Sorption Thermodynamics of CO2, H2O, and CH3OH in a Glassy Polyetherimide: A Molecular Perspective




Abstract
:
1. Introduction
2. Materials and Methods
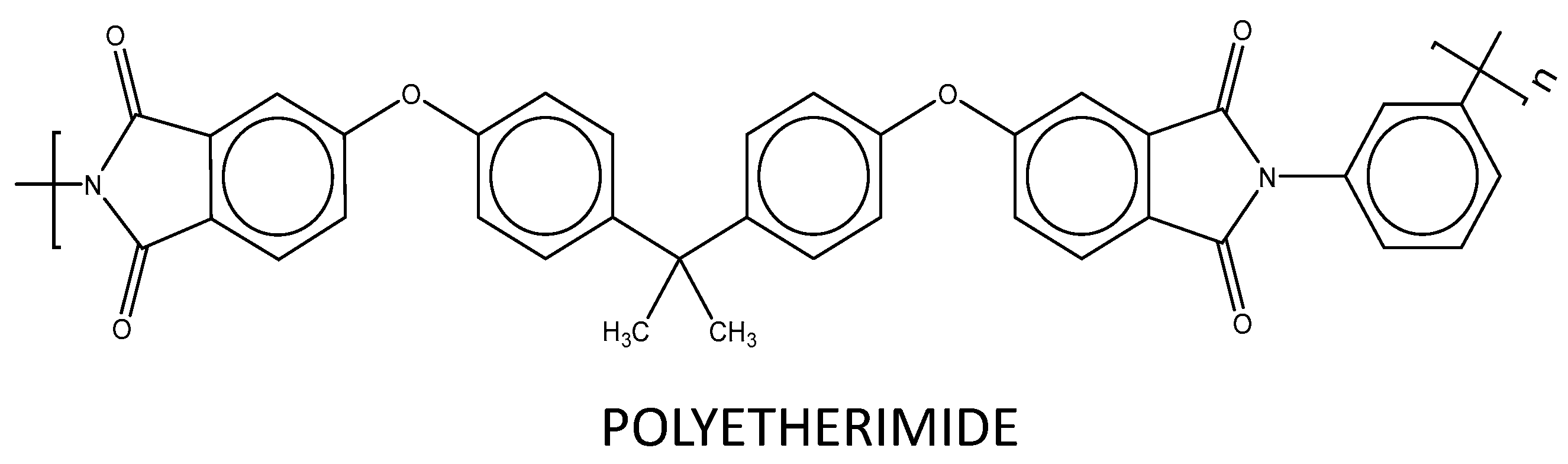
2.1. Materials
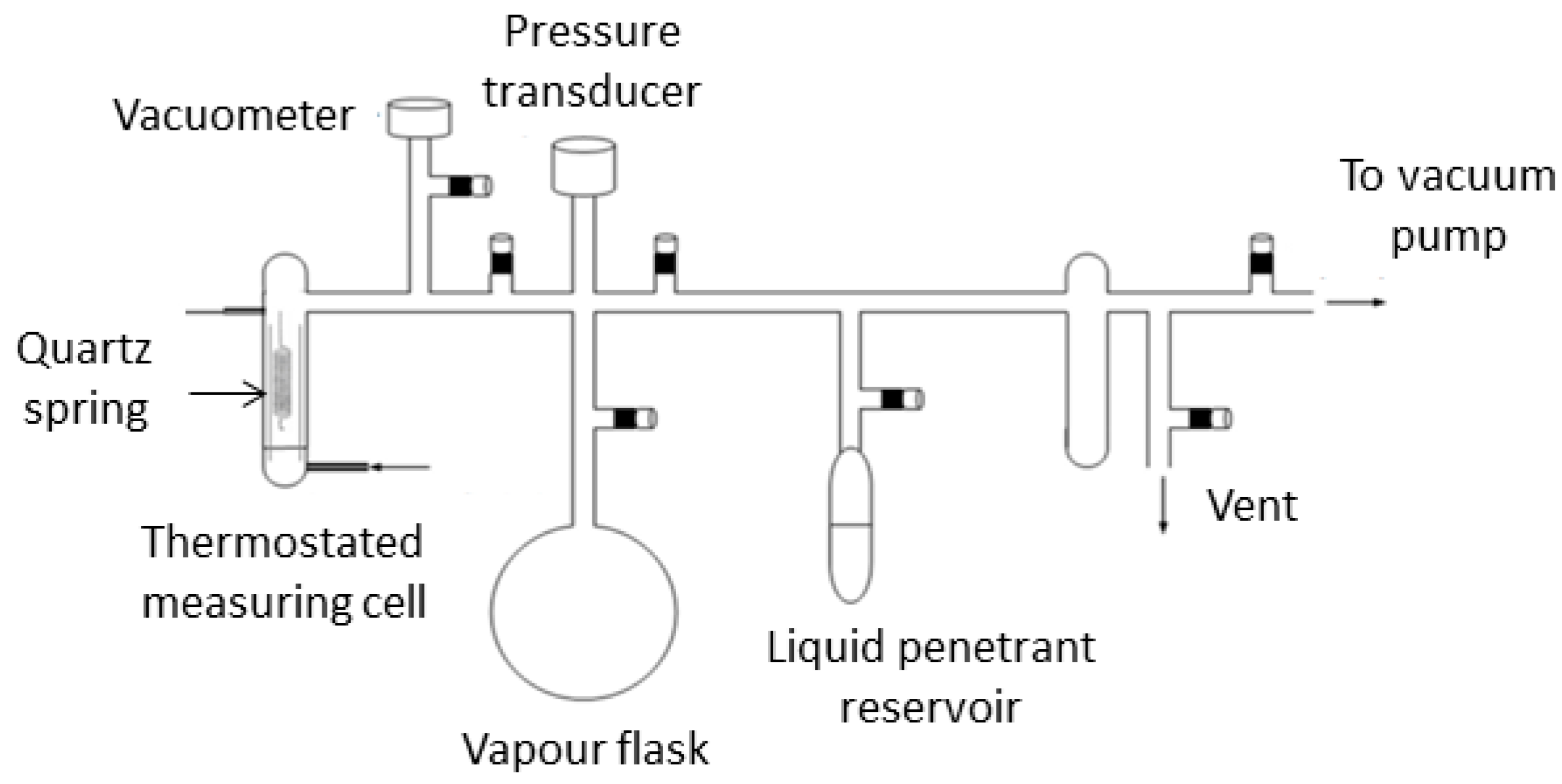
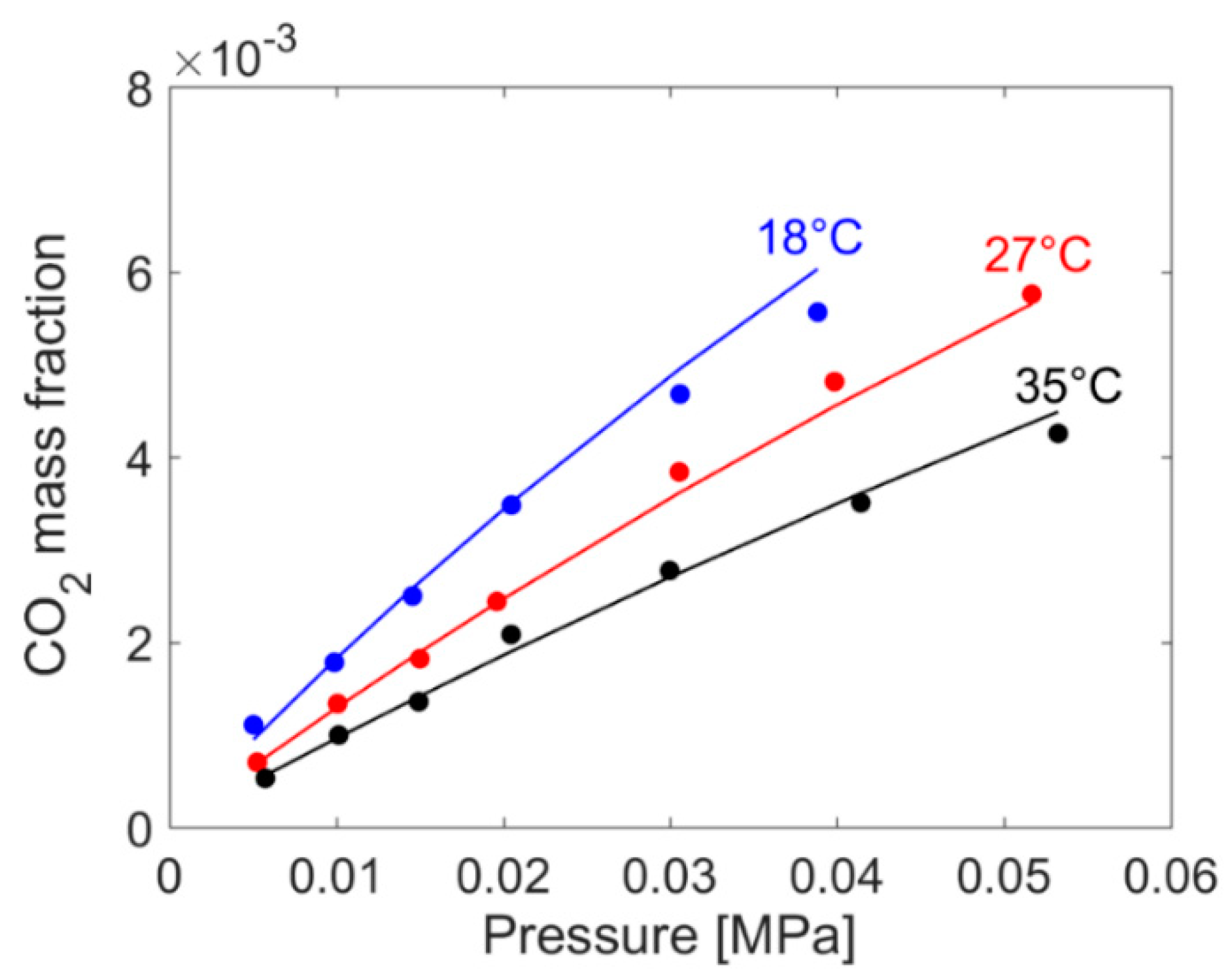
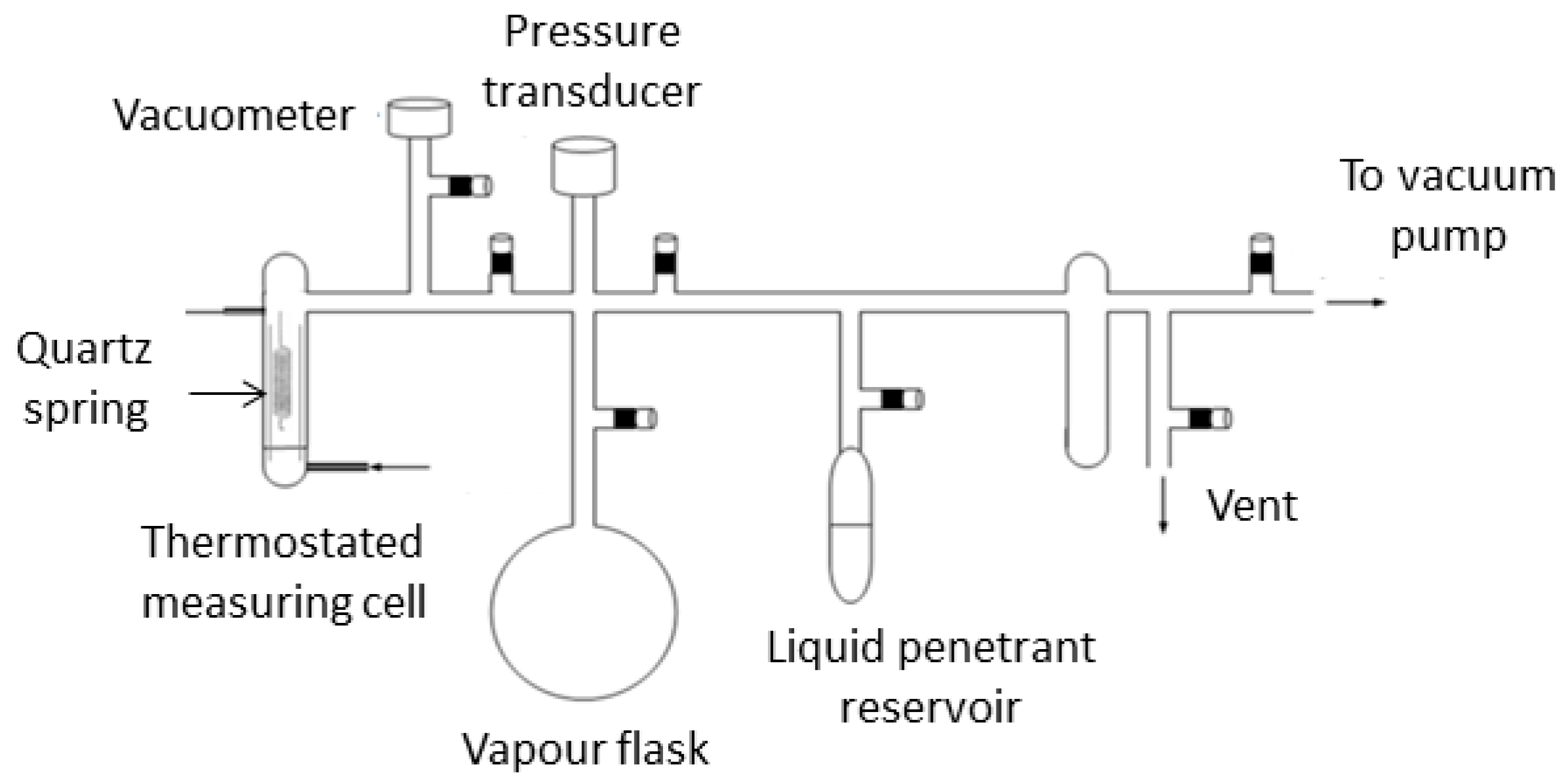
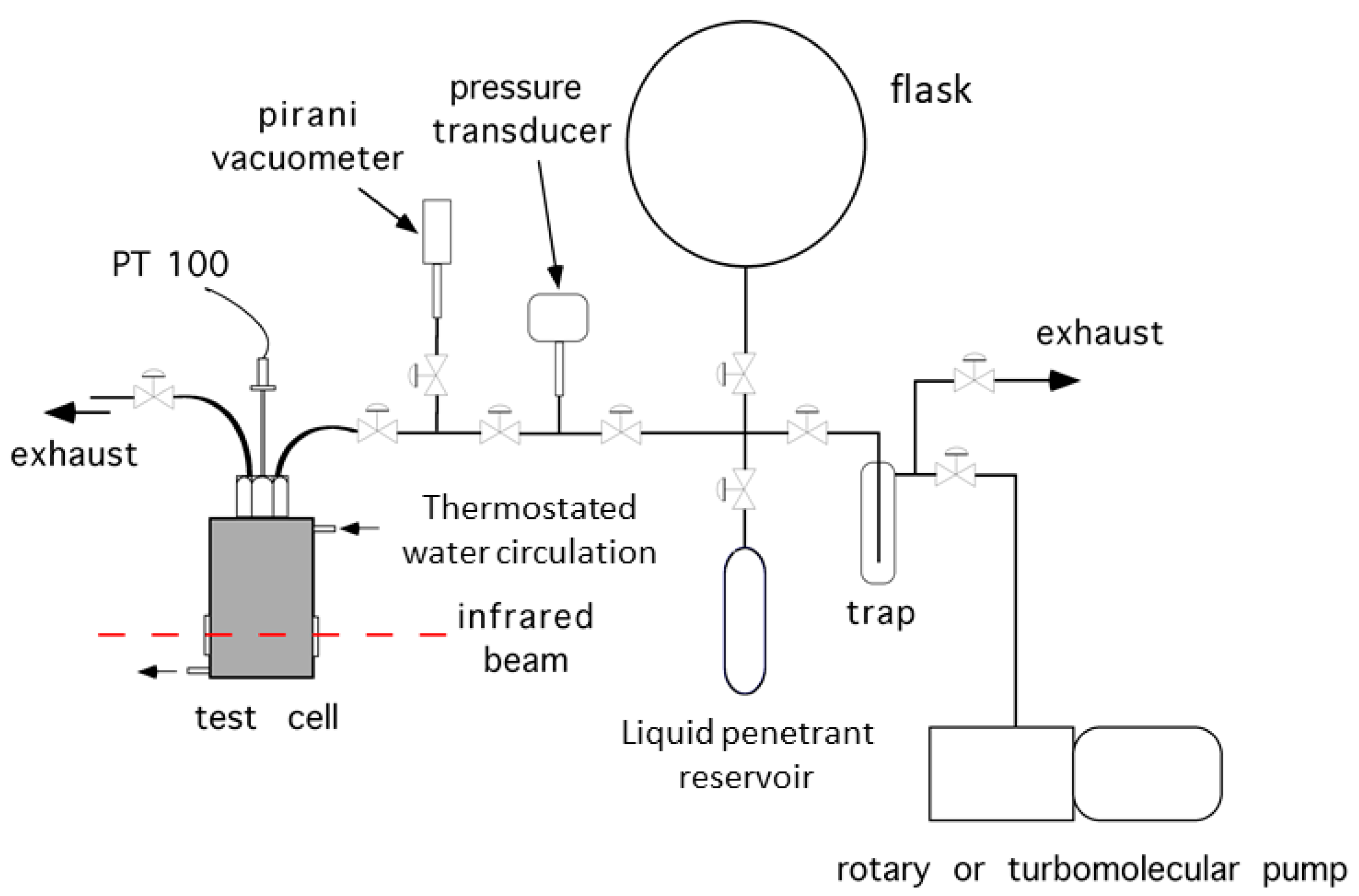
2.2. Gravimetric Measurements
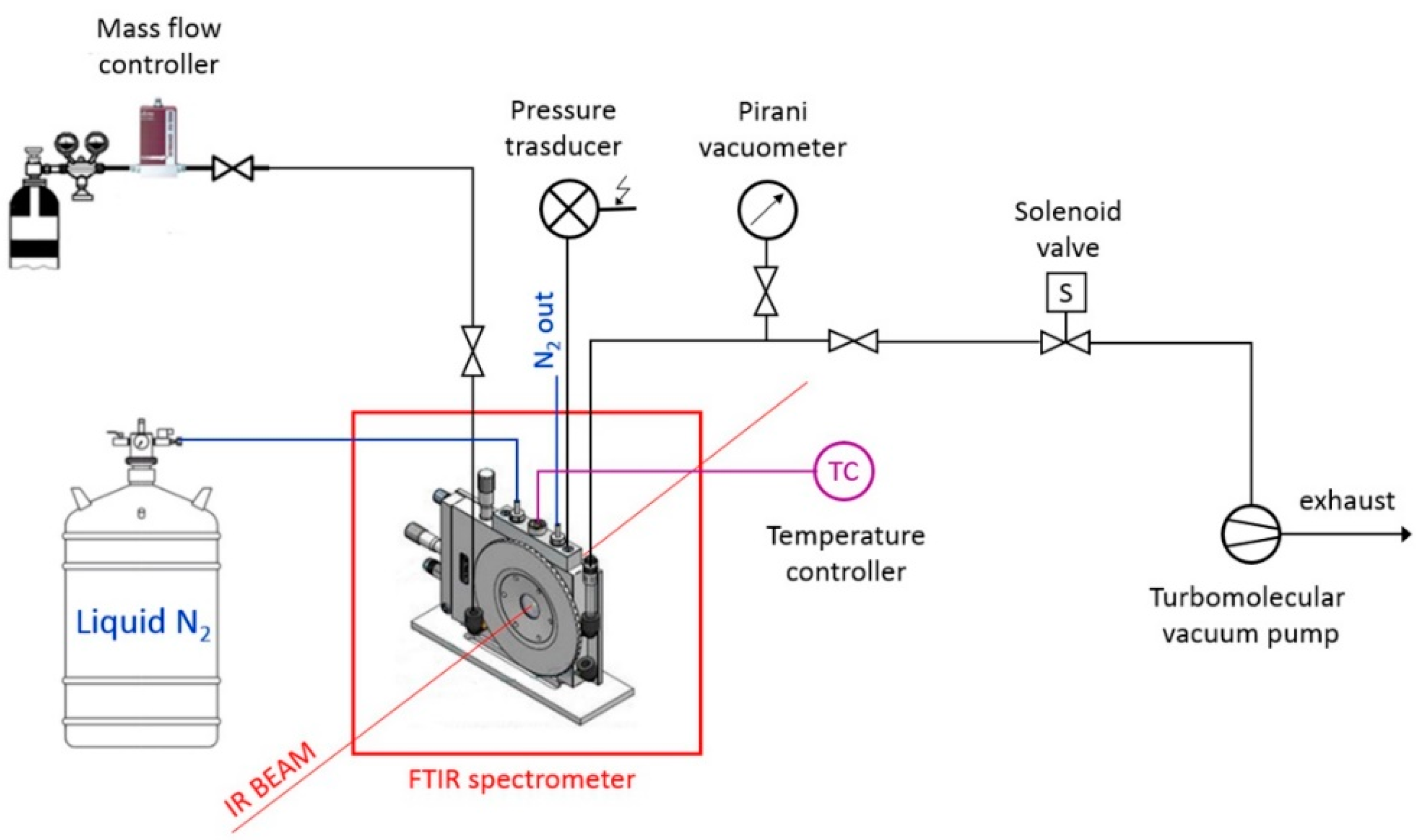
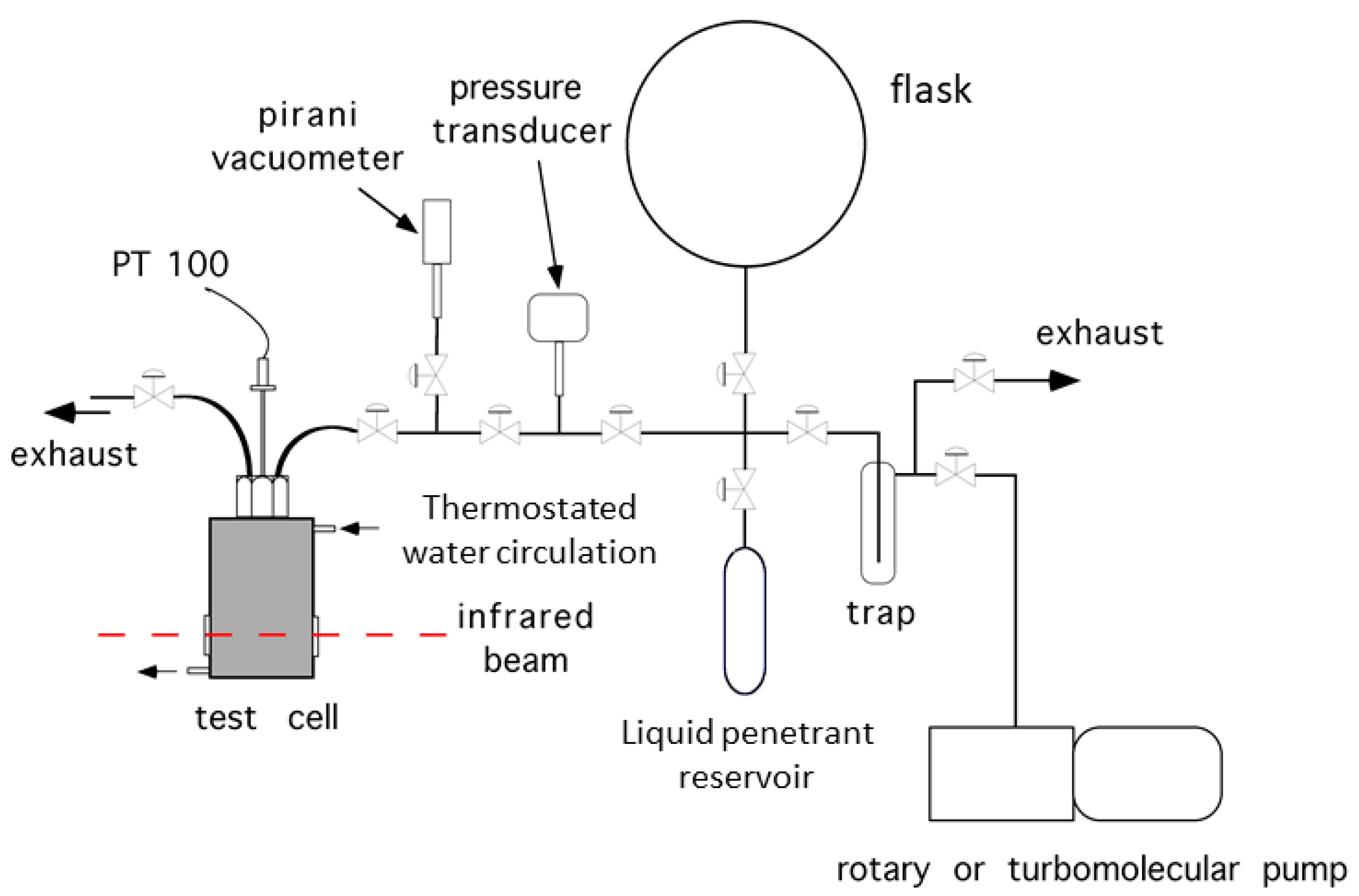
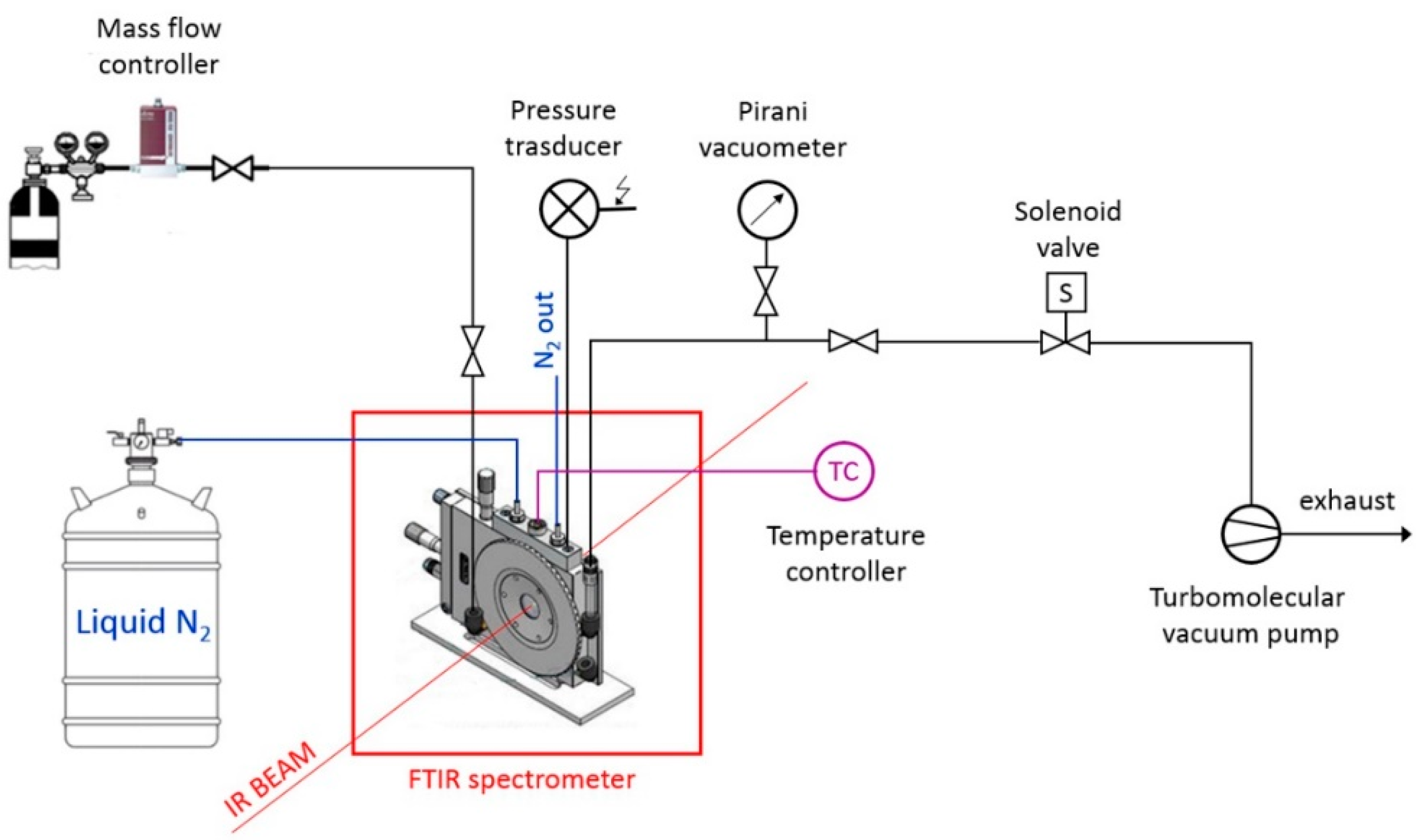
2.3. In Situ FTIR Spectroscopy
3. Theoretical Background: Modeling of Sorption Thermodynamics
4. Results and Discussion
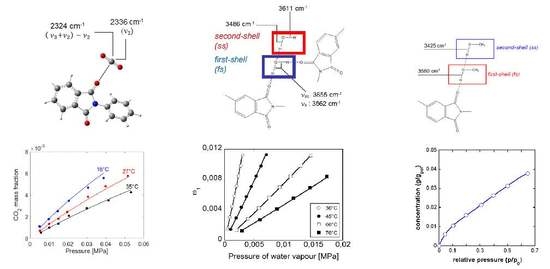
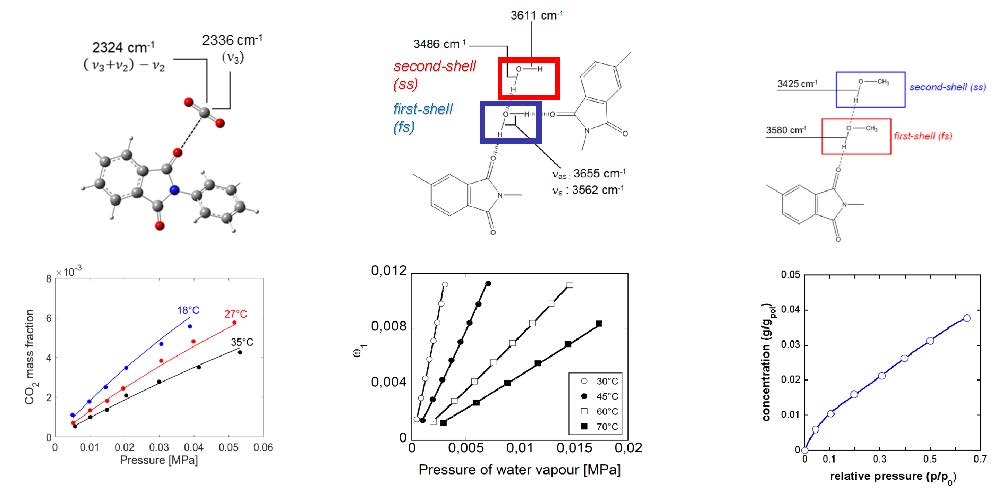
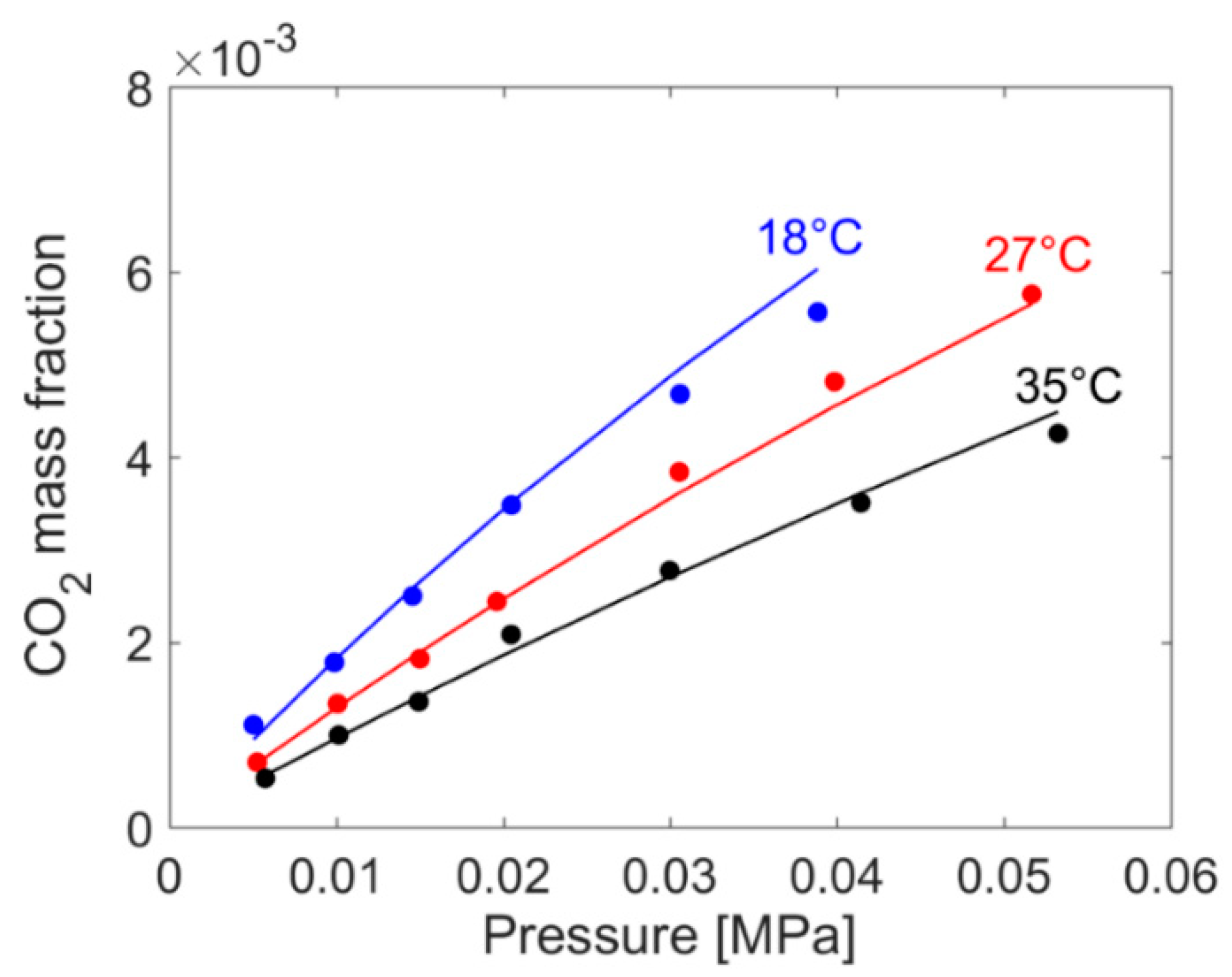
4.1. PEI–CO2 System
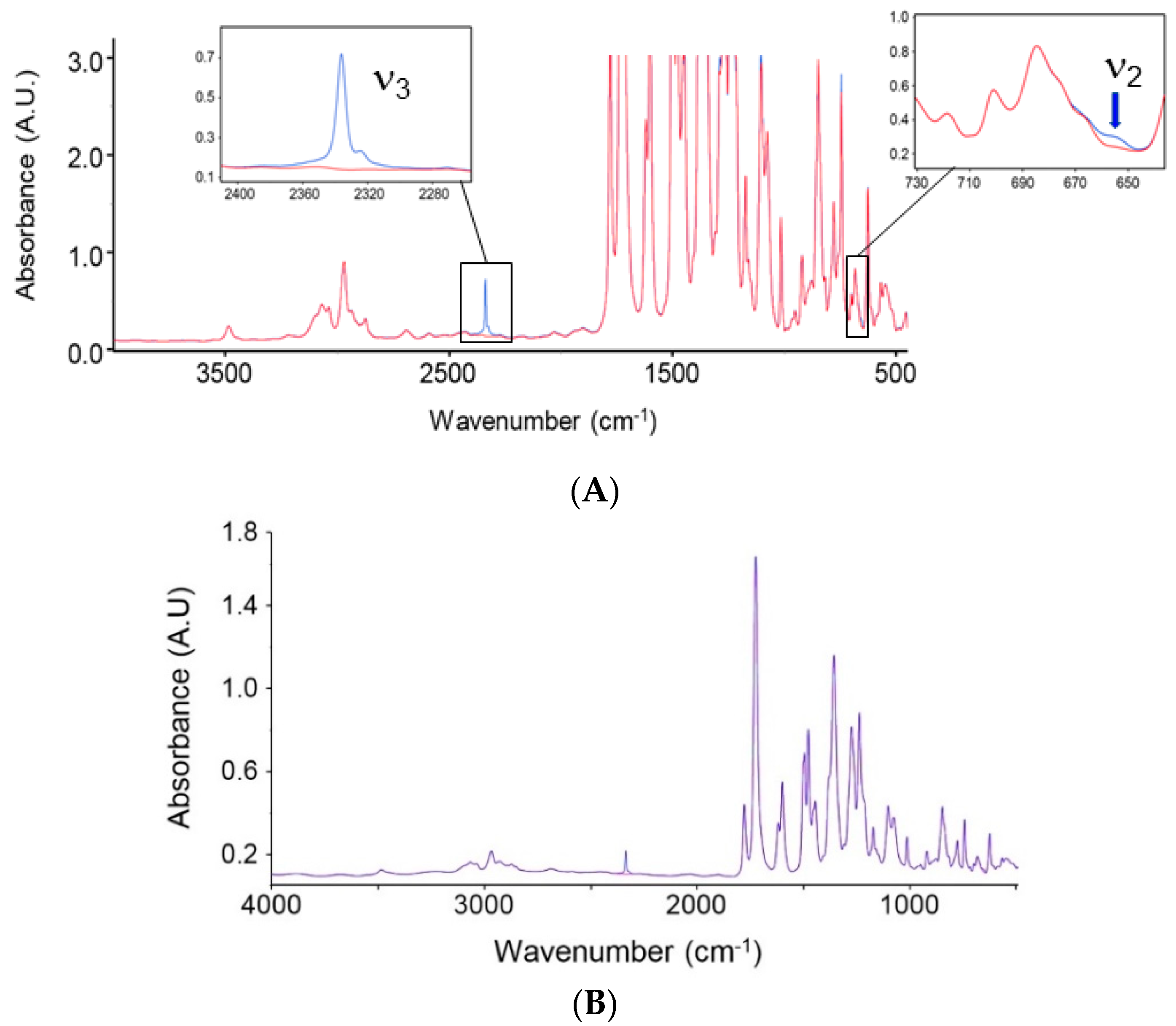
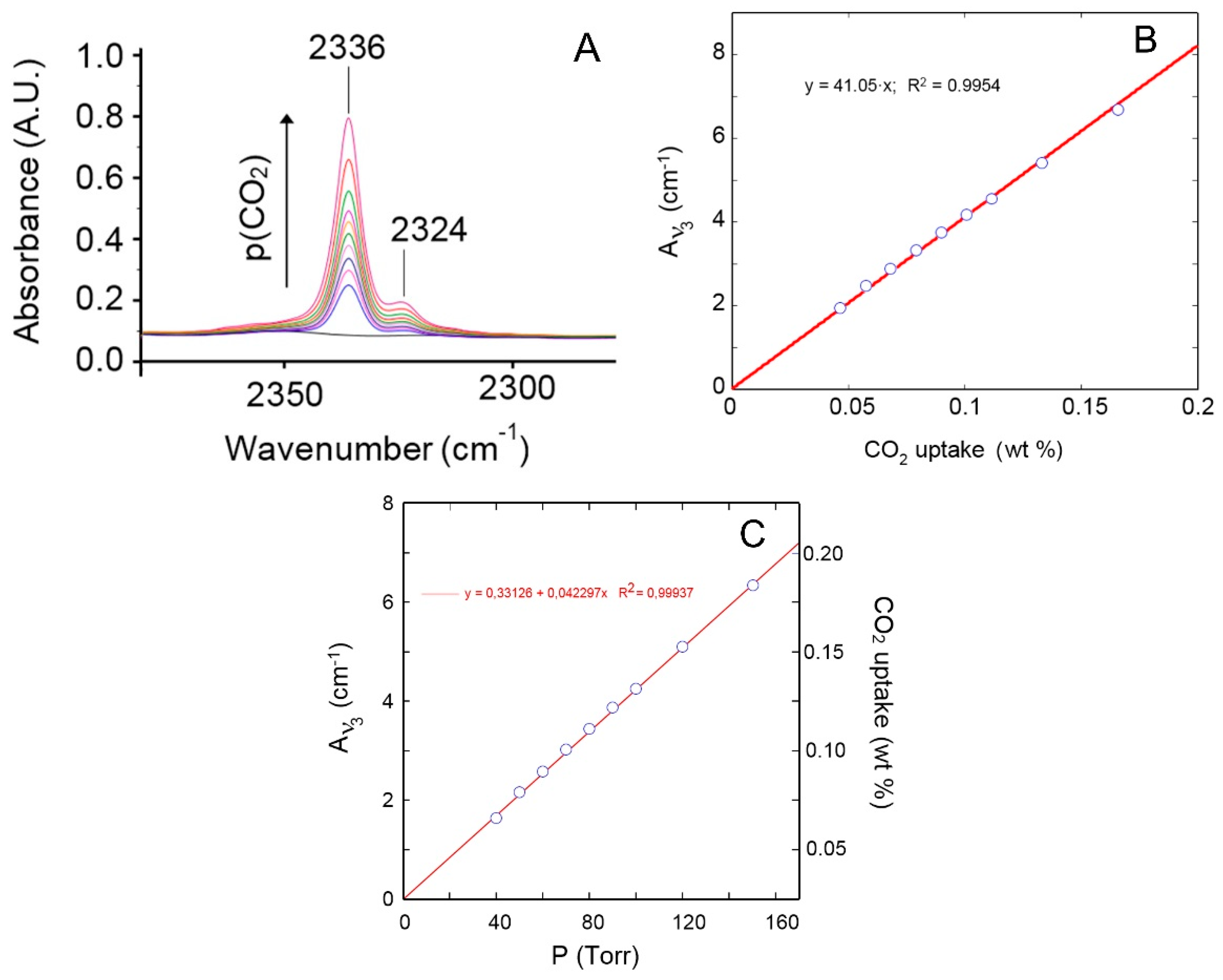
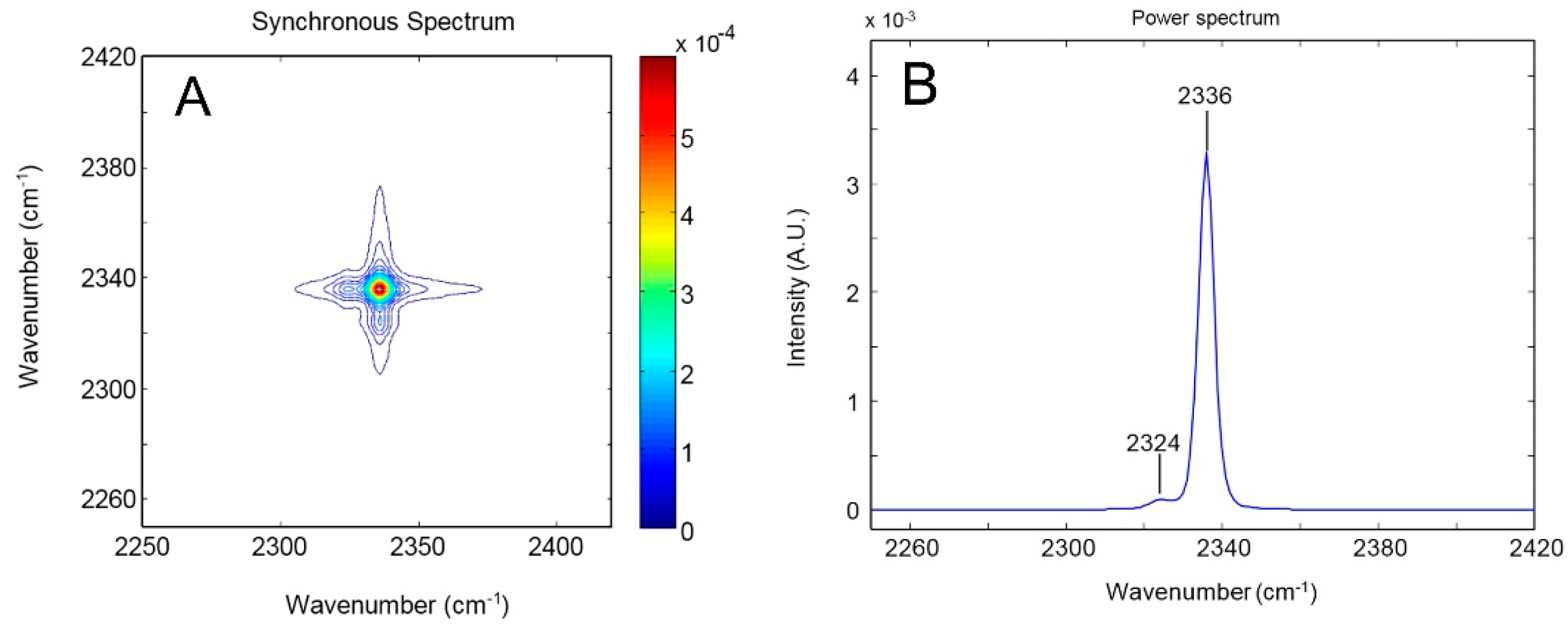
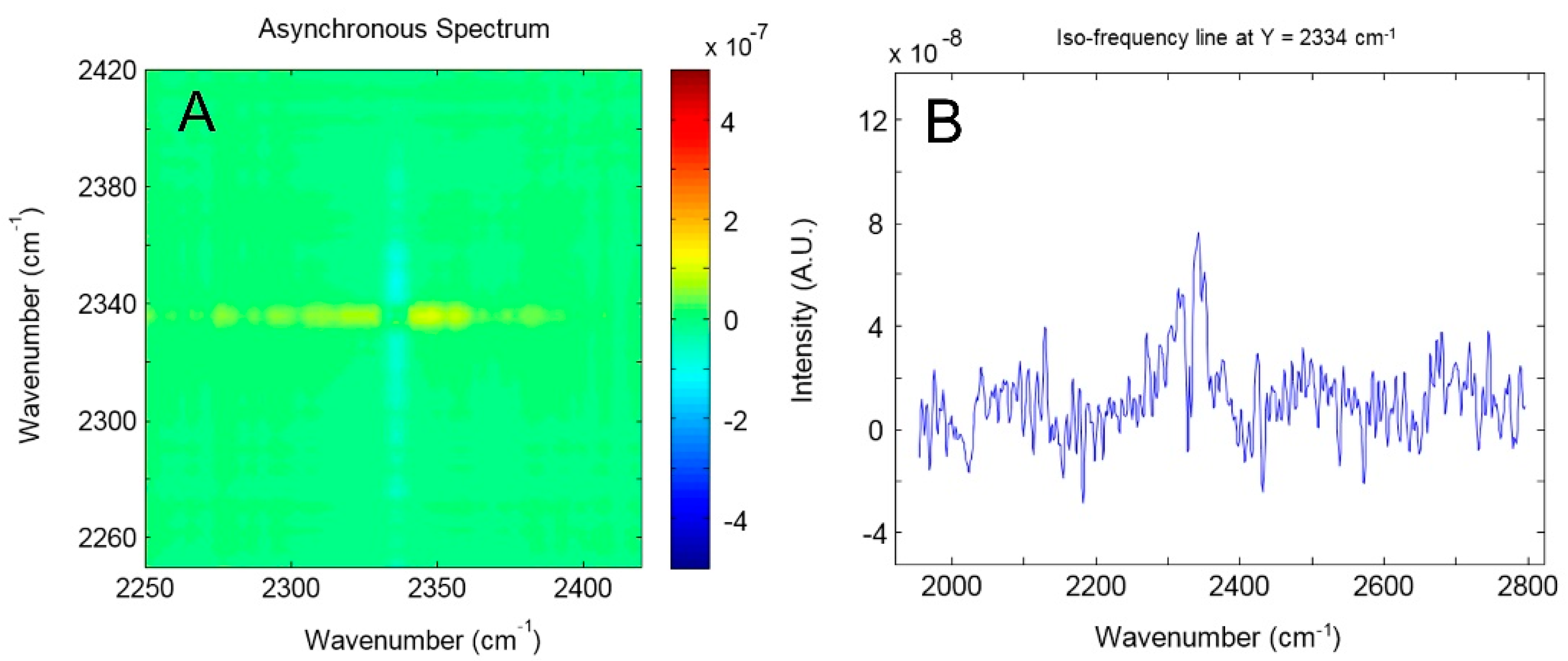
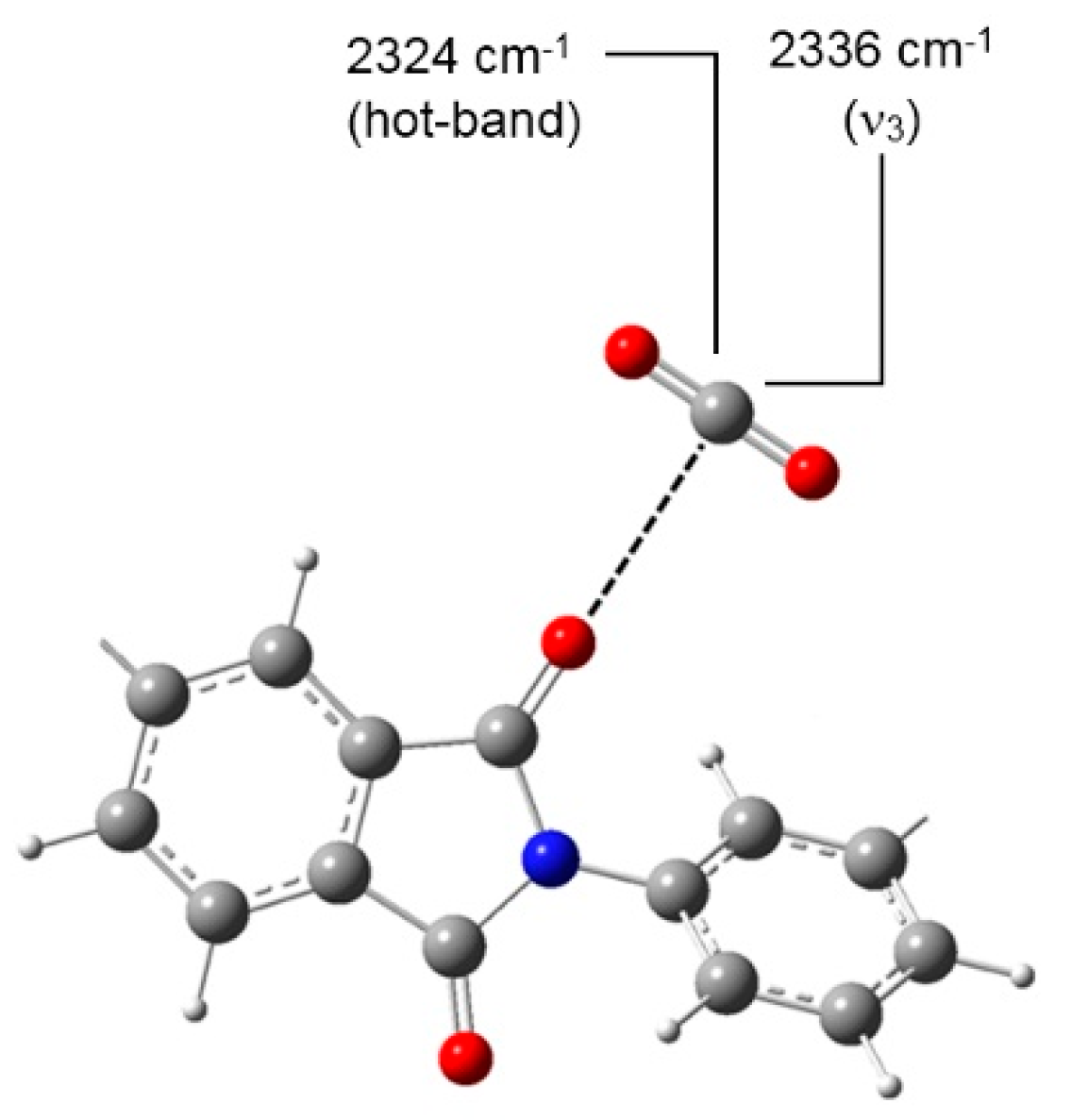
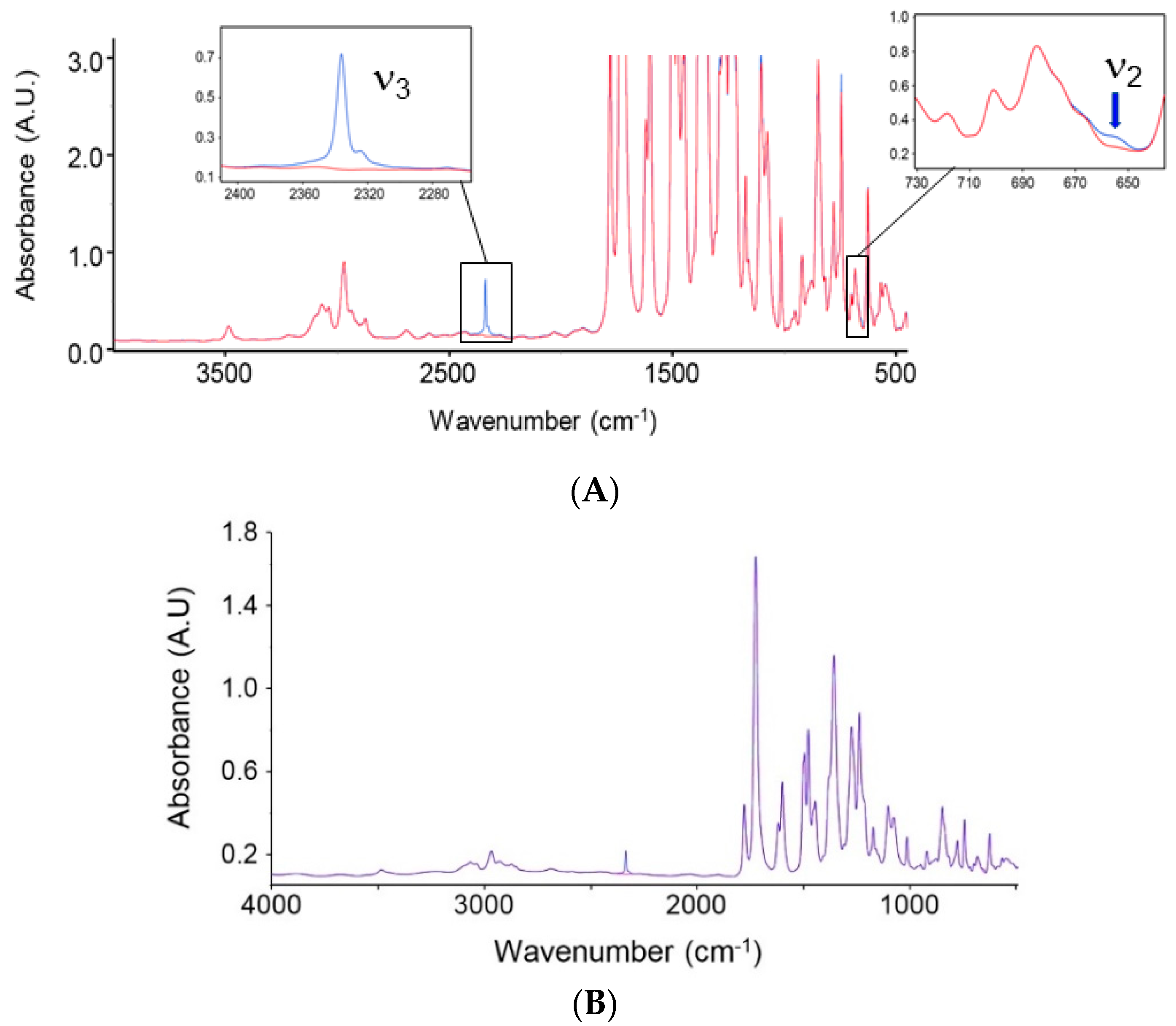
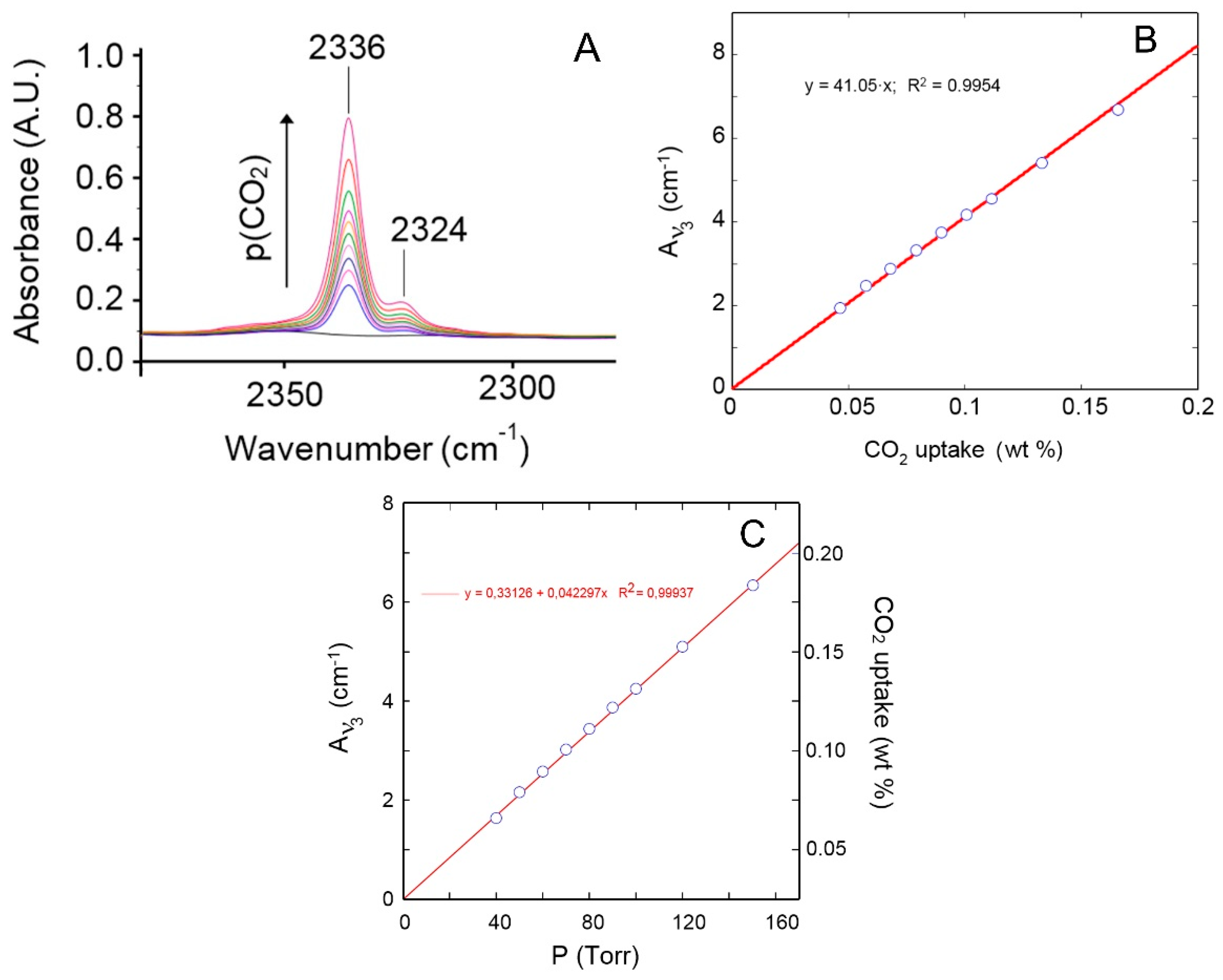
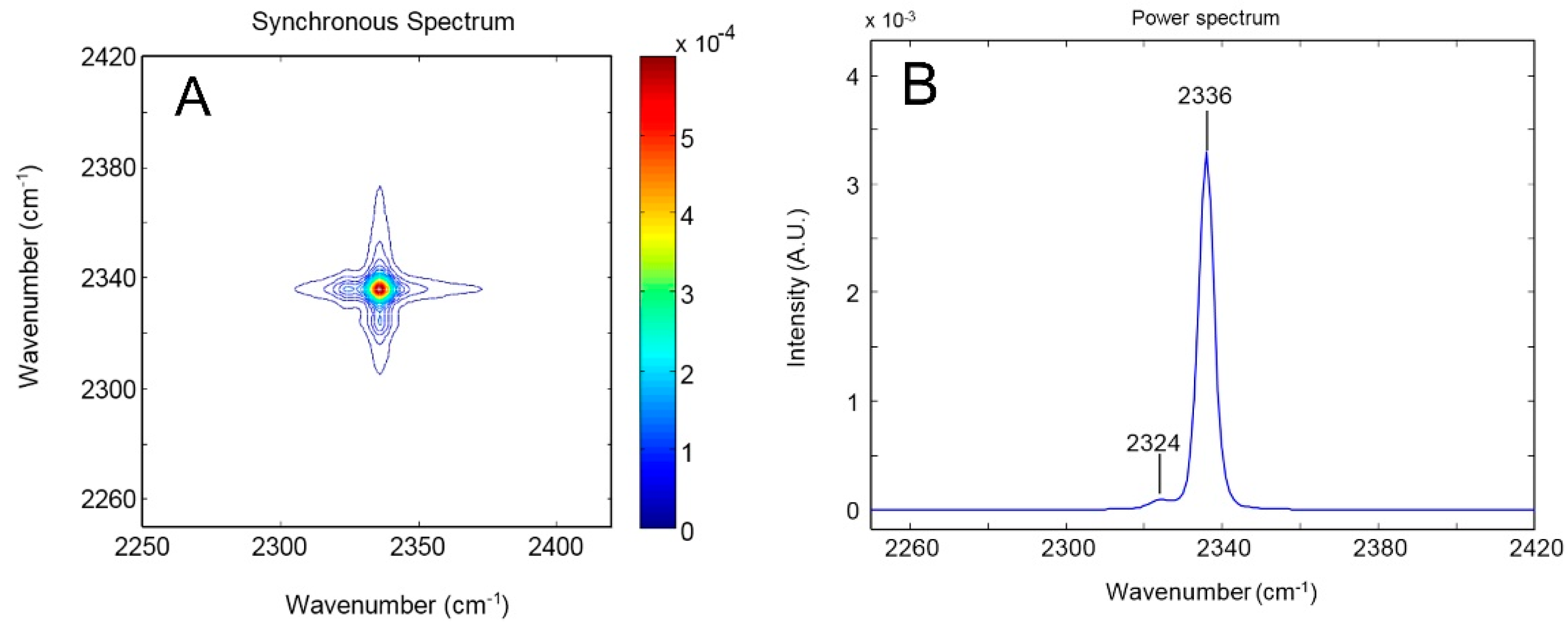
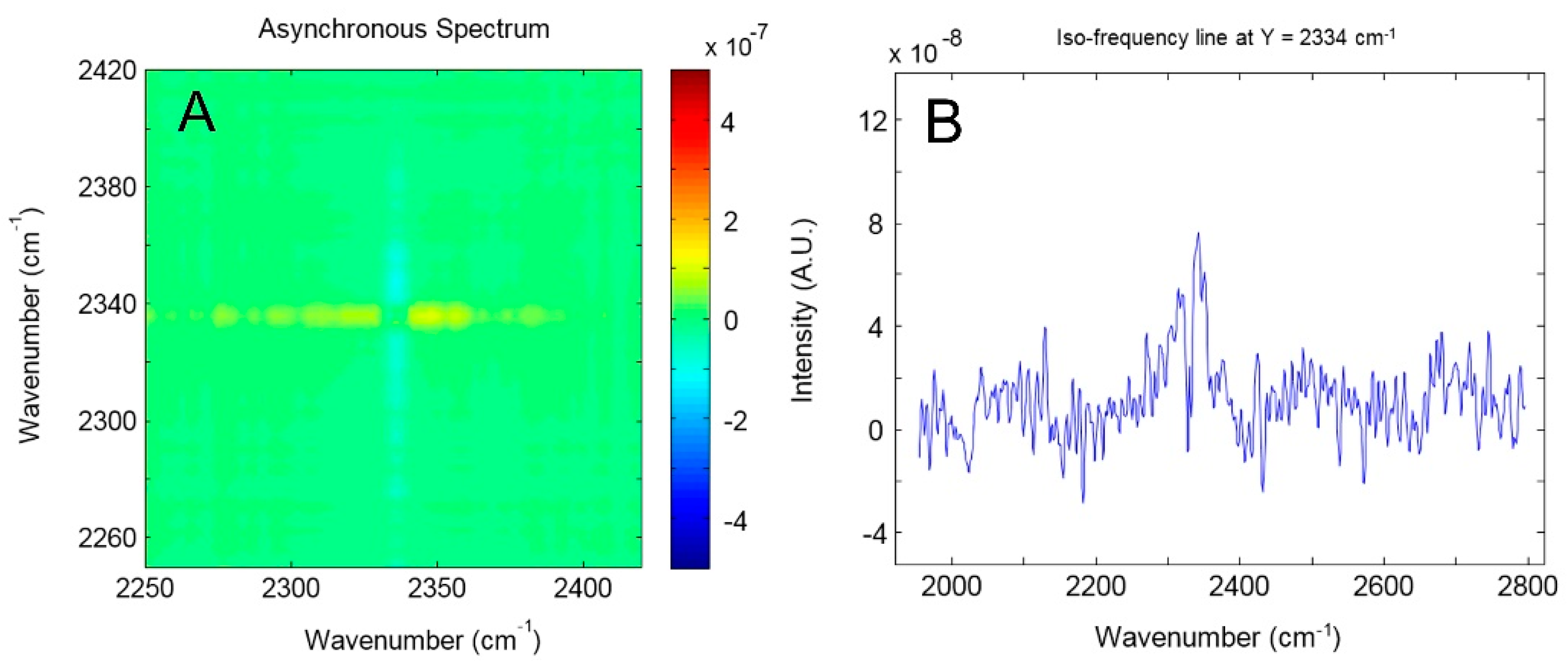
4.1.1. FTIR Analysis: Absorbance, Difference, and Two-Dimensional Correlation Spectra
4.1.2. Modeling Sorption Thermodynamics
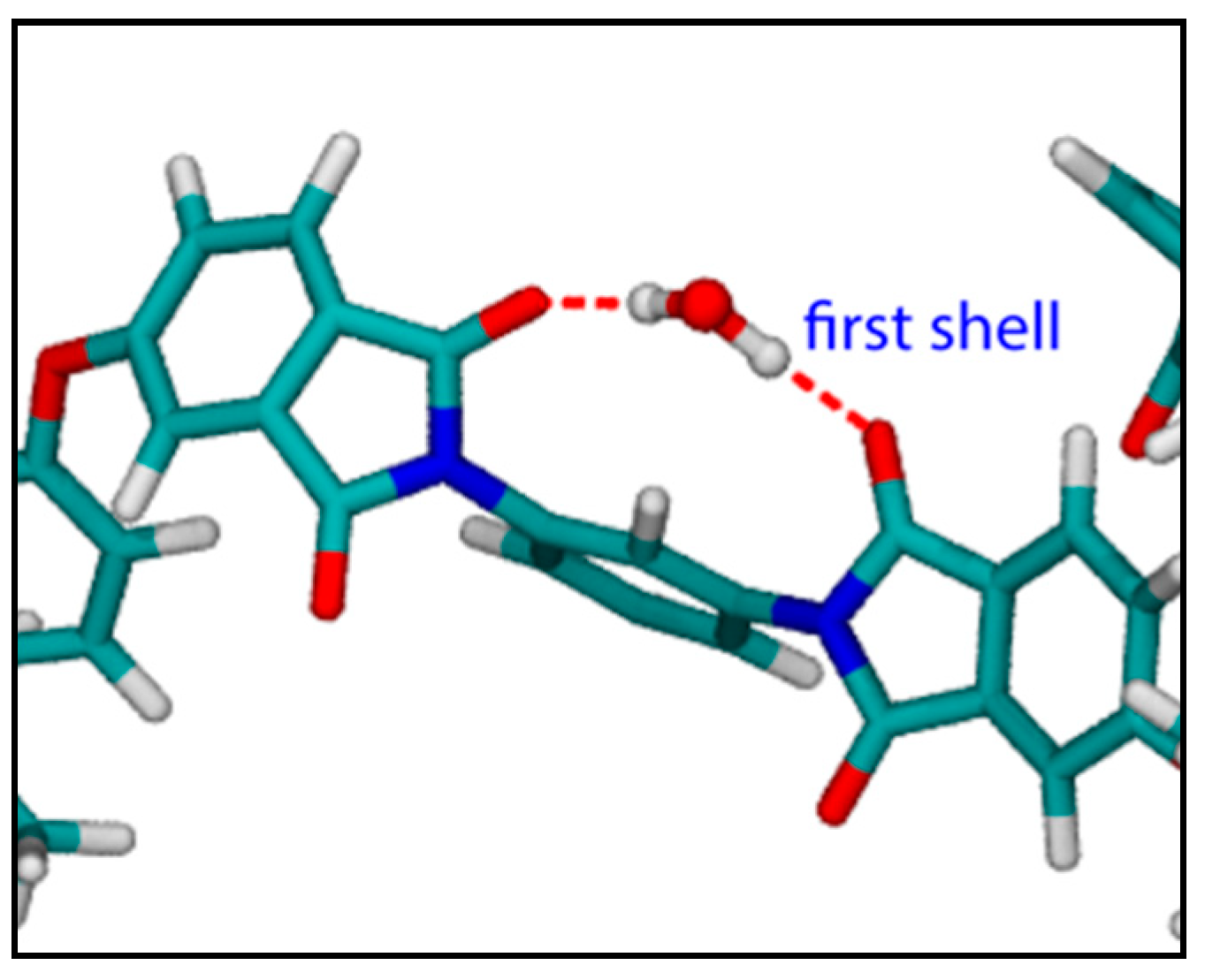
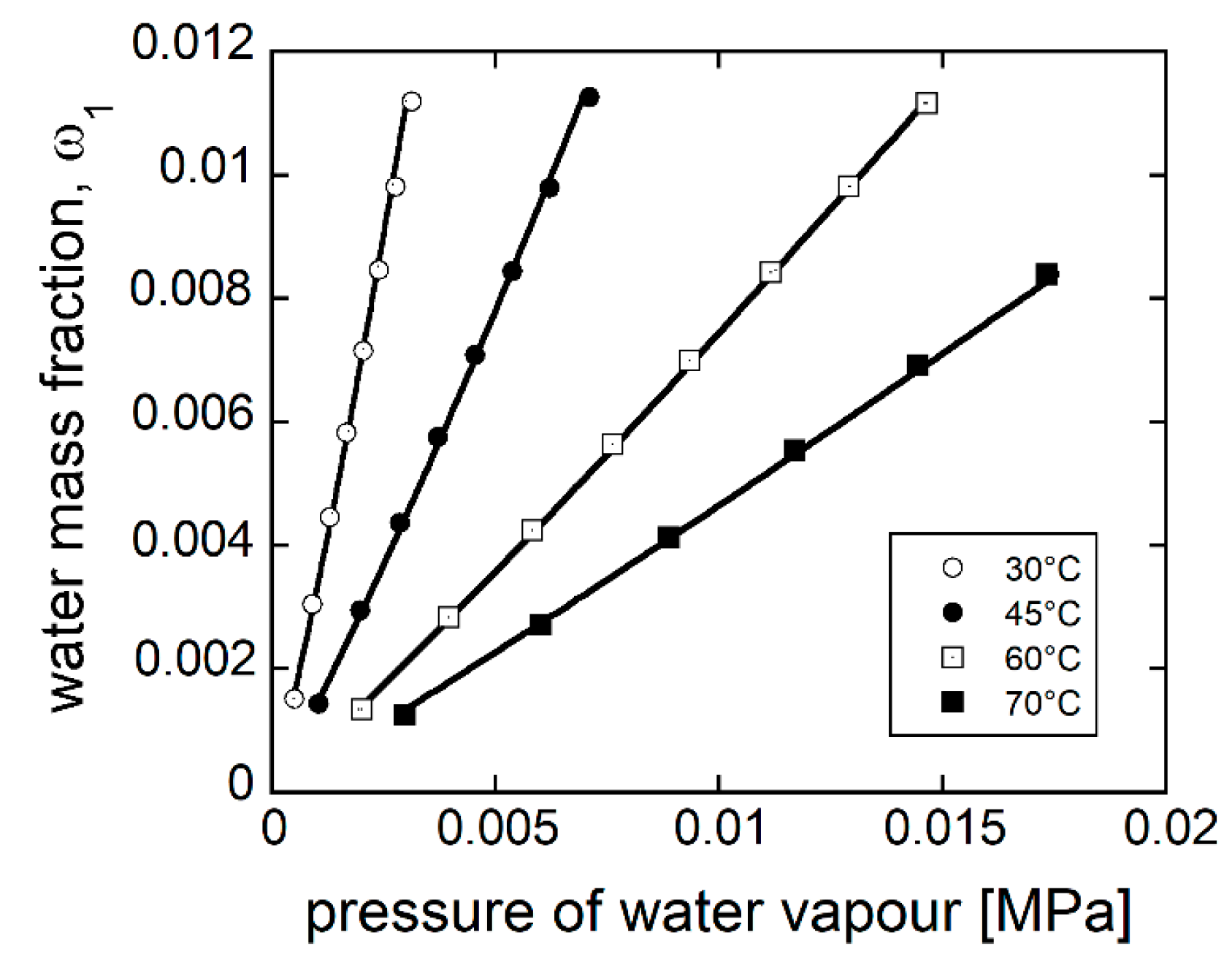
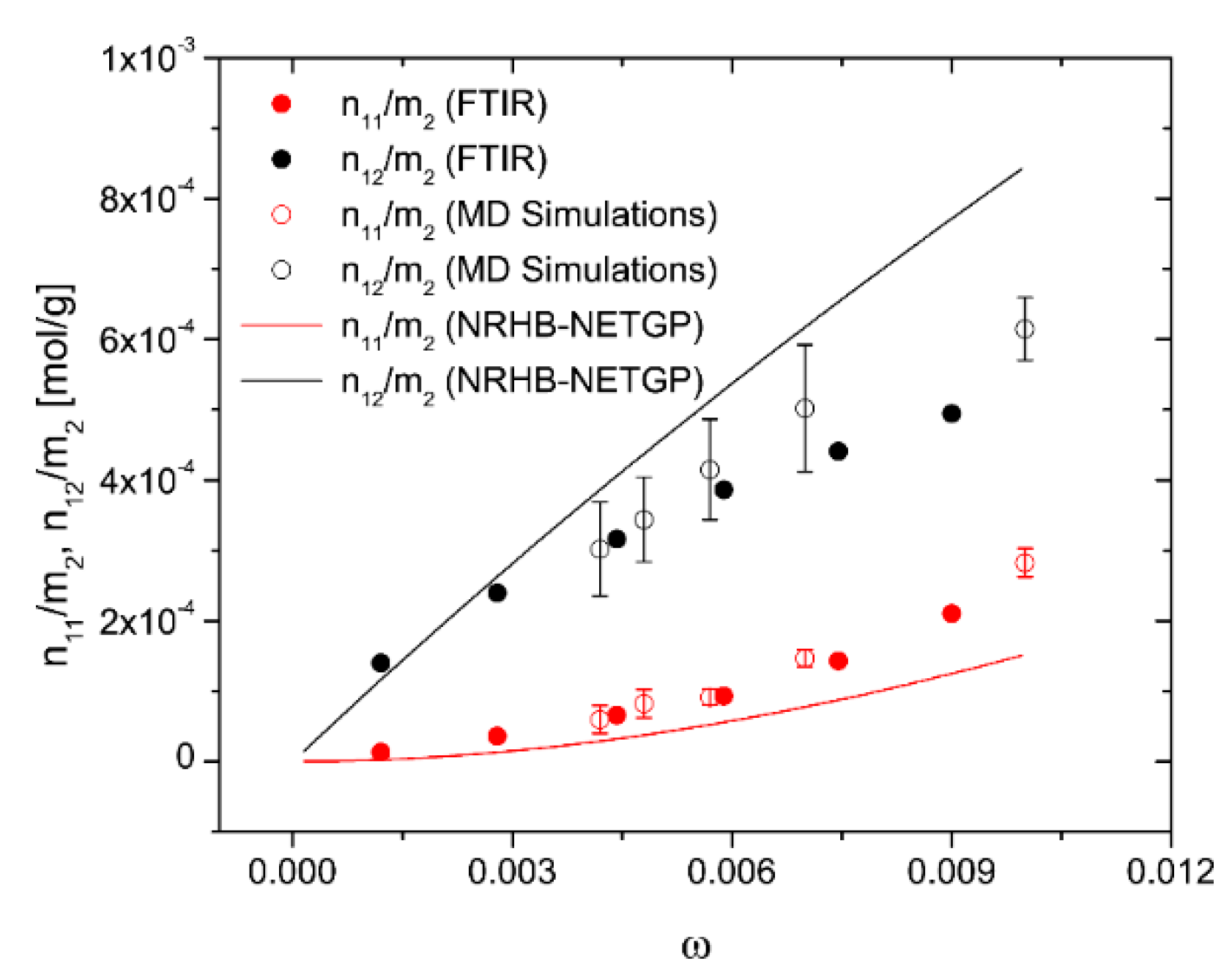
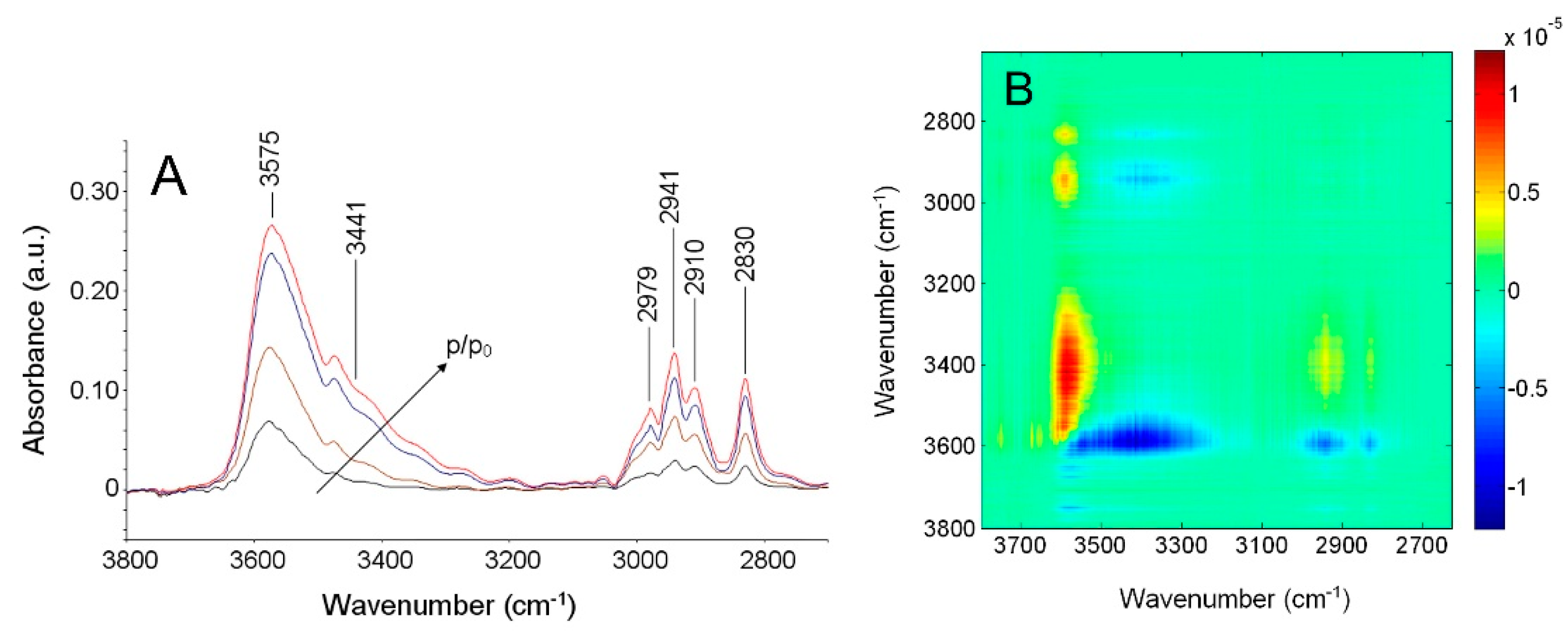
4.2. PEI–H2O System
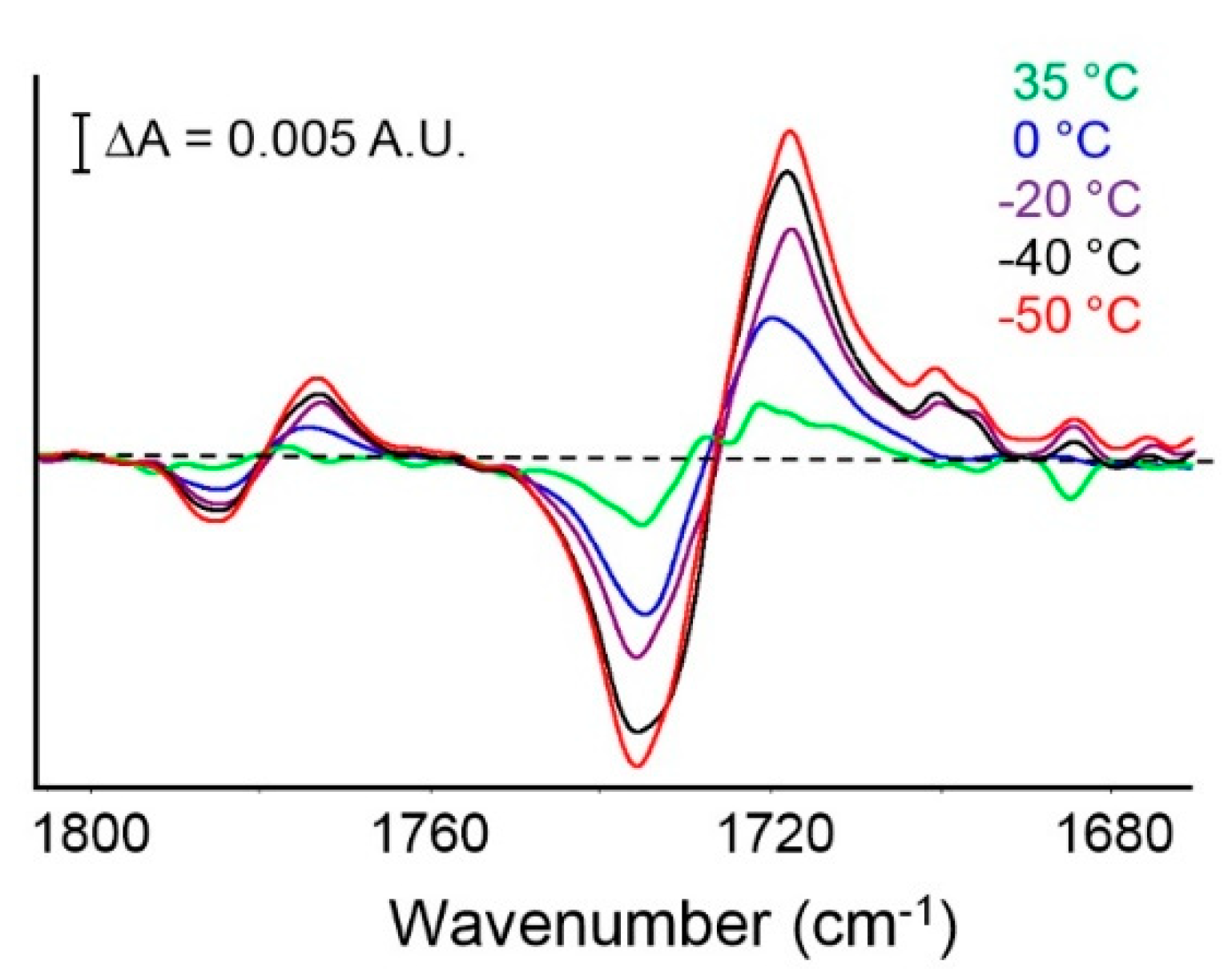
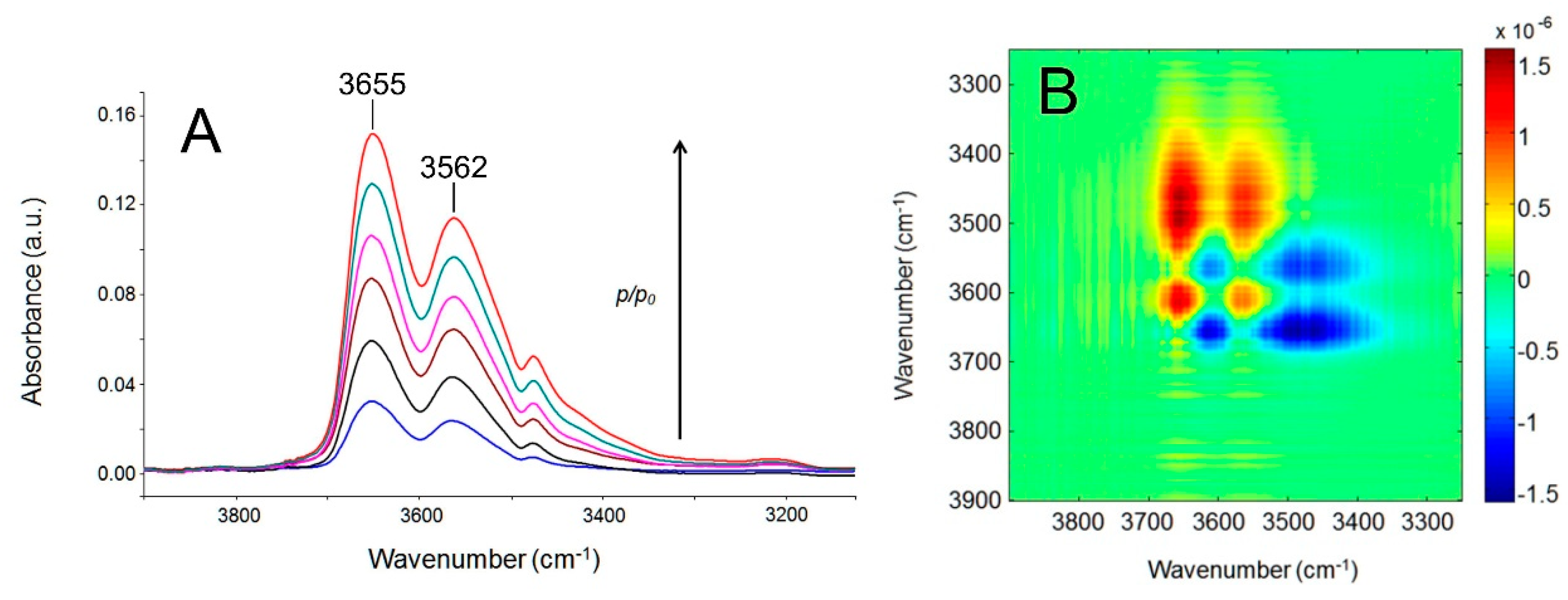
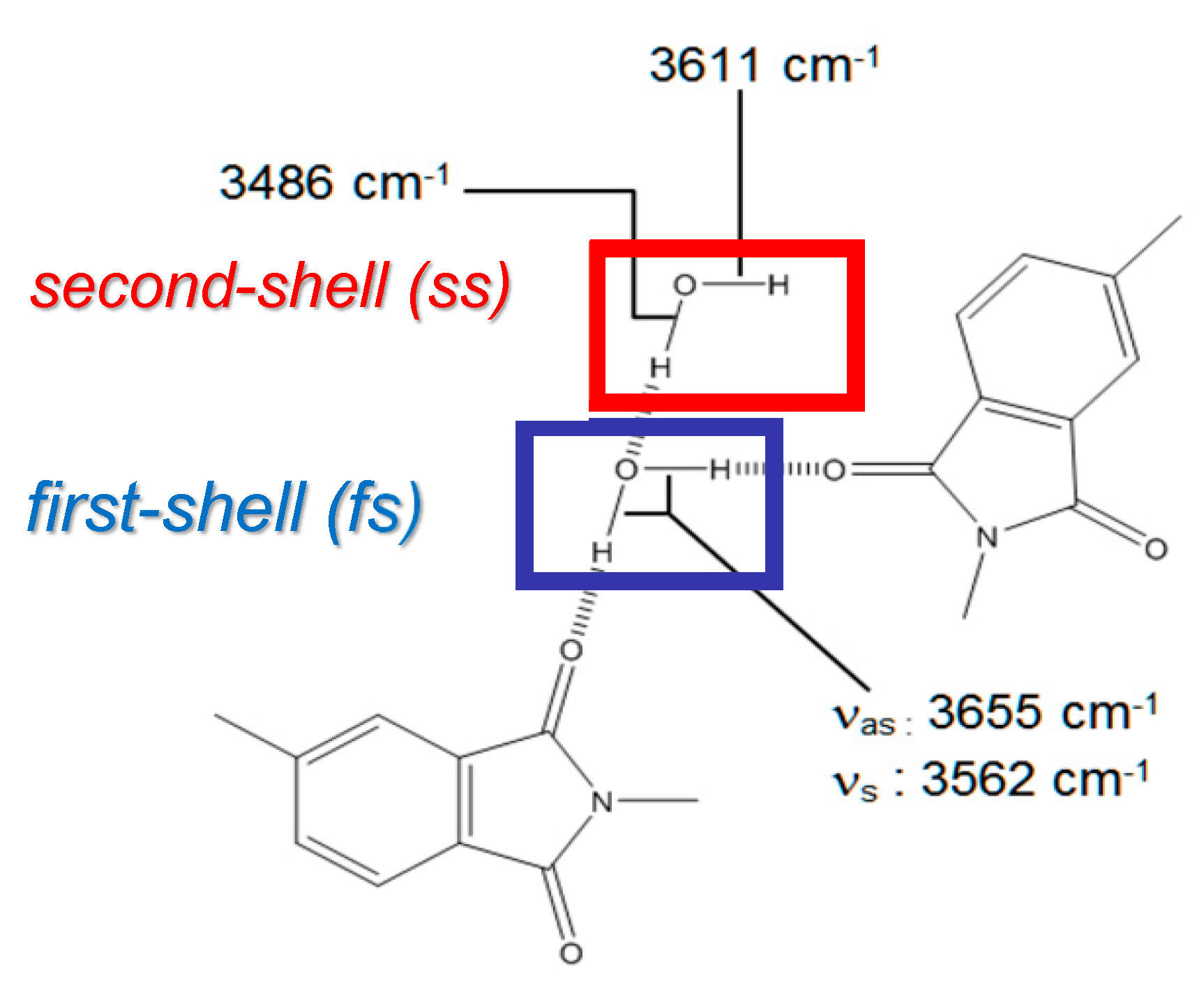
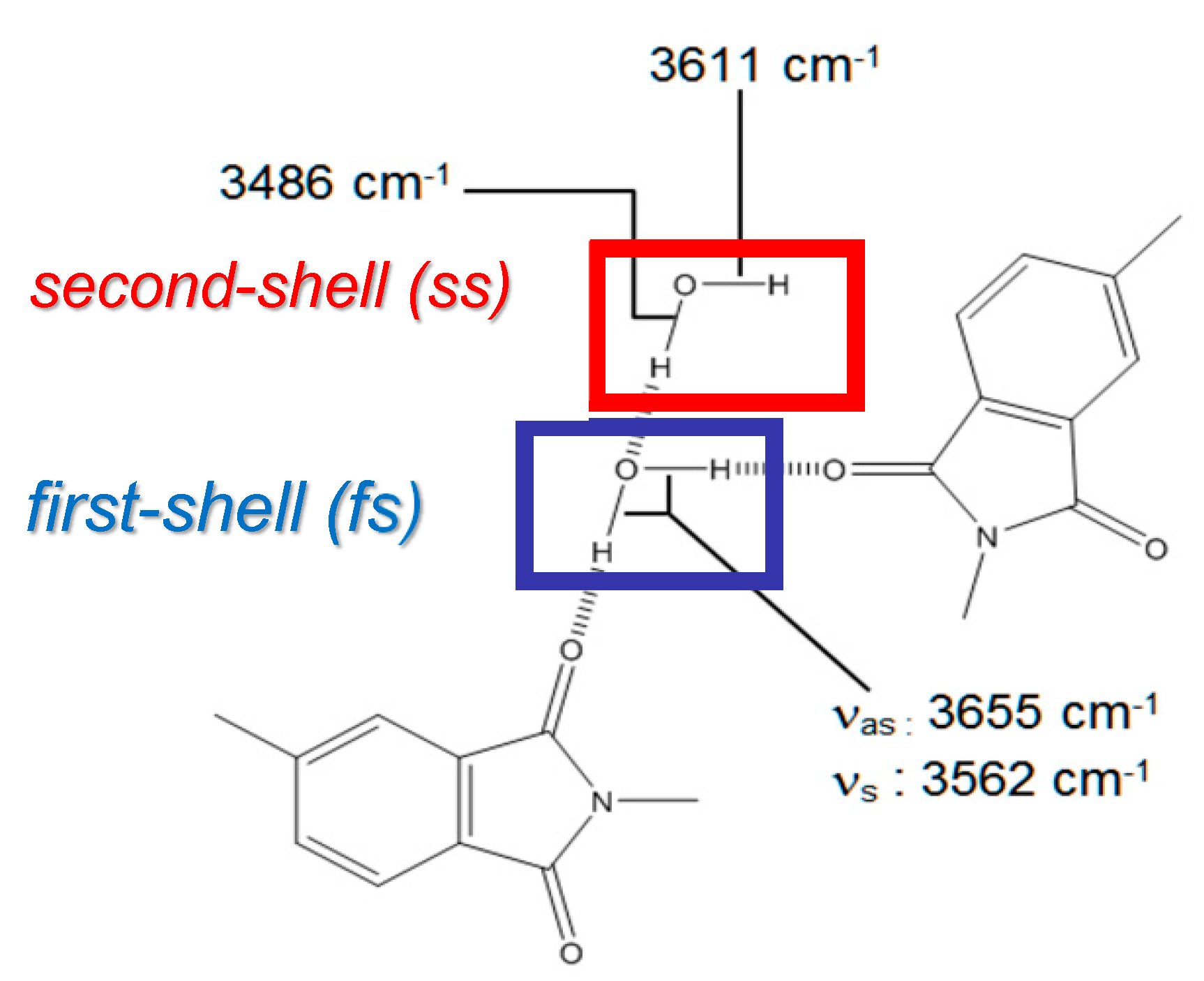
4.2.1. FTIR Analysis: Absorbance, Difference, and Two-Dimensional Correlation Spectra
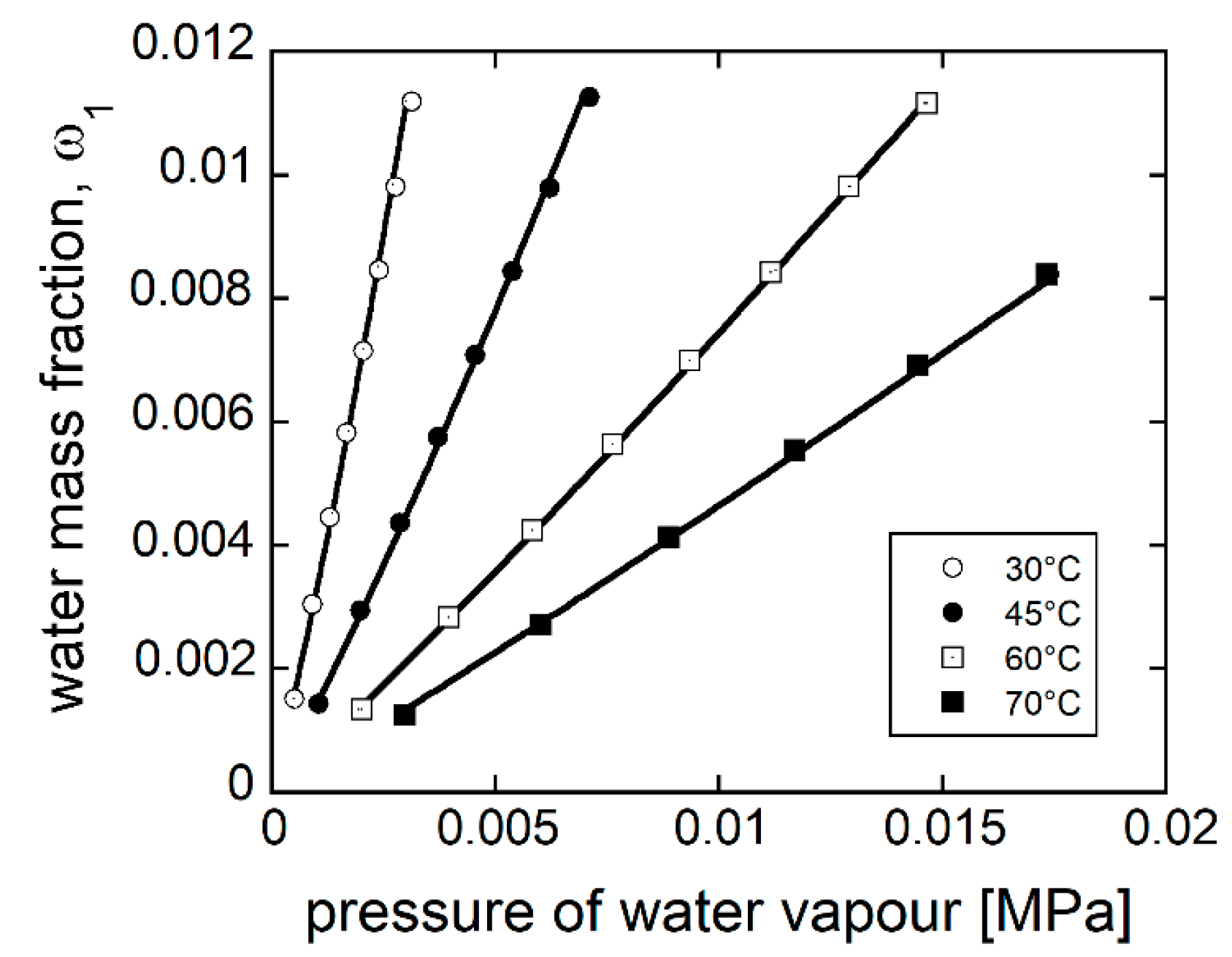
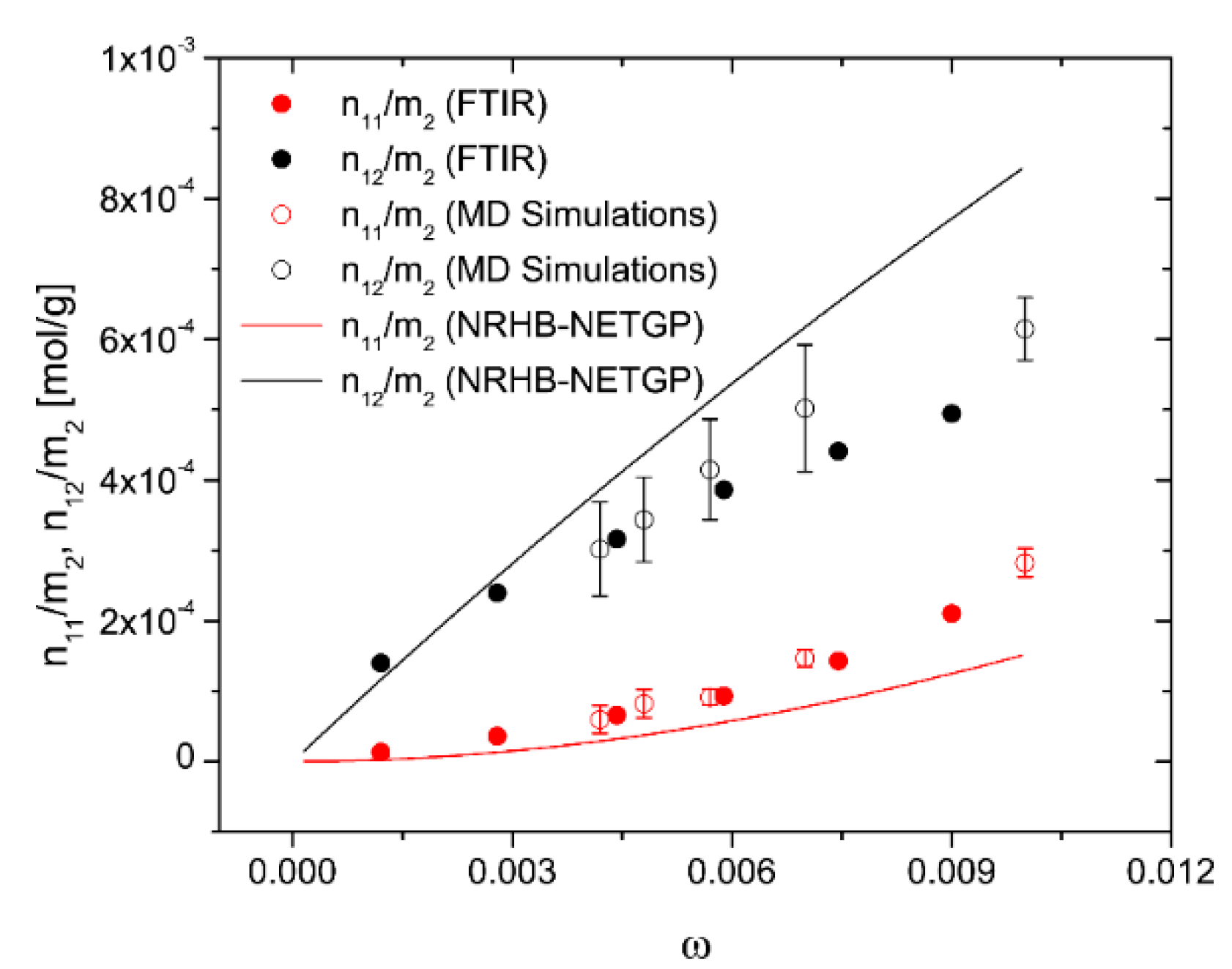
4.2.2. Modeling Sorption Thermodynamics
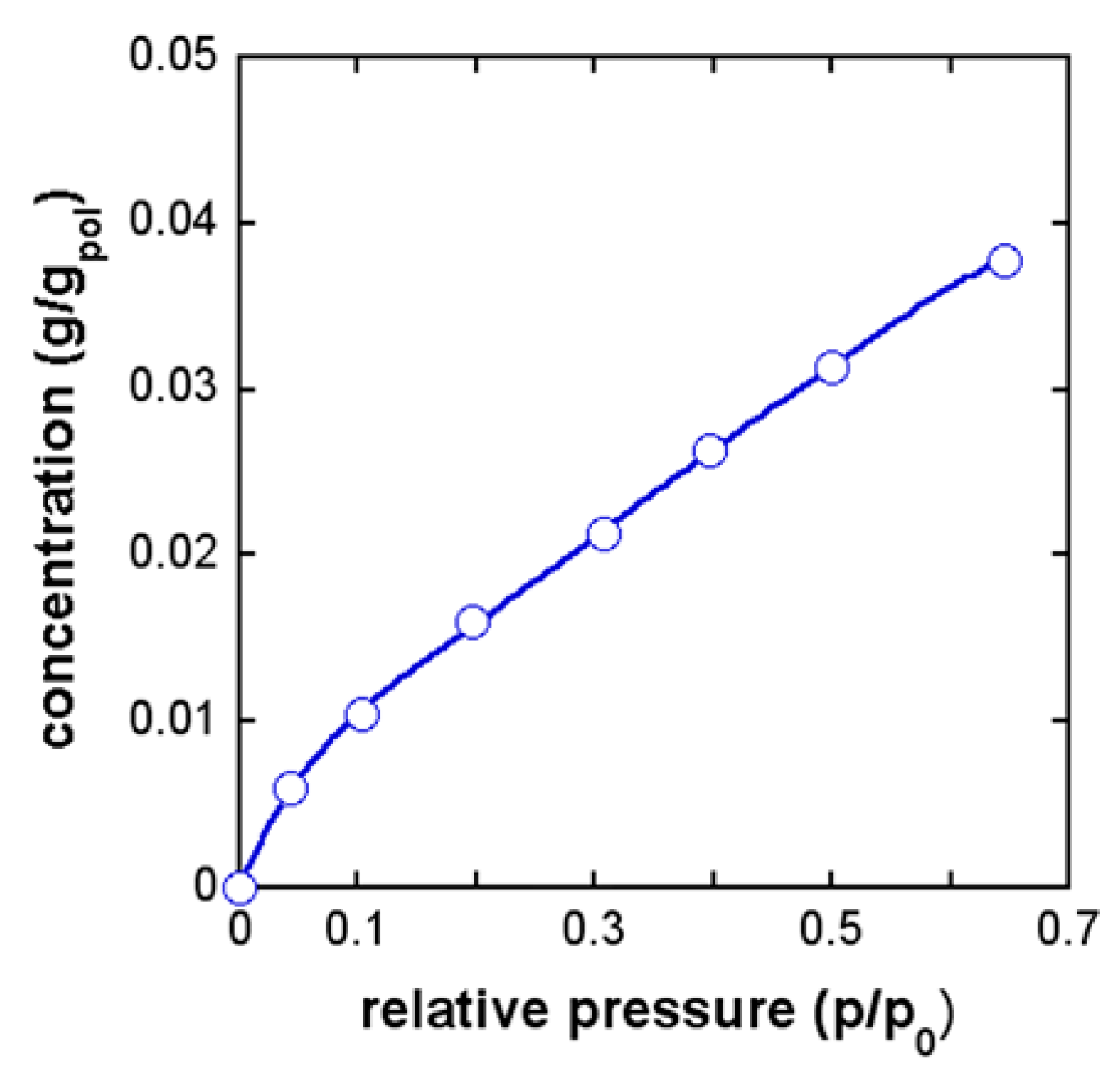
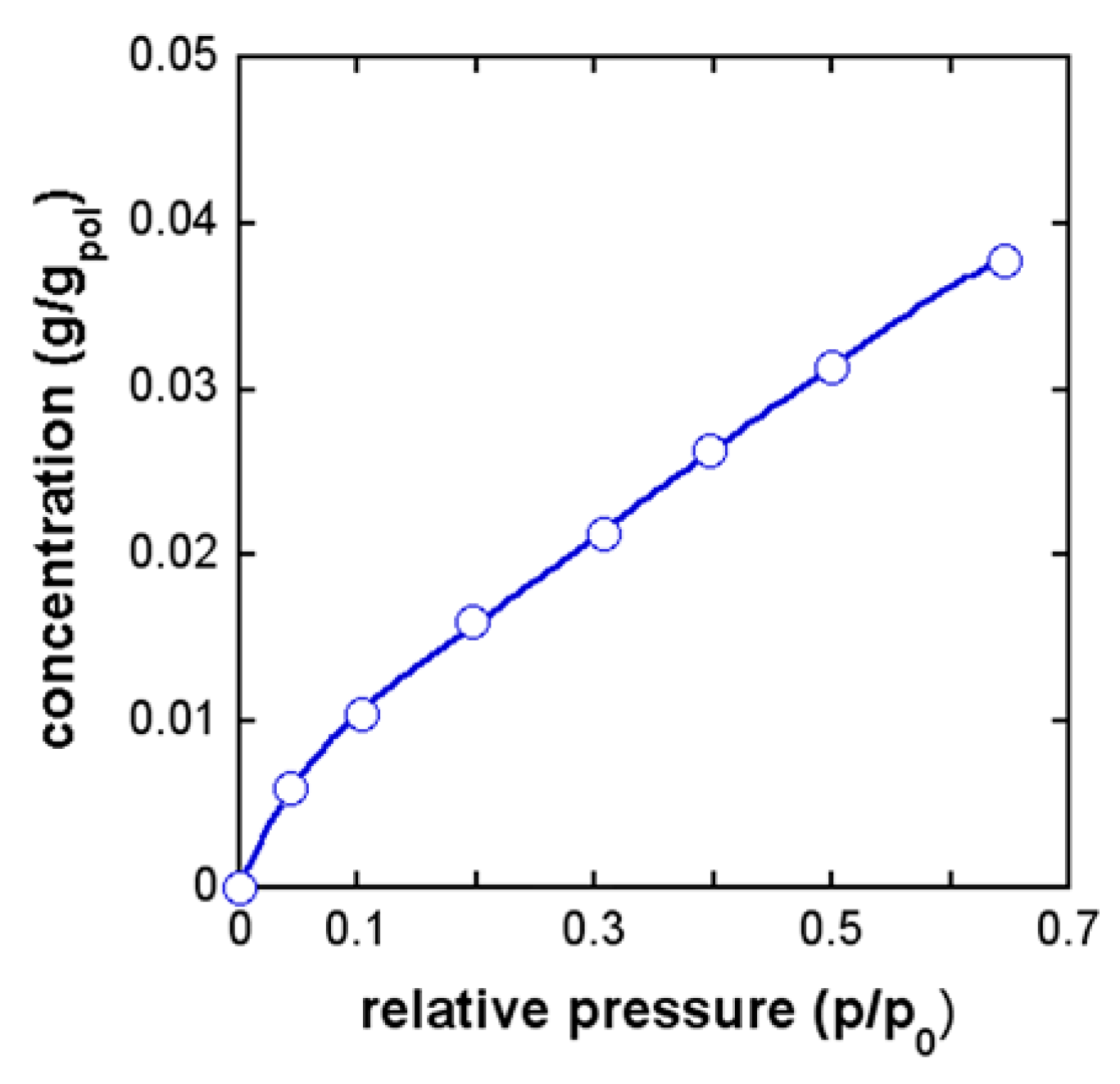
4.3. PEI–CH3OH System
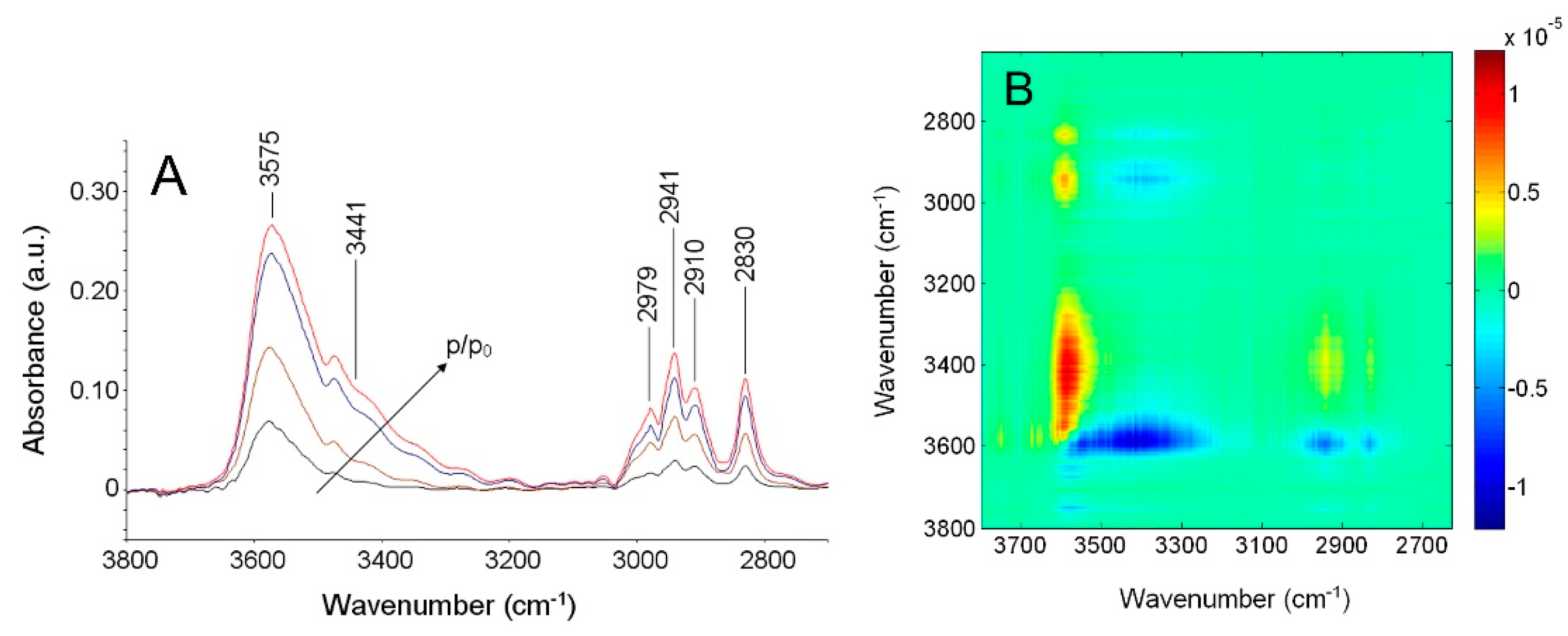
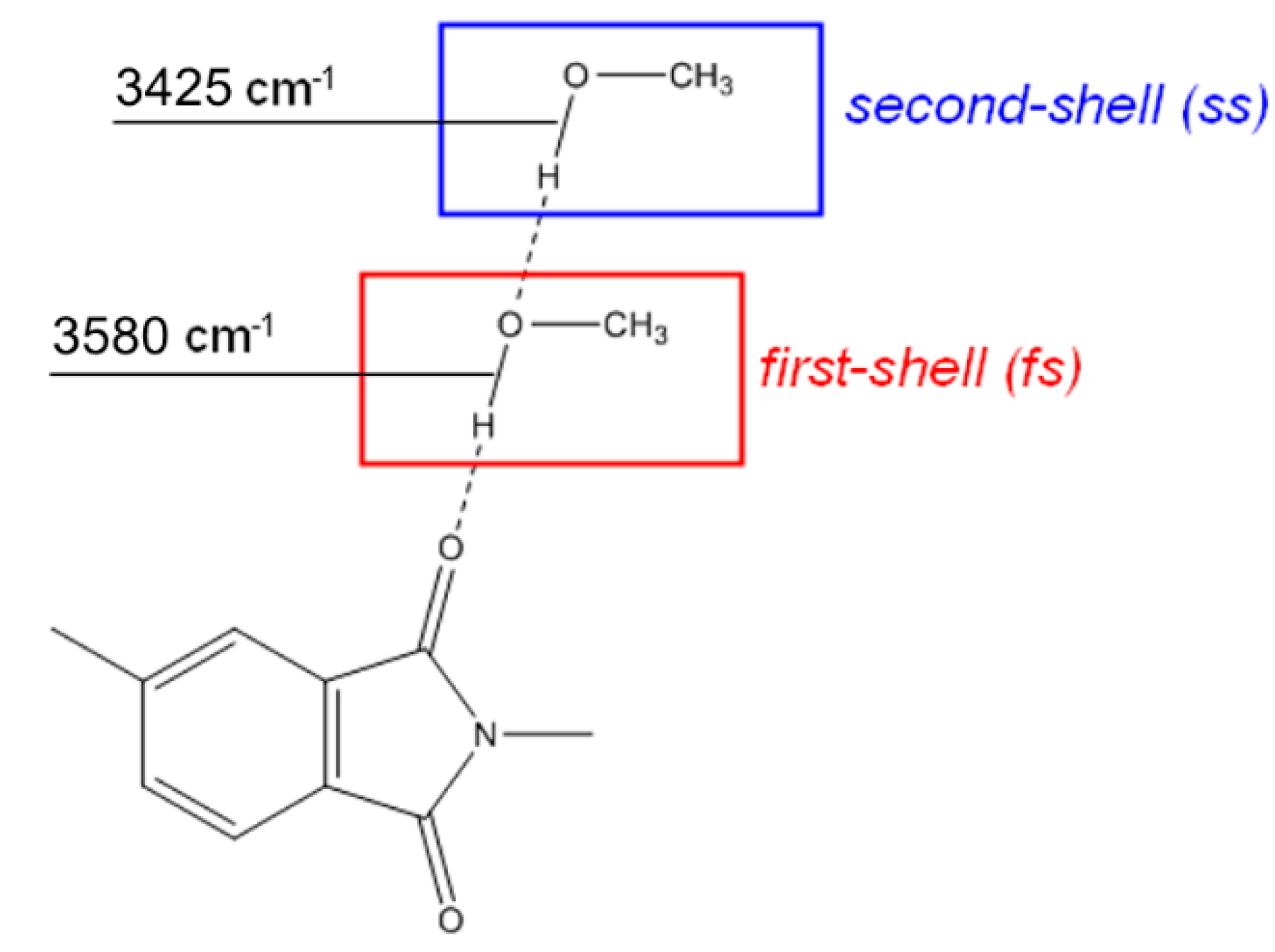
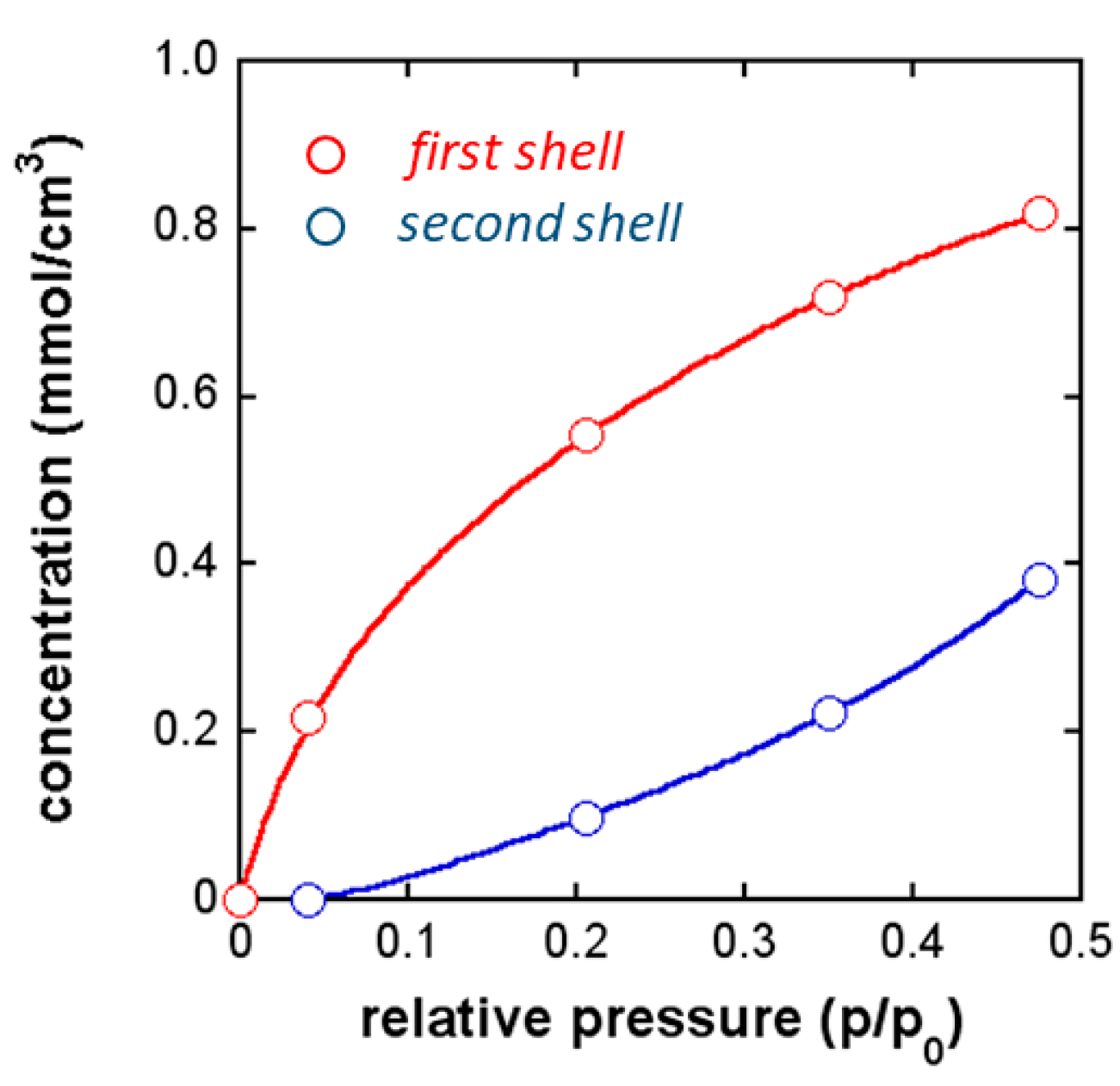
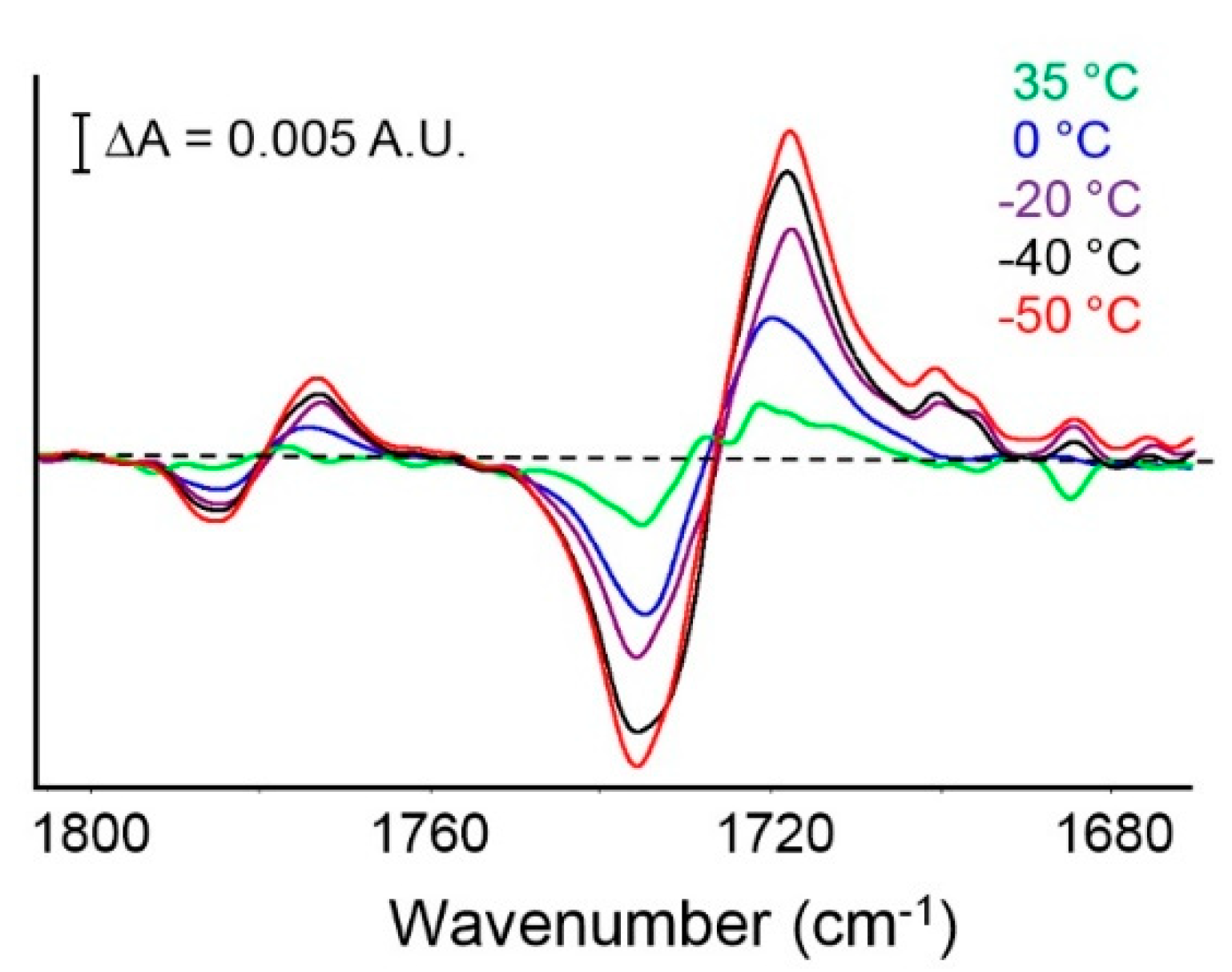
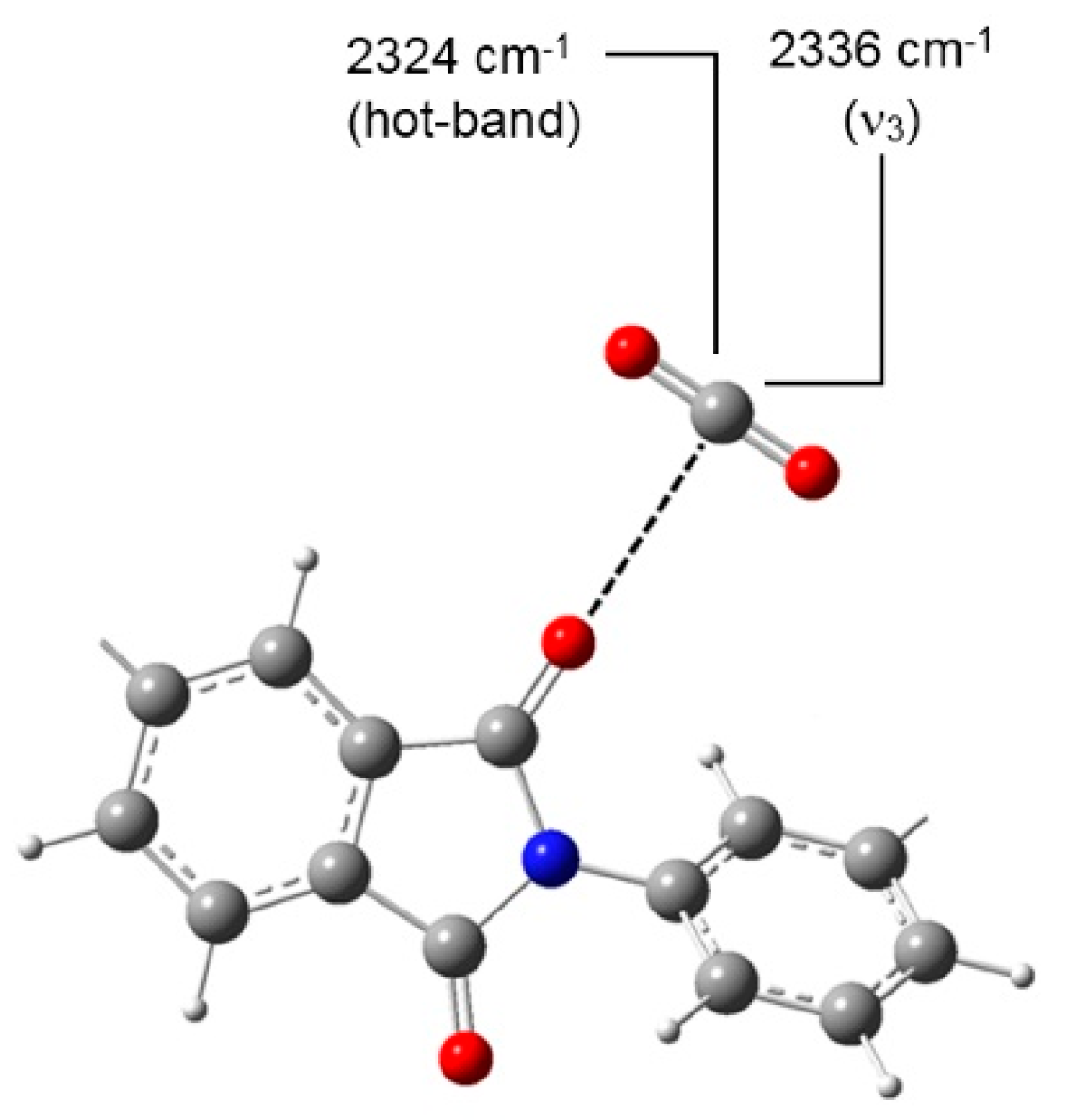
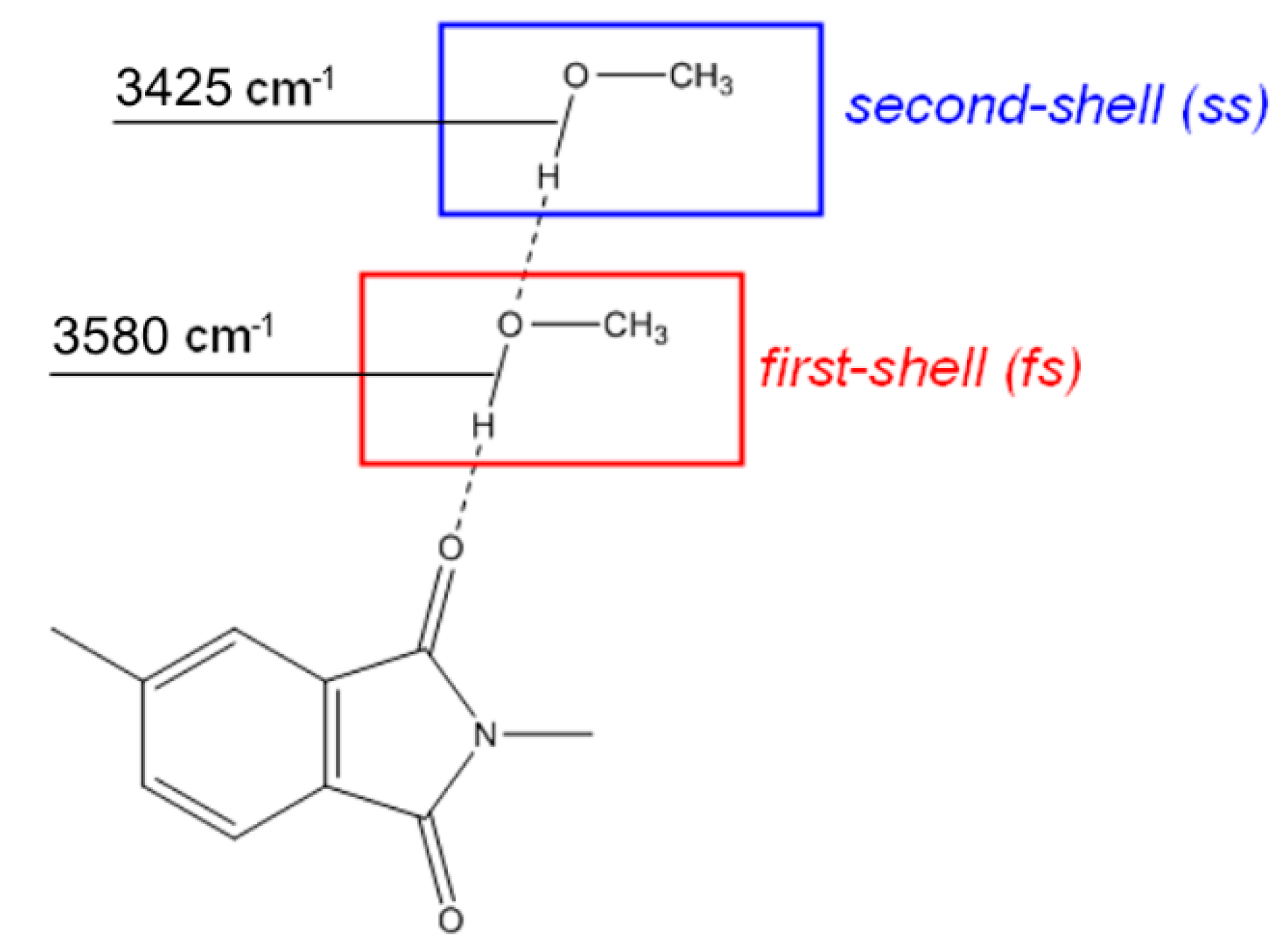
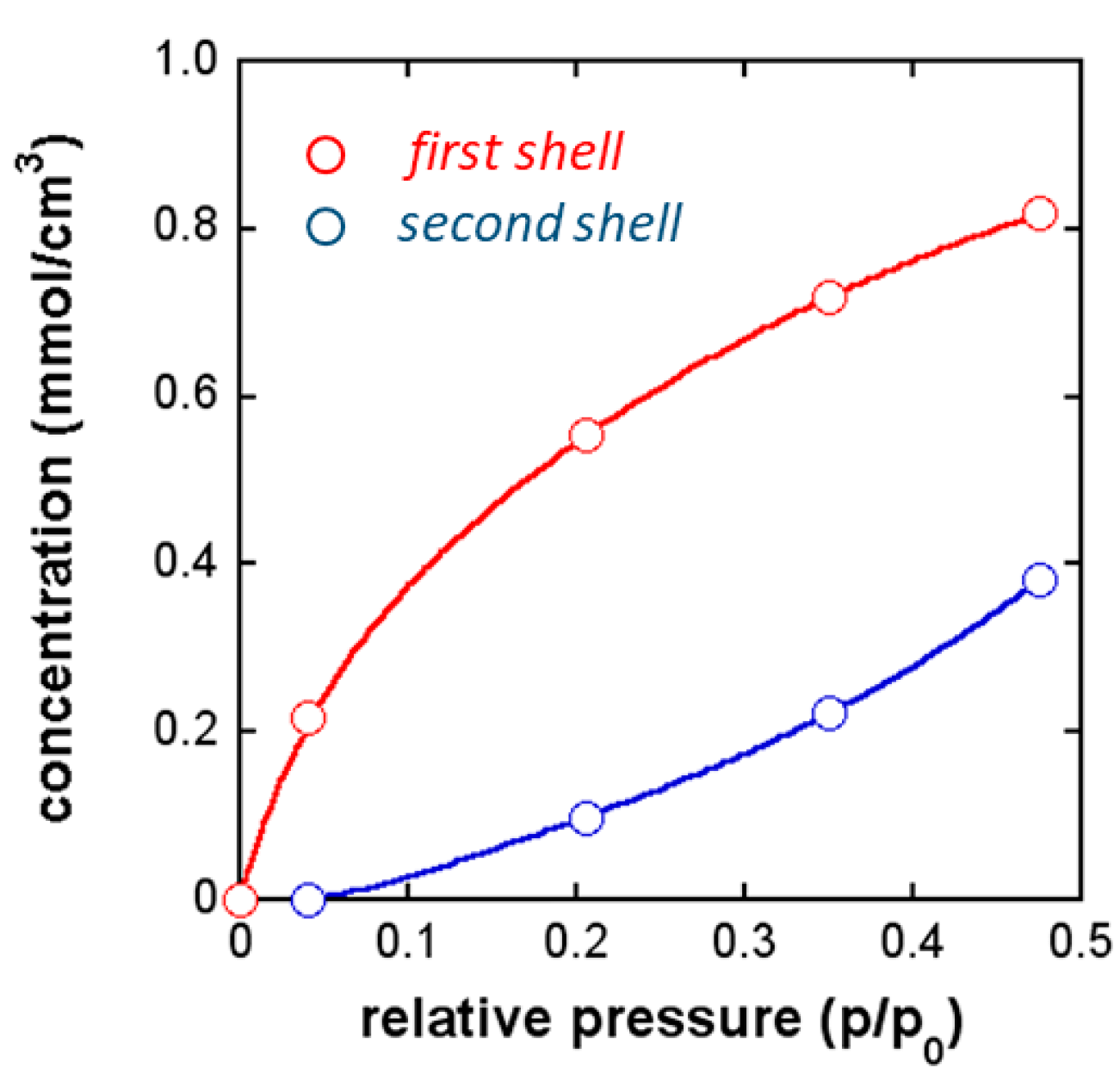
FTIR Analysis: Absorbance, Difference, and Two-Dimensional Correlation Spectra
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
A.1. The Equilibrium NRHB Model
A.2. Extension of Equilibrium Theories to Non-Equilibrium Glassy Polymers
References
- Kontogeorgis, G.M.; Folas, G.K. Thermodynamic Models for Industrial Applications: From Classical and Advanced Mixing Rules to Association Theories, 1st ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2010; ISBN 978-0-470-69726-9. [Google Scholar]
