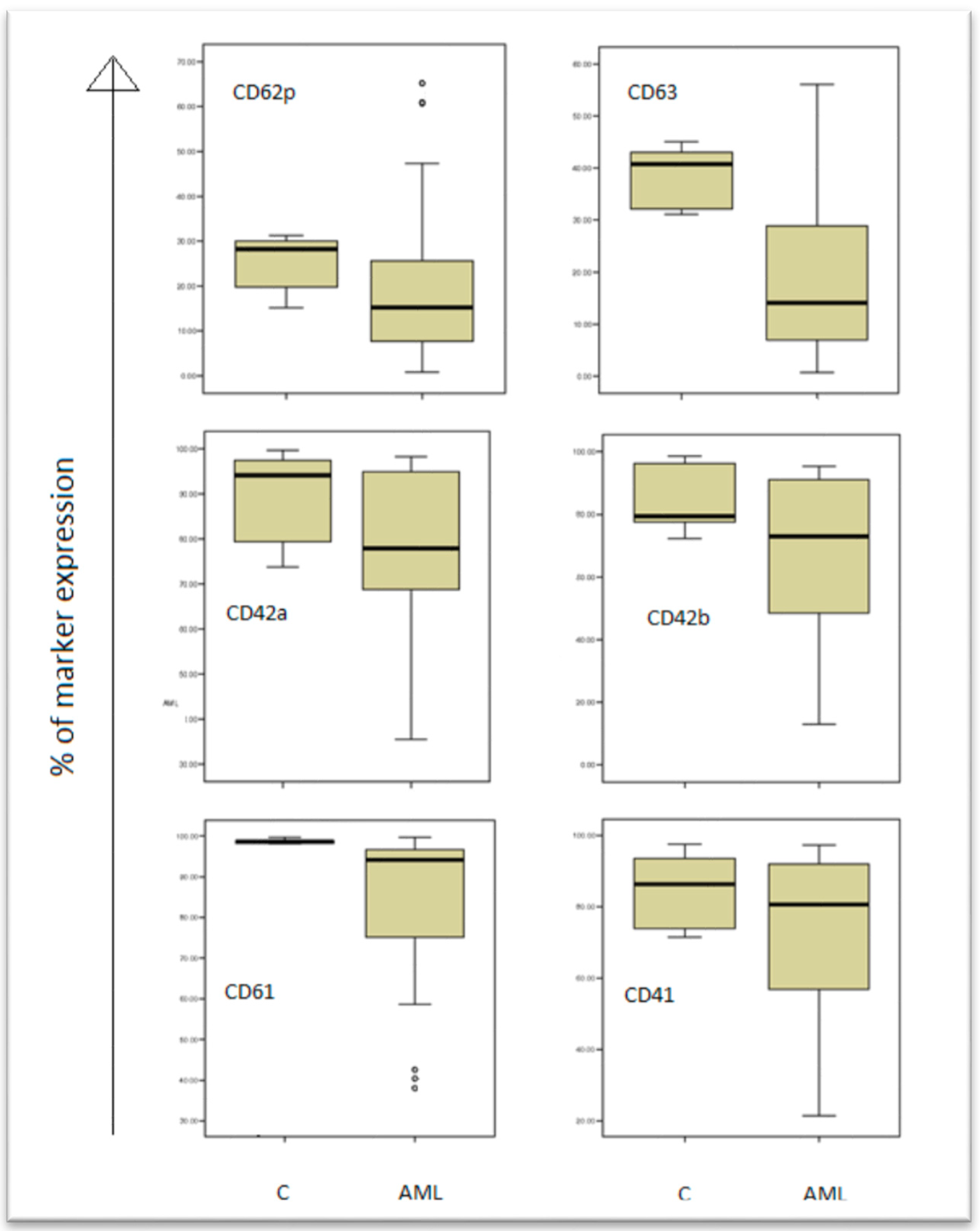
Platelets’ antigen expressions (percentage fluorescence above a threshold set to include only 1% of platelets incubated with control) had an abnormal, non-homogenous distribution in patient group (skewness and kurtosis almost equal to ±1). The medians and the 25th–75th percentiles were used to appreciate and compare distributions in patients and human control groups (
Table 2,
Figure 2).
The results are presented in box plots indicating the median as the horizontal line, 25th–75th percentiles in the group distributions as boxes, and 2.5–97.5% cumulative frequencies as whiskers. Outliners (identified by the 1.5 × inter-quartile range (IQR) criterion) are plotted as empty squares: CD62P = p-selectin; CD63 = granulophysin.
The control arm (C), and patients arm (AML) are shown in the boxplots, and the expression of all markers (CD61, CD41, CD42a, CD42b, CD62p, CD63) are decreased in AML patients comparing to controls.