- Panayiotou, C.G.; Birdi, K.S. Handbook of Surface and Colloid Chemistry, 3rd ed.; CRC Press Taylor and Francis Group: New York, NY, USA, 2009. [Google Scholar]
- Iwamoto, R.; Kusanagi, H. Hydration Structure and Mobility of the Water Contained in Poly(ethylene terephthalate). J. Phys. Chem. A 2015, 119, 2885–2894. [Google Scholar] [CrossRef] [PubMed]
- Sammon, C.; Mura, C.; Yarwood, J.; Everall, N.; Swart, R.; Hodge, D. FTIR−ATR Studies of the Structure and Dynamics of Water Molecules in Polymeric Matrixes. A Comparison of PET and PVC. J. Phys. Chem. B 1998, 102, 3402–3411. [Google Scholar] [CrossRef]
- Davis, E.M.; Elabd, Y.A. Water Clustering in Glassy Polymers. J. Phys. Chem. B 2013, 117, 10629–10640. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Langkilde, F.W.; Zografi, G. Fourier transform Raman spectroscopic study of the interaction of water vapor with amorphous polymers. J. Pharm. Sci. 2001, 90, 888–901. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.G.; Sarti, G.C. Solubility of Gases and Liquids in Glassy Polymers. Annu. Rev. Chem. Biomol. 2011, 2, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Belova, N.A.; Safronov, A.P.; Yampolskii, Y.P. Inverse Gas Chromatography and the Thermodynamics of Sorption in Polymers. Polym. Sci. Ser. A 2012, 54, 859–873. [Google Scholar] [CrossRef]
- Galizia, M.; Chi, W.S.; Smith, Z.P.; Merkel, T.C.; Baker, R.W.; Freeman, B.D. Polymers and Mixed Matrix Membranes for Gas and Vapor Separation: A Review and Prospective Opportunities. Macromolecules 2017, 50, 7809–7843. [Google Scholar] [CrossRef]
- Glass, J.E. Adsorption characteristics of water-soluble polymers. I. Poly(vinyl alcohol) and poly(vinylpyrrolidone) at the aqueous-air interface. J. Phys. Chem. 1968, 72, 4450–4458. [Google Scholar] [CrossRef]
- Zhao, Z.-J.; Wang, Q.; Zhang, L.; Wu, T. Structured Water and Water—Polymer Interactions in Hydrogels of Molecularly Imprinted Polymers. J. Phys. Chem. B 2008, 112, 7515–7521. [Google Scholar] [CrossRef]
- Chiessi, E.; Cavalieri, F.; Paradossi, G. Water and Polymer Dynamics in Chemically Cross-Linked Hydrogels of Poly(vinyl alcohol): A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2007, 111, 2820–2827. [Google Scholar] [CrossRef]
- Eslami, H.; Müller-Plathe, F. Molecular Dynamics Simulation of Water Influence on Local Structure of Nanoconfined Polyamide-6,6. J. Phys. Chem. B 2011, 115, 9720–9731. [Google Scholar] [CrossRef] [PubMed]