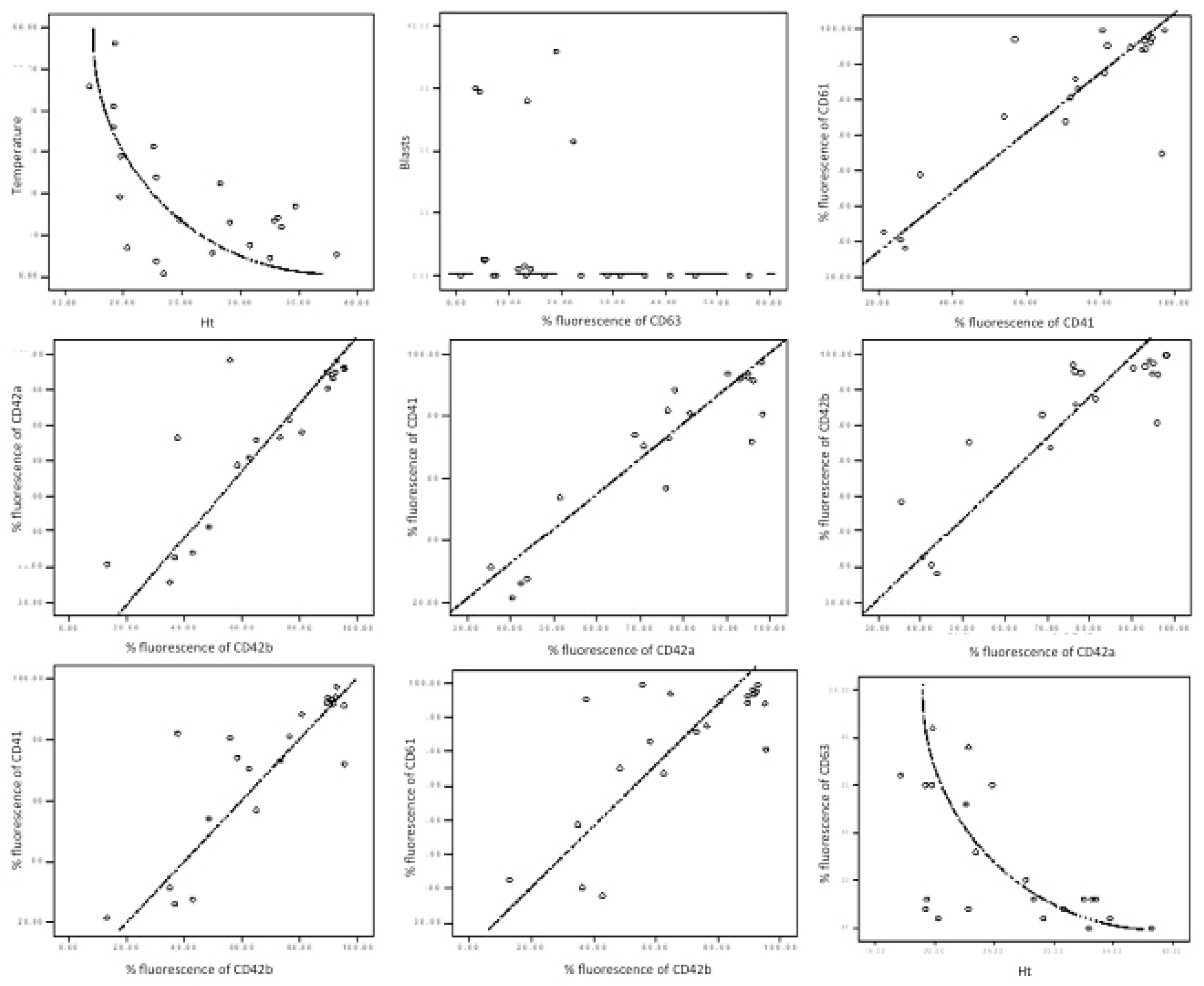
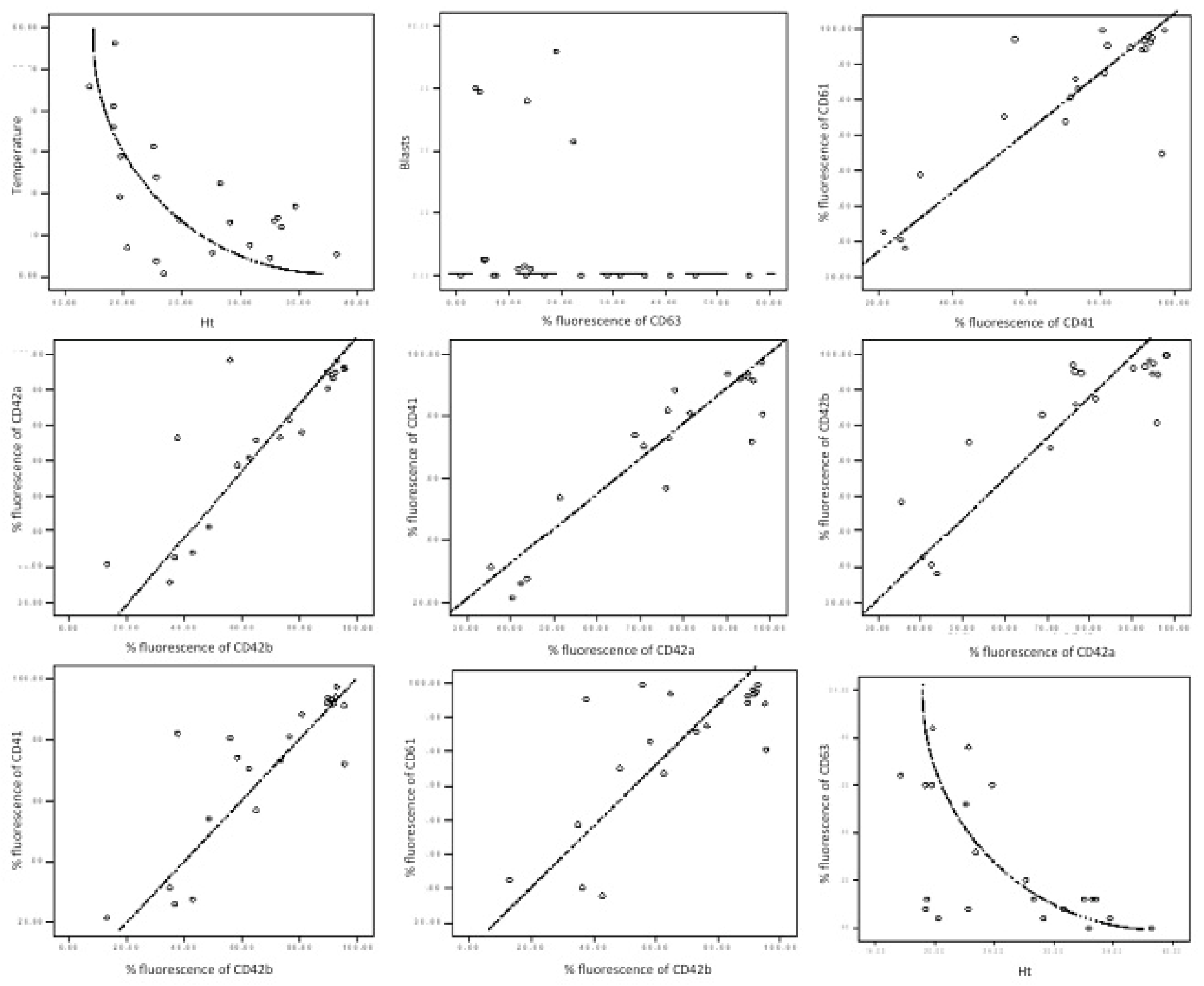
3.1. Activation of Platelets in AML
The
p-selectin level was found to be significantly reduced (15.26% versus 28.23%,
p = 0.038) in patients versus controls. The CD63 (granulophysin) level correlates with the CD62P (
p selectin) level (
Table 3 and
Figure 3: Rho = 0.69,
p value < 0.01), and it had a decreased level in AML patients versus controls (14.11% vs. 40.78%).