- Mensitieri, G.; Iannone, M. Modelling accelerated ageing in polymer composites. In Ageing of Composites, 1st ed.; Martin, R., Ed.; Woodhead Publishing, Ltd.: Cambridge, UK; CRC Press: Boca Raton, FL, USA, 2008; pp. 224–281. ISBN 9781845693527. [Google Scholar]
- Li, D.; Yao, J.; Wang, H. Thin Films and Membranes with Hierarchical Porosity. In Encyclopedia of Membrane Science and Technology, 1st ed.; Hoek, E.M.V., Tarabara, V.V., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; ISBN 978-0-470-90687-3. [Google Scholar]
- Bernstein, R.; Kaufman, Y.; Freger, V. Membrane Characterization. In Encyclopedia of Membrane Science and Technology, 1st ed.; Hoek, E.M.V., Tarabara, V.V., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; ISBN 978-0-470-90687-3. [Google Scholar]
- Brunetti, A.; Barbieri, G.; Drioli, E. Gas Separation, Applications. In Encyclopedia of Membrane Science and Technology, 1st ed.; Hoek, E.M.V., Tarabara, V.V., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2013; ISBN 978-0-470-90687-3. [Google Scholar]
- Koros, W.J.; Burgess, S.K.; Chen, Z. Polymer Transport Properties. In Encyclopedia of Polymer Science and Technology, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015; ISBN 9781118633892. [Google Scholar]
- Koros, W.J.; Fleming, G.K.; Jordan, S.M.; Kim, T.H.; Hoehn, H.H. Polymeric membrane materials for solution-diffusion based permeation separations. Prog. Polym. Sci. 1988, 13, 339–401. [Google Scholar] [CrossRef]
- Scherillo, G.; Petretta, M.; Galizia, M.; La Manna, P.; Musto, P.; Mensitieri, G. Thermodynamics of water sorption in high performance glassy thermoplastic polymers. Front. Chem. 2014, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Scherillo, G.; Galizia, M.; Musto, P.; Mensitieri, G. Water Sorption Thermodynamics in Glassy and Rubbery Polymers: Modeling the Interactional Issues Emerging from FTIR Spectroscopy. Ind. Eng. Chem. Res. 2013, 52, 8674–8691. [Google Scholar] [CrossRef]
- Scherillo, G.; Sanguigno, L.; Galizia, M.; Lavorgna, M.; Musto, P.; Mensitieri, G. Non-equilibrium compressible lattice theories accounting for hydrogen bonding interactions: Modelling water sorption thermodynamics in fluorinated polyimides. Fluid Phase Equilibr. 2012, 334, 166–188. [Google Scholar] [CrossRef]
- Musto, P. Two-Dimensional FTIR Spectroscopy Studies on the Thermal-Oxidative Degradation of Epoxy and Epoxy−Bis(maleimide) Networks. Macromolecules 2003, 36, 3210–3221. [Google Scholar] [CrossRef]
- Panayiotou, C.; Pantoula, M.; Stefanis, E.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures. 1. Pure Fluids. Ind. Eng. Chem. Res. 2004, 43, 6592–6606. [Google Scholar] [CrossRef]