A non-linear negative correlation was found between CD63 fluorescence percentage and haematocrit level (
Table 3,
Figure 3: Rho = −0.598,
p value = 0.003).
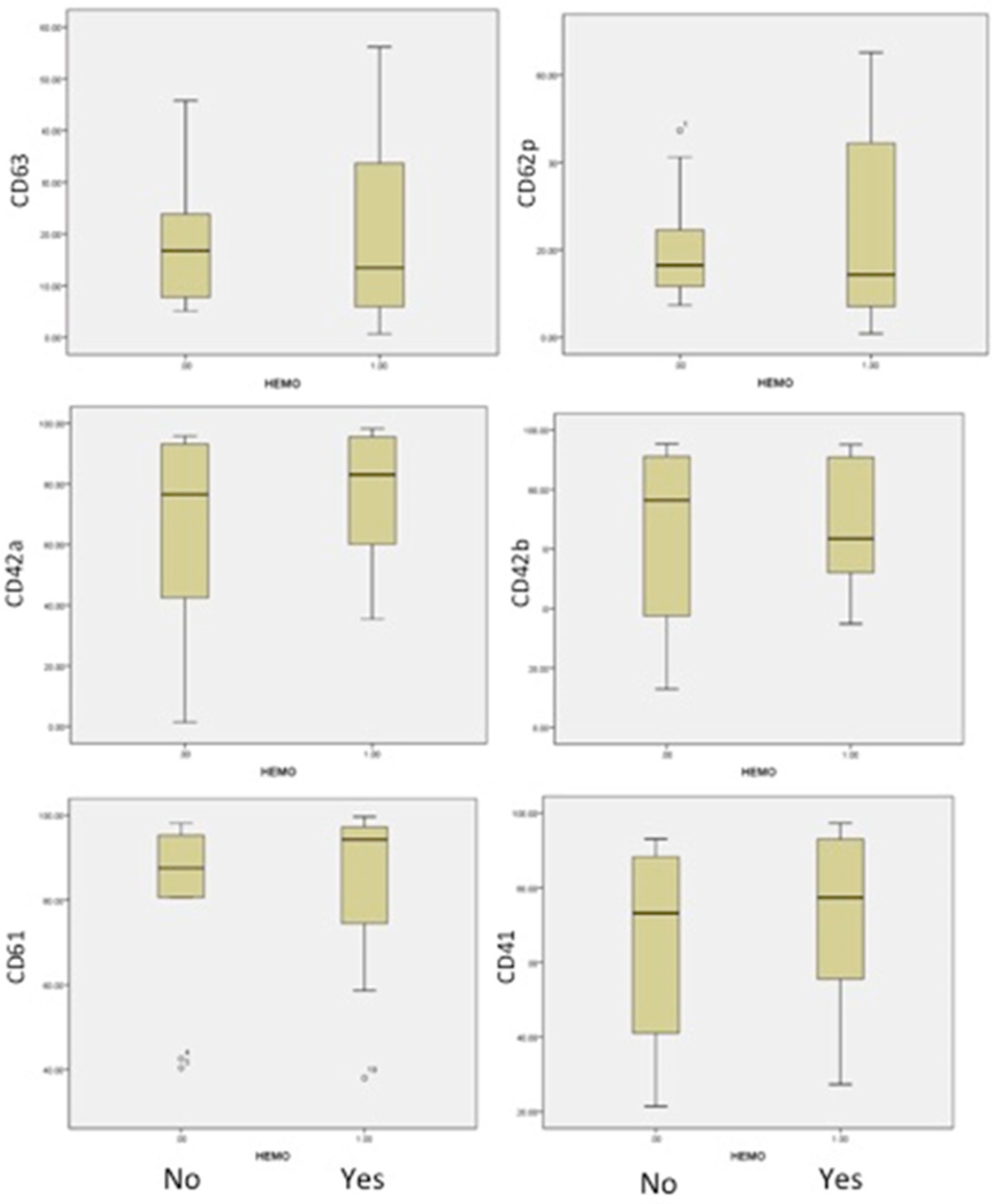
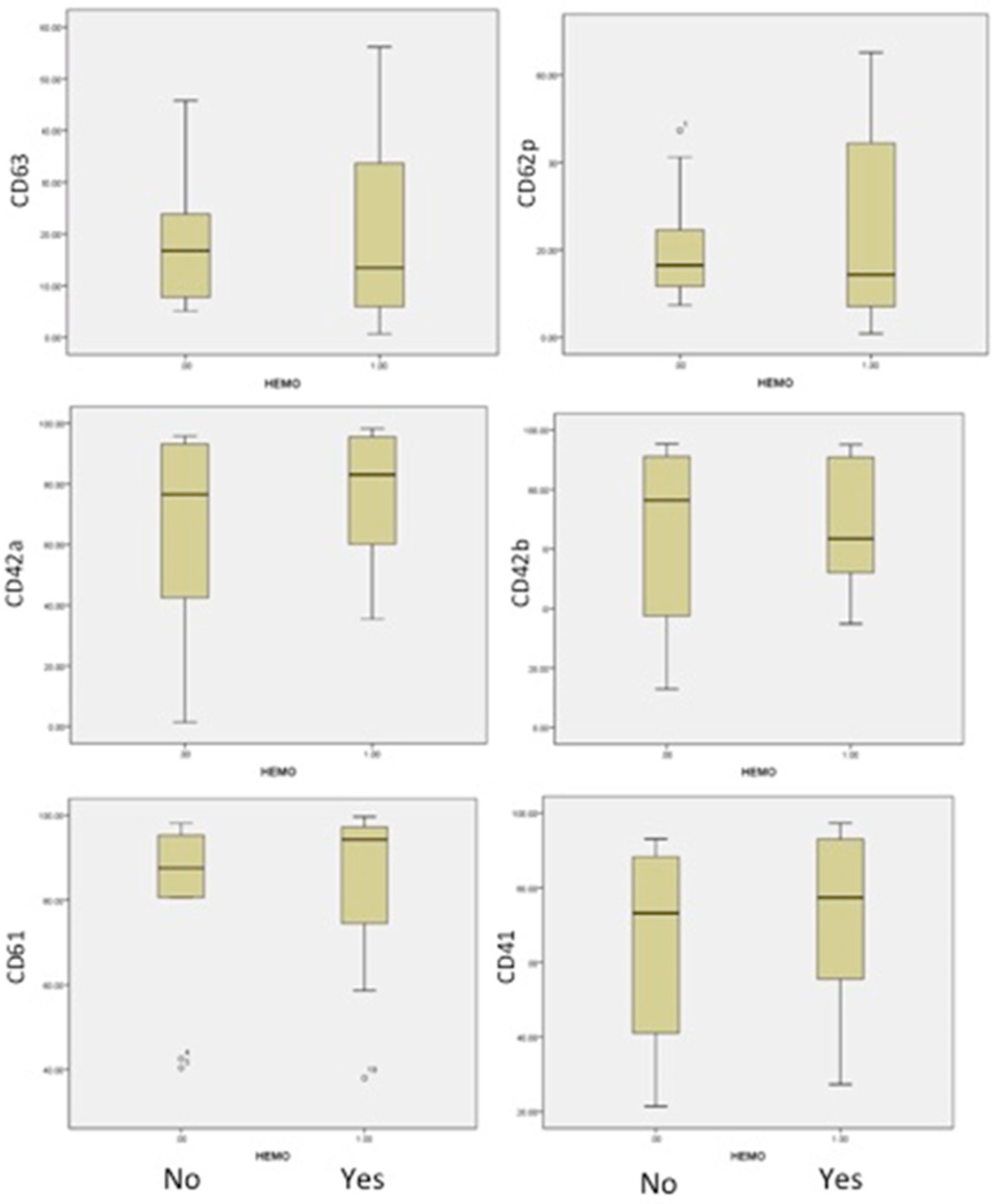
The analysis of AML patients, with or without bleeding, was conducted in correlation with the number of platelets and their antigen expressions. The results are presented in
Table 4. The medians and 25th–75th percentiles were used to appreciate and compare distributions in both groups (
Figure 4). The median age was closely similar in both groups: 46 and 43 years old.
3.2. Von Willebrand Receptor in AML
The CD42b expression decreases in patients versus controls (p = 0.047), while the anti-CD42a antigen antibody, which reacts with the whole CD42a/CD42b complex, presents no particular difference in the study group versus the control group.
3.3. Fibrinogen Receptor
Gp IIb-IIIa, β3 integrin (CD61), the most frequent platelet surface antigen, presents a significant decrease in fluorescence percentage levels (94.08% vs. 98.67%,
p value < 0.01) in patients versus controls (
Table 3).
An interesting relation between Gp IIb-IIIa and Gp Ib-IX occurred (
Table 3). Even if the connection between the two protein complexes had a variable strength (Rho: 0.570–0.783), the
p value < 0.01 indicated that, in patients with AML, the expression of these two platelet determinants was related, likely as a global defect of signal transduction.
3.4. Comparison between Patients with Bleeding and without Bleeding