- Panayiotou, C.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures. 2. Multicomponent Mixtures. Ind. Eng. Chem. Res. 2007, 46, 2628–2636. [Google Scholar] [CrossRef]
- Doghieri, F.; Sarti, G.C. Nonequilibrium Lattice Fluids: A Predictive Model for the Solubility in Glassy Polymers. Macromolecules 1996, 29, 7885–7896. [Google Scholar] [CrossRef]
- Sarti, G.C.; Doghieri, F. Predictions of the solubility of gases in glassy polymers based on the NELF model. Chem. Eng. Sci. 1998, 53, 3435–3447. [Google Scholar] [CrossRef]
- Robeson, L.M. Polymer membranes for gas separation. Curr. Opin. Solid. State Mater. Sci. 1999, 4, 549–555. [Google Scholar] [CrossRef]
- Seo, J.; Jang, W.; Han, H. Thermal, optical, and water sorption properties in composite films of poly(ether imide) and bismaleimides: Effect of chemical structure. J. Appl. Polym. Sci. 2009, 113, 777–783. [Google Scholar] [CrossRef]
- Karimi, M.; Albrecht, W.; Heuchel, M.; Kish, M.H.; Frahn, J.; Weigel, T.; Hofmann, D.; Modarress, H.; Lendlein, A. Determination of water/polymer interaction parameter for membrane-forming systems by sorption measurement and a fitting technique. J. Membr. Sci. 2005, 265, 1–12. [Google Scholar] [CrossRef]
- Merdas, I.; Thominette, F.; Verdu, J. Humid ageing of polyetherimide. Chemical and physical interactions with water. In Polyimides and Other High Temperature Polymer: Synthesis, Characterization and Applications; Mittal, K.L., Ed.; VSP: Utrecht, The Netherlands, 2003; Volume 2, pp. 255–266. ISBN 90-6764-378-5. [Google Scholar]
- Okamoto, K.-I.; Tanihara, N.; Watanabe, H.; Tanaka, K.; Kita, H.; Nakamura, A.; Kusuki, Y.; Nakagawa, K. Sorption and diffusion of water vapor in polyimide films. J. Polym. Sci. Pol. Phys. 1992, 30, 1223–1231. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, L.; Matsuura, T.; Chung, T.S.; Goh, S.H. Investigation of the fundamental differences between polyamide-imide (PAI) and polyetherimide (PEI) membranes for isopropanol dehydration via pervaporation. J. Membr. Sci. 2008, 318, 217–226. [Google Scholar] [CrossRef]
- Wang, Y.; Chung, T.S.; Neo, B.W.; Gruender, M. Processing and engineering of pervaporation dehydration of ethylene glycol via dual-layer polybenzimidazole (PBI)/polyetherimide (PEI) membranes. J. Membr. Sci. 2011, 378, 339–350. [Google Scholar] [CrossRef]
- Zhao, W.; Shi, B. Removal of Volatile Organic Compounds from Water by Pervaporation Using Polyetherimide-Polyethersulfone Blend Hollow Fiber Membranes. Sep. Sci. Technol. 2009, 44, 1737–1752. [Google Scholar] [CrossRef]
- Qiu, W.; Kosuri, M.; Zhou, F.; Koros, W.J. Dehydration of ethanol–water mixtures using asymmetric hollow fiber membranes from commercial polyimides. J. Membr. Sci. 2009, 327, 96–103. [Google Scholar] [CrossRef]
- Huang, R.Y.M. (Ed.) Pervaporation Membrane Separation Processes; Membrane Science and Technology Series; Elsevier Science Publishers: Amsterdam, The Netherlands, 1991; Volume 1, ISBN O-444-88227-8. [Google Scholar]
- Semenova, S.I. Separation properties of polyimides. In Polyimide Membranes-Applications, Fabrications, and Properties; Ohya, H., Kudryavsev, V.V., Semenova, S.I., Eds.; Kodansha: Tokyo, Japan, 1996; pp. 103–178. ISBN 4-06-208189-X. [Google Scholar]
- Feng, X.; Huang, R.Y.M. Pervaporation and performance of asymmetric polyetherimide membranes for isopropanol dehydration by pervaporation. J. Membr. Sci. 1996, 109, 165–172. [Google Scholar] [CrossRef]
- Yanagishita, H.; Maejima, C.; Kitamoto, D.; Nakane, T. Preparation of asymmetric polyimide membrane for water/ethanol separation in pervaporation by the phase inversion process. J. Membr. Sci. 1994, 86, 231–240. [Google Scholar] [CrossRef]
- Huang, R.Y.M.; Feng, X. Dehydration of isopropanol by pervaporation using aromatic polyetherimide membranes. Sep. Sci. Technol. 1993, 28, 2035–2048. [Google Scholar] [CrossRef]
- Yanagishita, H.; Kitamoto, D.; Nakane, T. Separation of alcohol aqueous solution by pervaporation using asymmetric polyimide membrane. High Perform. Polym. 1995, 7, 275–281. [Google Scholar] [CrossRef]
- Okamoto, K.; Tanihara, N.; Watanabe, H.; Tanaka, K.; Kita, H.; Nakamura, A.; Kusuki, Y.; Nakagawa, K. Vapor permeation and pervaporation separation of water-ethanol through polyimide membranes. J. Membr. Sci. 1992, 68, 53–63. [Google Scholar] [CrossRef]
- Chen, W.J.; Martin, C.R. Highly methanol selective membranes for the pervaporation separation of methyl-t-butyl ether/methanol mixtures. J. Membr. Sci. 1995, 104, 101–108. [Google Scholar] [CrossRef]
- Nakagawa, K.; Asakura, Y.; Nakanishi, S.; Hoshino, H.; Kouda, H.; Kusuki, Y. Separation of vapor mixtures of water and alcohol by aromatic polyimide hollow fibers. Kobunshi Ronbunshu 1989, 46, 405–411. [Google Scholar] [CrossRef]
- De Nicola, A.; Correa, A.; Milano, G.; La Manna, P.; Musto, P.; Mensitieri, G.; Scherillo, G. Local Structure and Dynamics of Water Absorbed in Polyetherimide: A Hydrogen Bonding Anatomy. J. Phys. Chem. B 2017, 121, 3162–3176. [Google Scholar] [CrossRef] [PubMed]
- Musto, P.; Galizia, M.; La Manna, P.; Pannico, M.; Mensitieri, G. Diffusion and Molecular Interactions in a Methanol/Polyimide System Probed by Coupling time-resolved FTIR Spectroscopy with Gravimetric Measurements. Front. Chem. 2014, 2, 1–9. [Google Scholar] [CrossRef]