An important difference was identified in patients with bleeding, in comparison to patients without bleeding, in the form of a decrease in Gp Ib-IX expression (CD42a/CD42b) and the activation of less platelets (CD62P and CD63), as shown in
Figure 4. This suggested a more severe acquired Bernard–Soulier syndrome and the blocking of platelet activation, independent of patient age or the number of platelets. The expression of fibrinogen receptor (CD61/CD41) was found with a higher level of expression, even if a lower level of expression in this receptor was found in all AML patients compared to healthy human volunteers. Our findings are strong evidence that haemorrhagic events in acute myeloid leukemias are more related to the impaired function of platelets.
3.5. Clinical and Laboratory Characteristics
Proven sepsis represents a significant statistic predictor of haemorrhage and correlates with the increased clinically significant bleeding risk the following day, as well as with the refractoriness risk to platelet concentrate transfusion. Our data revealed a statistically significant correlation between the haematocrit level and body temperature (ro: 0.559, p value: 0.05). A cutaneous haemorrhage was non-statistically correlated with the number of platelets. No correlation could be established between coagulation factors or uraemia markers and the other determinants. In the control group, no significant correlation was found between the studied variables.
We could not analyse the correlations between the different expression of platelets markers and AML subgroups because of the small number of patients in each subgroup.
4. Discussion
The bleeding predisposition in AML patients is the result of a platelet production defect leading to thrombocytopenia and the qualitative changes of the megakaryocytes and platelets, respectively [
38,
39]. Recent studies reported the more significant role of platelet dysfunction than the number of platelets for predicting haemorrhagic complications in AML patients [
3]. Impaired platelet aggregation tests and lower expressions of GPIIb-IIIa and
p selectin (CD62P) in platelets stimulated by TRAP (thrombin-receptor stimulating peptide) represent predictive factors for bleedings in AML [
3].
Early studies that concentrated on the relationship between haemorrhage and thrombocytopenia concluded that bleeding was considered a frequent complication of induction chemotherapy or stem-cell transplants in acute myeloid leukaemia.
The most frequently observed platelet dysfunctions associated with haematological diseases are: inadequate platelet aggregation to ADP, epinephrine or collagen; low nucleotides secretion; low serotonin secretion; a decrease in thromboxane production and low PDGF and beta-thromboglobuline production [
42,
43,
44,
45]; the overproduction of cAMP [
46]; and platelet signalling defects.
The reduced level of CD63 and CD62P in our patients indicated two possible events: (1) no activation process took place or (2) there was a brief activation followed by rapid p selectin secretion.
In addition to the above-mentioned studies, which were focused on functional platelets’ characteristics, our research performed on additional patient platelets that were not further stimulated, revealed the same low level of activation markers mentioned before. These results raise the hypothesis of the lack of platelets activation in vivo, a comparable state with the one found in vitro by the other experiments. Furthermore, the activation of platelets represents a vital first step in the clotting cascade.
The haematocrit role in platelets activation was previously investigated using in vitro stimulations [
47]. Eugster M and Reinhart W found an inverse correlation between haematocrit and a necessary amount of time for the formation of the platelet plug. Our results show an inversely proportional relation between the haematocrit and granulophysin expression on platelets, which may be a false statistical result with no biological signification or a future research perspective.
Specific heterozygous mutations affecting the hematopoietic transcription factor CBFA2 were previously reported to be associated with a familial platelet disorder predisposing to acute myeloid leukaemia [
48]. Our study correlated the percentage of the peripheral blast cells with platelet-specific markers. Although peripheral blasts seemed statistically related to CD63, this conclusion could not be graphically validated (
Figure 3). This fact imposes a larger number of cases and, potentially, a different analytic strategy.
4.1. Von Willebrand Receptor in AML
The receptor for the von Willebrand Factor (CD42b) is one of the most major adhesive receptors expressed on the surface of circulating platelets, able to interact with several ligands, including the adhesive protein von Willebrand factor, the coagulation factors (thrombin, factors XI and XII), and the membrane glycoproteins (
p-selectin and Mac-1) [
46]. The reduced expression of CD42b in our patients might be interpreted as a particular reduction in the α chain of GpIb due to the downregulation during platelet activation [
49,
50,
51]. Interestingly, Leinoe et al. [
21] investigated if this decrease in CD42b in platelets under TRAP activation may occur because a downregulation of GpIb due to platelet activation in vivo. What we can say is that the same findings in vivo suggest that a broken activation mechanism could be involved in the expression of both
p-selectin and granulophysin and the decreasing of GpIb, considered to be a regulatory mechanism, which may witness this effort to activate a defective platelet.
4.2. Fibrinogen Receptor
The resting form of the CD41/CD61 complex binds to immobilized fibrinogen, and, upon platelet activation, the complex becomes a receptor for soluble fibrinogen, fibronectin, the von Willebrand Factor, vitronectin and thrombospondin [
52]. This receptor is also involved in platelet aggregation [
53,
54]. The anti CD41 antigen antibody reacts with GpIIb in the intact GpIIb-IIIa complex, blocking fibrinogen and platelet aggregation induced by thrombin, collagen and ADP. The reduction in the CD61 fluorescence level may be interpreted as a selective decrease in the β3 subunit of GpIIIa, which may be a cause of the aggregation defect. A defective β3 integrin phosphorylation, associated with altered outside-in signalling, was observed [
55]. This may lead to an increase in the haemorrhagic syndrome of AML patients.
In AML, the low expression of GP1b on platelets could be explained by action of elastase released from myeloid blasts [
56]. The interaction between platelets and AML blasts was proven by one study, and this interaction could produce functional changes and explain the presence of the dysfunctional platelet in AML patients [
19]. A correlation between the low expression of CD62P, CD63 and the fibrinogen receptor and the high risk for haemorrhage in the next 7 days of follow up for AML patients was reported recently in platelets stimulated by TRAP. In AML patients defective signalling pathways were found that could explain the low activation of GPIIbIIIa, an abnormal conformation of this activated receptor that influences its function [
3,
11]. In other studies, a correlation was observed between the high expression of
p selectin and GP IIbIIIa and haemorrhagic history [
20]. In AML patients, the MPL expression on blasts was correlated with the severity of neutropenia and thrombocytopenia, especially in AML patients with t(8;21) genetic aberrancy [
57].
4.3. Clinical and Laboratory Characteristics
The documented presence of the infection for the day before the study and/or the fever is associated with different types of bleeding, which can be clinically assessed as mild, significant or major. Recently, Vinholt et al. did not obtain any significant correlation between the infection and haemorrhagic risk of AML patients [
3]. Gaydos et al. [
58] found correlations between platelet numbers and all types of bleedings (mild, clinically significant and major) in acute leukemia patients. Lawrence et al. [
59] obtained no correlation between bleeding and the number of platelets, or the minimal number of the platelets. Other studies, which focused on the same issue, analysed this in association with the clinical issue of prophylactic transfusion with platelet concentrate and stressed that the risk of an important haemorrhagic complication still exists [
60,
61]. The absence of any correlation between cutaneous haemorrhage and the number of platelets, in addition to the above-described aberrant expression of phenotypic surface platelets markers, emphasizes the role of functional and structural platelet defects in the haemorrhagic burst.