- Galizia, M.; La Manna, P.; Pannico, M.; Mensitieri, G.; Musto, P. Methanol diffusion in polyimides: A molecular description. Polymer 2014, 55, 1028–1039. [Google Scholar] [CrossRef]
- Cotugno, S.; Larobina, D.; Mensitieri, G.; Musto, P.; Ragosta, G. A novel spectroscopic approach to investigate transport processes in polymers: The case of water–epoxy system. Polymer 2001, 42, 6431–6438. [Google Scholar] [CrossRef]
- Mensitieri, G.; Cotugno, S.; Musto, P.; Ragosta, G.; Nicolais, L. Transport of water in high Tg Polymers: A comparison between interacting and non-interacting systems. In Polyimides and Other High Temperature Polymers: Synthesis, Characterization and Applications; Mittal, K.L., Ed.; VSP: Utrecht, The Netherlands, 2003; Volume 2, pp. 267–285. ISBN 90-6764-378-5. [Google Scholar]
- Sanchez, I.C.; Lacombe, R.H. An Elementary Molecular Theory of Classical Fluids. Pure Fluids. J. Phys. Chem. 1976, 80, 2352–2362. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. Statistical Thermodynamics of Fluid Mixtures. J. Phys. Chem. 1976, 80, 2568–2580. [Google Scholar]
- Sanchez, I.C.; Lacombe, R.H. Statistical Thermodynamics of Polymer Solutions. Macromolecules 1978, 11, 1145–1156. [Google Scholar] [CrossRef]
- Panayiotou, C.; Sanchez, I.C. Hydrogen Bonding in Fluids—An Equation-of-State Approach. J. Phys. Chem. 1991, 95, 10090–10097. [Google Scholar] [CrossRef]
- Veytsman, B.A. Are Lattice Models Valid for Fluids with Hydrogen Bonds? J. Phys. Chem. 1990, 94, 8499–8500. [Google Scholar] [CrossRef]
- Veytsman, B.A. Equation of State for Hydrogen-Bonded Systems. J. Phys. Chem. 1998, B 102, 7515–7517. [Google Scholar] [CrossRef]
- Prausnitz, J.M.; Lichtenthaler, R.N.; Gomes de Azevedo, E. Molecular Thermodynamics of Fluid-Phase Equilibria, 3rd ed.; Prentice Hall PTR: Upper Saddle River, NJ, USA, 1998; ISBN 978-0139777455. [Google Scholar]
- Guggenheim, E.A. Mixtures: The Theory of the Equilibrium Properties of Some Simple Classes of Mixtures, Solutions and Alloys; Oxford University Press: Oxford, UK, 1952. [Google Scholar]
- Panayiotou, C.; Vera, J.H. Statistical Thermodynamics of r-Mer Fluids and Their Mixtures. Polym. J. 1982, 14, 681–694. [Google Scholar] [CrossRef]
- You, S.S.; Yoo, K.P.; Lee, C.S. An Approximate Nonrandom Lattice Theory of Fluids: General Derivation and Application to Pure Fluids. Fluid Phase Equilibr. 1994, 93, 193–213. [Google Scholar] [CrossRef]
- Taimoori, M.; Panayiotou, C. The non-random distribution of free volume in fluids: Non-polar systems. Fluid Phase Equilibr. 2001, 192, 155–169. [Google Scholar] [CrossRef]
- Yeom, M.S.; Yoo, K.P.; Park, B.H.; Lee, C.S. A Nonrandom Lattice Fluid Hydrogen Bonding Theory for Phase Equilibria of Associating Systems. Fluid Phase Equilibr. 1999, 158, 143–149. [Google Scholar] [CrossRef]
- Vlachou, T.; Prinos, I.; Vera, J.H.; Panayiotou, C. Nonrandom Distribution of Free Volume in Fluids and their Mixtures: Hydrogen-Bonded Systems. Ind. Eng. Chem. Res. 2002, 41, 1057–1063. [Google Scholar] [CrossRef]
- Panayiotou, C. The QCHB Model of Fluids and their Mixtures. J. Chem. Thermodyn. 2003, 35, 349–381. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Kontogeorgis, G.M. Modeling the Vapor–Liquid Equilibria of Polymer–Solvent Mixtures: Systems with Complex Hydrogen Bonding Behavior. Fluid Phase Equilib. 2009, 280, 100–109. [Google Scholar] [CrossRef]
- Doghieri, F.; Quinzi, M.; Rethwisch, D.G.; Sarti, G.C. Predicting Gas Solubility in Glassy Polymers through Nonequilibrium EOS. In Advanced Materials for Membrane Separations; Pinnau, I., Freeman, B.D., Eds.; ACS Symposium ser. N. 876; American Chemical Society (ACS): Washington, DC, USA, 2004; pp. 74–90. ISBN 0-8412-3833-2. [Google Scholar]
- De Angelis, M.G.; Doghieri, F.; Sarti, G.C.; Freeman, B.D. Modeling gas sorption in amorphous Teflon through the non equilibrium thermodynamics for glassy polymers (NETGP) approach. Desalination 2006, 193, 82–89. [Google Scholar] [CrossRef]
- Pantoula, M.; von Schnitzler, J.; Eggers, R.; Panayiotou, C. Sorption and swelling in glassy polymer/carbon dioxide systems Part II—Swelling. J. Supercrit. Fluid 2007, 39, 426–434. [Google Scholar] [CrossRef]
- Musto, P.; Ragosta, G.; Mensitieri, G.; Lavorgna, M. On the Molecular Mechanism of H2O Diffusion into Polyimides: A Vibrational Spectroscopy Investigation. Macromolecules 2007, 40, 9614–9627. [Google Scholar] [CrossRef]
- Czarnecki, M.A. Two-Dimensional Correlation Spectroscopy: Effect of Normalization of the Dynamic Spectra. Appl. Spectrosc. 1999, 53, 1392–1397. [Google Scholar] [CrossRef]
- Noda, I.; Ozaki, Y. Two-Dimensional Correlation Spectroscopy: Applications in Vibrational and Optical Spectroscopy; John Wiley & Sons: Chichester, UK, 2005; ISBN 978-0-471-62391-5. [Google Scholar]
- Musto, P.; La Manna, P.; Pannico, M.; Mensitieri, G.; Gargiulo, N.; Caputo, D. Molecular interactions of CO2 with the CuBTC metal organic framework: An FTIR study based on two-dimensional correlation spectroscopy. J. Mol. Struct. 2018, 1166, 326–333. [Google Scholar] [CrossRef]
- Rothshild, W.G. Mobility of Small Molecules in Viscous Media. Macromolecules 1968, 1, 43–47. [Google Scholar]