5. Conclusions
In our group of patients, we found a significant decrease in activation markers such as p-selectin and granulophysin, associated with the decrease in GpIb and β3 integrin, demonstrating that, in AML, there could be multiple defects of signalling transduction, leading to defects of adhesion, aggregation and the secretion of platelets, consequently leading to haemorrhagic syndrome independent of the number of platelets in the peripheral blood. These data are complementary to other already published results in demonstrating the multiple platelet defects of AML patients, suggesting that the risk of bleeding is directly correlated to the level of platelet defect.
In conclusion, the platelet behaviour in our study group offers important clues for postulating that flow cytometry analysis could be a very important method to identify those patients that are potential candidates to haemorrhagic disorders.
A limitation of our study is the small number of patients in the study group and the lack of genetic aberrancies data in AML patients, which could be of clinical interest in further research. Additionally, the study was not designed as a longitudinal study, and a follow up study could be of interest for more information regarding haemorrhagic risk.
Author Contributions
H.B.: conception and design, flow cytometry data acquisition and writing of “Design and methods”, and
Section 4, critical and responsible revision of the draft, guarantee of the intellectual content and final approval; A.M.V.: conception and design; S.S., V.M.P.: design, statistical analysis, writing of the
Section 1 and
Section 3, drafting the
Section 4, preparing the article for submission; I.D.: flow cytometry data acquisition, critical and responsible revision of the draft; D.C., I.V., C.C., C.M., A.N. and M.O.: documentation process, gathering the clinical data, contribution to drafting the
Section 1 and
Section 4. All authors have read and agreed to the published version of the manuscript.
Funding
The current article was written based on the data collected during a National Research Grants in the Program “Excellence in Research” (CEEX)—MUL-TRO nr. 62/2005 and PARTE MPN Project nr. 141PED/2017-2018—funded by the Romanian Ministry of Research and Development. The study was carried out at the Departments of Haematology, Emergency University Hospital, “Carol Davila” University of Medicine and Pharmacy, Bucharest, Romania, and Colentina Clinical Hospital, Bucharest, Romania.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board no 29342/11 October 2005 and no 532/20 January 2017 of Romanian Ministry of Research and Development.
Informed Consent Statement
The study was reviewed and approved by the Ethics Committee of the University Emergency Hospital in Bucharest, Romania. The approval codes are 29342/11 October 2005 for first study. The second study approved for checking and improvement of methods has 532/20 January 2017. Data were accessed anonymously. All methods were carried out in accordance with the relevant guidelines and regulations.
Data Availability Statement
All data are available, either analysed as figures and tables presented in the current manuscript; or as raw data upon request from any external collaborator or reviewer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haase, D.; Feuring-Buske, M.; Konemann, S.; Fonatsch, C.; Troff, C.; Verbeek, W.; Pekrun, A.; Hiddemann, W.; Wormann, B. Evidence for malignant transformation in acute myeloid leukemia at the level of early hematopoietic stem cells by cytogenetic analysis of CD34+ subpopulations. Blood 1995, 86, 2906–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimwade, D.; Enver, T. Acute promyelocytic leukemia: Where does it stem from? Leukemia 2004, 18, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just Vinholt, P.; Højrup Knudsen, G.; Sperling, S.; Frederiksen, H.; Nielsen, C. Platelet function tests predict bleeding in patients with acute myeloid leukemia and thrombocytopenia. Am. J. Hematol. 2019, 94, 891–901. [Google Scholar] [CrossRef]
- Hatzl, S.; Uhl, B.; Hinterramskogler, M.; Leber, S.; Eisner, F.; Haring, M.; Jud, P. Acute myeloid leukemia with severe coagulation disorder and concomitant central nervous system bleeding—A clinical diagnostic case report. Int. Fed. Clin. Chem. Lab. Med. 2018, 29, 146–151. [Google Scholar]
- Choudhry, A.; DeLoughery, T.G. Bleeding and thrombosis in acute promyelocytic leukemia. Am. J. Hematol. 2012, 87, 596–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matolcsy, A.; Kálmán, E.; Pajor, L.; Kónya, T.; Weber, E. Morphologic and flow cytometric analysis of circulating megakaryoblasts in chronic myeloid leukaemia. Leuk. Res. 1991, 15, 887–897. [Google Scholar] [CrossRef]
- Wong, K.F.; Chan, J.K.C. Are ‘dysplastic’ and hypogranular megakaryocytes specific markers for myelodysplastic syndrome? Br. J. Haematol. 1991, 77, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Friedman, I.A.; Schwartz, S.O.; Leithold, S.L. Platelet Function Defects With Bleeding: Early Manifestation of Acute Leukemia. Arch. Intern. Med. 1964, 113, 177–185. [Google Scholar] [CrossRef]
- Cowan, D.H.; Graham, R.C., Jr. Structural-functional relationships in platelets in acute leukemia and related disorders. Ser. Haematol. 1975, 8, 68–100. [Google Scholar]
- Slichter, S.J. Relationship between platelet count and bleeding risk in thrombocytopenic patients. Transfus. Med. Rev. 2004, 18, 153–167. [Google Scholar] [CrossRef]
- Leinoe, E.B.; Hoffmann, M.H.; Kjaersgaard, E.; Nielsen, J.D.; Bergmann, O.J.; Klausen, T.W.; Johnsen, H.E. Prediction of haemorrhage in the early stage of acute myeloid leukaemia by flow cytometric analysis of platelet function. Br. J. Haematol. 2005, 128, 526–532. [Google Scholar] [CrossRef]
- Qian, X.; Wen-jun, L. Platelet changes in acute leukemia. Cell Biochem. Biophys. 2013, 67, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Weyden, M.B.V.D.; Clancy, R.L.; Howard, M.A.; Firkin, B.G. Qualitative Platelet Defects with Reduced Life-Span in Acute Leukaemia. Aust. N. Z. J. Med. 1972, 2, 339–345. [Google Scholar] [CrossRef]
- Maldonado, J.E.; Pierre, R.V. The platelets in preleukemia and myelomonocytic leukemia. Ultrastructural cytochemistry and cytogenetics. Mayo Clin. Proc. 1975, 50, 573–587. [Google Scholar] [PubMed]
- Ganguly, P.; Sutherland, S.B.; Bradford, H.R. Defective binding of thrombin to platelets in myeloid leukaemia. Br. J. Haematol. 1978, 39, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, B.E.; Cooper, P.C.; Brown, P.R.; Pickering, C.; Winfield, D.A.; Preston, F.E. The platelet defect in acute myeloid leukaemia. J. Clin. Pathol. 1984, 37, 1339–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinholt, P.J. The role of platelets in bleeding in patients with thrombocytopenia and hematological disease. Clin. Chem. Lab. Med. 2019, 57, 1808–1817. [Google Scholar] [CrossRef]
- Matzdorff, A.C.; Kühnel, G.; Kemkes-Matthes, B.; Pralle, H. Quantitative assessment of platelets, platelet microparticles, and platelet aggregates with flow cytometry. J. Lab. Clin. Med. 1998, 131, 507–517. [Google Scholar] [CrossRef]
- Foss, B.; Ulvestad, E.; Hervig, T.; Bruserud, Ø. Effects of cytarabine and various anthracyclins on platelet activation: Characterization of in vitro effects and their possible clinical relevance in acute myelogenous leukemia. Int. J. Cancer 2002, 97, 106–114. [Google Scholar] [CrossRef]
- Psaila, B.; Bussel, J.B.; Frelinger, A.L.; Babula, B.; Linden, M.D.; Li, Y.; Barnard, M.R.; Tate, C.; Feldman, E.J.; Michelson, A.D. Differences in platelet function in patients with acute myeloid leukemia and myelodysplasia compared to equally thrombocytopenic patients with immune thrombocytopenia. J. Thromb. Haemost. 2011, 9, 2302–2310. [Google Scholar] [CrossRef] [Green Version]
- Leinoe, E.B.; Hoffmann, M.H.; Kjaersgaard, E.; Johnsen, H.E. Multiple platelet defects identified by flow cytometry at diagnosis in acute myeloid leukaemia. Br. J. Haematol. 2004, 127, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Sandes, A.F.; Yamamoto, M.; Matarraz, S.; Chauffaille Mde, L.; Quijano, S.; López, A.; Oguro, T.; Kimura, E.Y.; Orfao, A. Altered immunophenotypic features of peripheral blood platelets in myelodysplastic syndromes. Haematologica 2012, 97, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Webert, K.; Cook, R.J.; Sigouin, C.S.; Rebulla, P.; Heddle, N.M. The risk of bleeding in thrombocytopenic patients with acute myeloid leukemia. Haematologica 2006, 91, 1530–1537. [Google Scholar] [PubMed]
- Böck, M.; Muggenthaler, K.H.; Schmidt, U.; Heim, M.U. Influence of antibiotics on posttransfusion platelet increment. Transfusion 1996, 36, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Slichter, S.J.; Davis, K.; Enright, H.; Braine, H.; Gernsheimer, T.; Kao, K.J.; Kickler, T.; Lee, E.; McFarland, J.; McCullough, J.; et al. Factors affecting posttransfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood 2005, 105, 4106–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann, A.M.; Sengul, H.; Lehmann, H.; Schwartz, C.; Goodman, S. Do basic laboratory tests or clinical observations predict bleeding in thrombocytopenic oncology patients? A reevaluation of prophylactic platelet transfusions. Transfus. Med. Rev. 2002, 16, 34–45. [Google Scholar] [CrossRef]
- Livio, M.; Gotti, E.; Marchesi, D.; Mecca, G.; Remuzzi, G.; de Gaetano, G. Uraemic bleeding: Role of anaemia and beneficial effect of red cell transfusions. Lancet 1982, 2, 1013–1015. [Google Scholar] [CrossRef]
- Fernandez, F.; Goudable, C.; Sie, P.; Ton-That, H.; Durand, D.; Suc, J.M.; Boneu, B. Low haematocrit and prolonged bleeding time in uraemic patients: Effect of red cell transfusions. Br. J. Haematol. 1985, 59, 139–148. [Google Scholar] [CrossRef]
- Blajchman, M.A.; Bordin, J.O.; Bardossy, L.; Heddle, N.M. The contribution of the haematocrit to thrombocytopenic bleeding in experimental animals. Br. J. Haematol. 1994, 86, 347–350. [Google Scholar] [CrossRef]
- Small, M.; Lowe, G.D.; Cameron, E.; Forbes, C.D. Contribution of the haematocrit to the bleeding time. Haemostasis 1983, 13, 379–384. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; Volume 2. [Google Scholar]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Segeren, C.M.; van‘t Veer, M.B. The FAB classification for acute myeloid leukaemia—Is it outdated? Neth. J. Med. 1996, 49, 126–131. [Google Scholar] [CrossRef] [Green Version]
- CD61. Beckmann Coulter Catalog. Available online: https://www.beckman.com/reagents/coulter-flow-cytometry/antibodies-and-kits/single-color-antibodies/cd61 (accessed on 29 October 2021).