- Stolov, A.A.; Morozov, A.I.; Remizov, A.B. Infrared spectra and spinning diffusion of methyl bromide in solutions. Vib. Spectosc. 1997, 15, 211–218. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Grenner, A.; Economou, I.G.; Kontogeorgis, G.M. Evaluation of the Nonrandom Hydrogen Bonding (NRHB) Theory and the Simplified Perturbed-Chain Statistical Associating Fluid Theory (sPC-SAFT). 2. Liquid-Liquid Equilibria and Prediction of Monomer Fraction in Hydrogen Bonding Systems. Ind. Eng. Chem. Res. 2008, 47, 5651–5659. [Google Scholar] [CrossRef]
- Fredenslund, A.; Jones, R.L.; Prausnitz, J.M. Group-contribution estimation of activity coefficients in nonideal liquid mixtures. AIChE J. 1975, 21, 1086–1099. [Google Scholar] [CrossRef]
- Sorensen, M.J. Group Contribution Estimation Methods. In Models for Thermodynamic and Phase Equilibria Calculations; Sandler, S.I., Ed.; Marcel Dekker: New York, NY, USA, 1994; ISBN 978-0824791308. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | Density (g/cm3) |
|---|---|
| 18 | 1.2700 ± 0.0006 |
| 27 | 1.2680 ± 0.0006 |
| 30 | 1.2673 ± 0.0006 |
| 35 | 1.2663 ± 0.0006 |
| 45 | 1.2641 ± 0.0006 |
| 60 | 1.2610 ± 0.0006 |
| 70 | 1.2589 ± 0.0006 |
| Substance | T * (K) | P * (MPa) | ρ * (g/cm3) |
|---|---|---|---|
| PEI | 893 | 545 | 1.354 |
| CO2 | 300 | 630 | 1.515 |
| Substance | s | ||||||
|---|---|---|---|---|---|---|---|
| PEI | 6775 ± 250 | 5.503 ± 0.15 | 0.7228 ± 0.08 | - | - | 0.743 a | - |
| H2O b | 5336.5 | −6.506 | 0.9703 | −16,100 | −14.7 | 0.861 | 0 |
| k12 | |||
|---|---|---|---|
| 0.121 ± 0.005 | −13,264 ± 200 | −6.107 ± 0.100 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mensitieri, G.; Scherillo, G.; La Manna, P.; Musto, P. Sorption Thermodynamics of CO2, H2O, and CH3OH in a Glassy Polyetherimide: A Molecular Perspective. Membranes 2019, 9, 23. https://doi.org/10.3390/membranes9020023
Mensitieri G, Scherillo G, La Manna P, Musto P. Sorption Thermodynamics of CO2, H2O, and CH3OH in a Glassy Polyetherimide: A Molecular Perspective. Membranes. 2019; 9(2):23. https://doi.org/10.3390/membranes9020023
Chicago/Turabian StyleMensitieri, Giuseppe, Giuseppe Scherillo, Pietro La Manna, and Pellegrino Musto. 2019. "Sorption Thermodynamics of CO2, H2O, and CH3OH in a Glassy Polyetherimide: A Molecular Perspective" Membranes 9, no. 2: 23. https://doi.org/10.3390/membranes9020023
APA StyleMensitieri, G., Scherillo, G., La Manna, P., & Musto, P. (2019). Sorption Thermodynamics of CO2, H2O, and CH3OH in a Glassy Polyetherimide: A Molecular Perspective. Membranes, 9(2), 23. https://doi.org/10.3390/membranes9020023