- Schlossman, S.F. Leucocyte Typing V: White Cell Differentiation Antigens, Proceedings of the Fifth International Workshop and Conference, Boston, MA, USA, 3–7 November 1993; Oxford University Press: Oxford, NY, USA, 1995. [Google Scholar]
- McEver, R.P. Properties of GMP-140, an inducible granule membrane protein of platelets and endothelium. Blood Cells 1990, 16, 73–80. [Google Scholar]
- Palabrica, T.; Lobb, R.; Furie, B.C.; Aronovitz, M.; Benjamin, C.; Hsu, Y.-M.; Sajer, S.A.; Furie, B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, M.; Frenette, P.S.; Saffaripour, S.; Johnson, R.C.; Hynes, R.O.; Wagner, D.D. Defects in hemostasis in P-selectin-deficient mice. Blood 1996, 87, 1238–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef]
- Israels, S.J.; McMillan-Ward, E.M. CD63 modulates spreading and tyrosine phosphorylation of platelets on immobilized fibrinogen. Thromb. Haemost. 2005, 93, 311–318. [Google Scholar] [CrossRef]
- Bene, M.C.; Castoldi, G.; Knapp, W.; Ludwig, W.D.; Matutes, E.; Orfao, A.; van’t Veer, M.B. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995, 9, 1783–1786. [Google Scholar]
- Raman, B.K.S.; Van Slyck, E.J.; Riddle, J.; Sawdyk, M.A.; Abraham, J.P.; Saeed, S.M. Platelet Function and Structure in Myeloproliferative Disease, Myelodysplastic Syndrome, and Secondary Thrombocytosis. Am. J. Clin. Pathol. 1989, 91, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Russell, N.H.; Salmon, J.; Keenan, J.P.; Bellingham, A.J. Platelet adenine nucleotides and arachidonic acid metabolism in the myeloproliferative disorders. Thromb. Res. 1981, 22, 389–397. [Google Scholar] [CrossRef]
- Cowan, D.H.; Haut, M.J. Platelet function in acute leukemia. J. Lab. Clin. Med. 1972, 79, 893–905. [Google Scholar]
- Cowan, D.H.; Graham, R.C., Jr.; Baunach, D. The platelet defect in leukemia. Platelet ultrastructure, adenine nucleotide metabolism, and the release reaction. J. Clin. Investig. 1975, 56, 188–200. [Google Scholar] [CrossRef]
- Lecchi, A.; Femia, E.A.; La Marca, S.; Onida, F.; Artoni, A. Acquired platelet dysfunction and overproduction of platelet cyclic AMP in two patients with myeloid malignancies. Platelets 2019, 30, 1053–1056. [Google Scholar] [CrossRef]
- Eugster, M.; Reinhart, W.H. The influence of the haematocrit on primary haemostasis in vitro. Thromb. Haemost. 2005, 94, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Buijs, A.; Poddighe, P.; van Wijk, R.; van Solinge, W.; Borst, E.; Verdonck, L.; Hagenbeek, A.; Pearson, P.; Lokhorst, H. A novel CBFA2 single-nucleotide mutation in familial platelet disorder with propensity to develop myeloid malignancies. Blood 2001, 98, 2856–2858. [Google Scholar] [CrossRef] [Green Version]
- Canobbio, I.; Balduini, C.; Torti, M. Signalling through the platelet glycoprotein Ib-V-IX complex. Cell. Signal. 2004, 16, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D. Thrombin-induced down-regulation of the platelet membrane glycoprotein Ib-IX complex. Semin. Thromb. Hemost. 1992, 18, 18–27. [Google Scholar] [CrossRef]
- Cahill, M.R.; Macey, M.G.; Newland, A.C. Fixation with formaldehyde induces expression of activation dependent platelet membrane glycoproteins, P selectin (CD62) and GP53 (CD63). Br. J. Haematol. 1993, 84, 527–529. [Google Scholar] [CrossRef]
- Bennett, J.S. Structure and function of the platelet integrin αIIbβ3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurden, P. Bidirectional trafficking of membrane glycoproteins following platelet activation in suspension. Thromb. Haemost. 1997, 78, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Caen, J.P.; Nurden, A.T. Glanzmann’s thrombasthenia: The spectrum of clinical disease. Blood 1990, 75, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glembotsky, A.C.; Bluteau, D.; Espasandin, Y.R.; Goette, N.P.; Marta, R.F.; Marin Oyarzun, C.P.; Korin, L.; Lev, P.R.; Laguens, R.P.; Molinas, F.C.; et al. Mechanisms underlying platelet function defect in a pedigree with familial platelet disorder with a predisposition to acute myelogenous leukemia: Potential role for candidate RUNX1 targets. J. Thromb. Haemost. 2014, 12, 761–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, K.A. An acquired form of Bernard Soulier syndrome associated with acute myeloid leukemia. Saudi Med. J. 2005, 26, 1095–1098. [Google Scholar] [PubMed]
- Rauch, P.J.; Ellegast, J.M.; Widmer, C.C.; Fritsch, K.; Goede, J.S.; Valk, P.J.; Löwenberg, B.; Takizawa, H.; Manz, M.G. MPL expression on AML blasts predicts peripheral blood neutropenia and thrombocytopenia. Blood 2016, 128, 2253–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaydos, L.A.; Freireich, E.J.; Mantel, N. The quantitative relation between platelet count and hemorrhage in patients with acute leukemia. N. Engl. J. Med. 1962, 266, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.B.; Yomtovian, R.A.; Dillman, C.; Masarik, S.R.; Chongkolwatana, V.; Creger, R.J.; Manka, A.; Hammons, T.; Lazarus, H.M. Reliability of automated platelet counts: Comparison with manual method and utility for prediction of clinical bleeding. Am. J. Hematol. 1995, 48, 244–250. [Google Scholar] [CrossRef]
- Rebulla, P.; Finazzi, G.; Marangoni, F.; Avvisati, G.; Gugliotta, L.; Tognoni, G.; Barbui, T.; Mandelli, F.; Sirchia, G. The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto. N. Engl. J. Med. 1997, 337, 1870–1875. [Google Scholar] [CrossRef]
- McCullough, J.; Vesole, D.H.; Benjamin, R.J.; Slichter, S.J.; Pineda, A.; Snyder, E.; Stadtmauer, E.A.; Lopez-Plaza, I.; Coutre, S.; Strauss, R.G.; et al. Therapeutic efficacy and safety of platelets treated with a photochemical process for pathogen inactivation: The Sprint Trial. Blood 2004, 104, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}