
The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy

 ,
,  ,
,  and
and 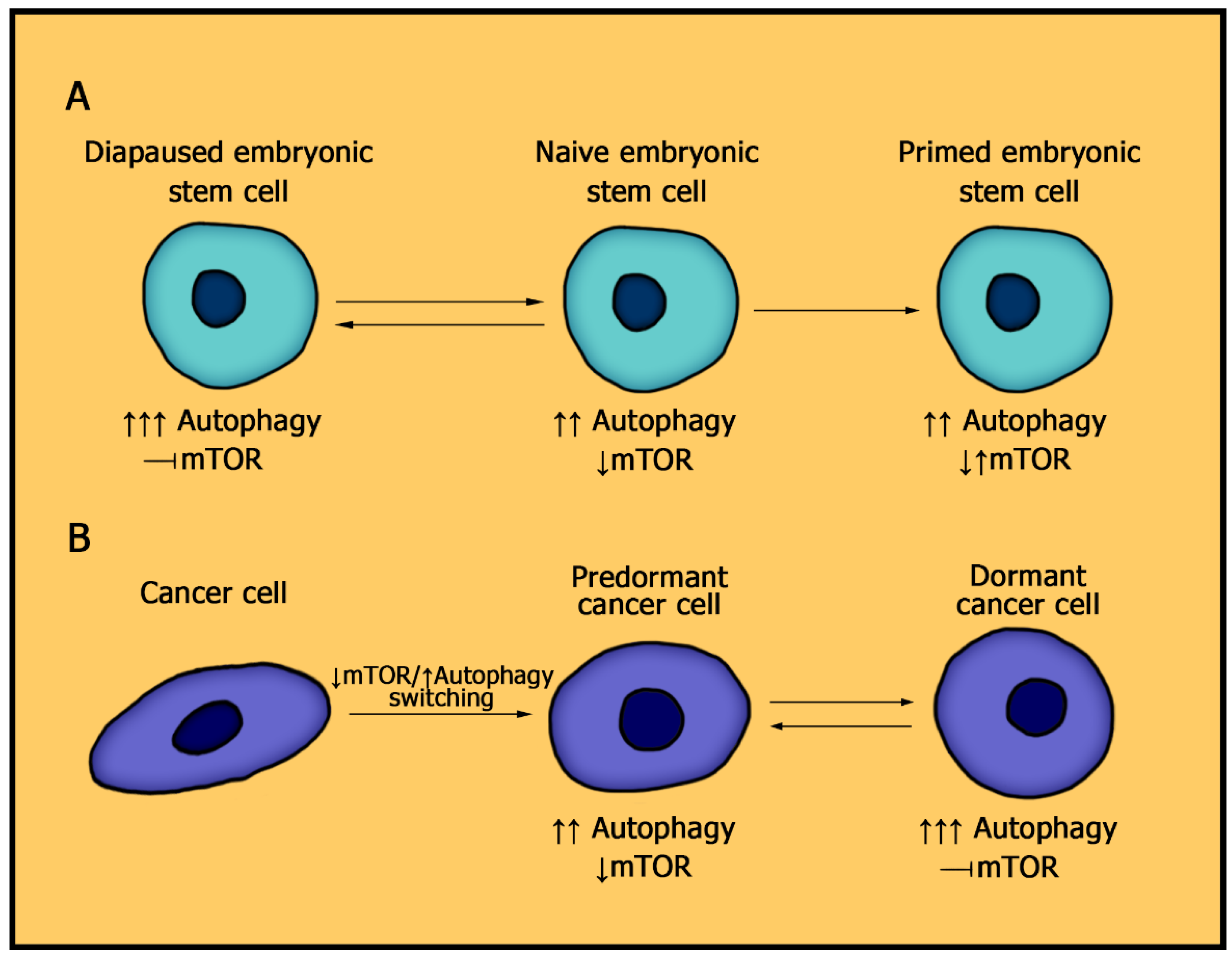{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The mTOR Pathway in ESCs
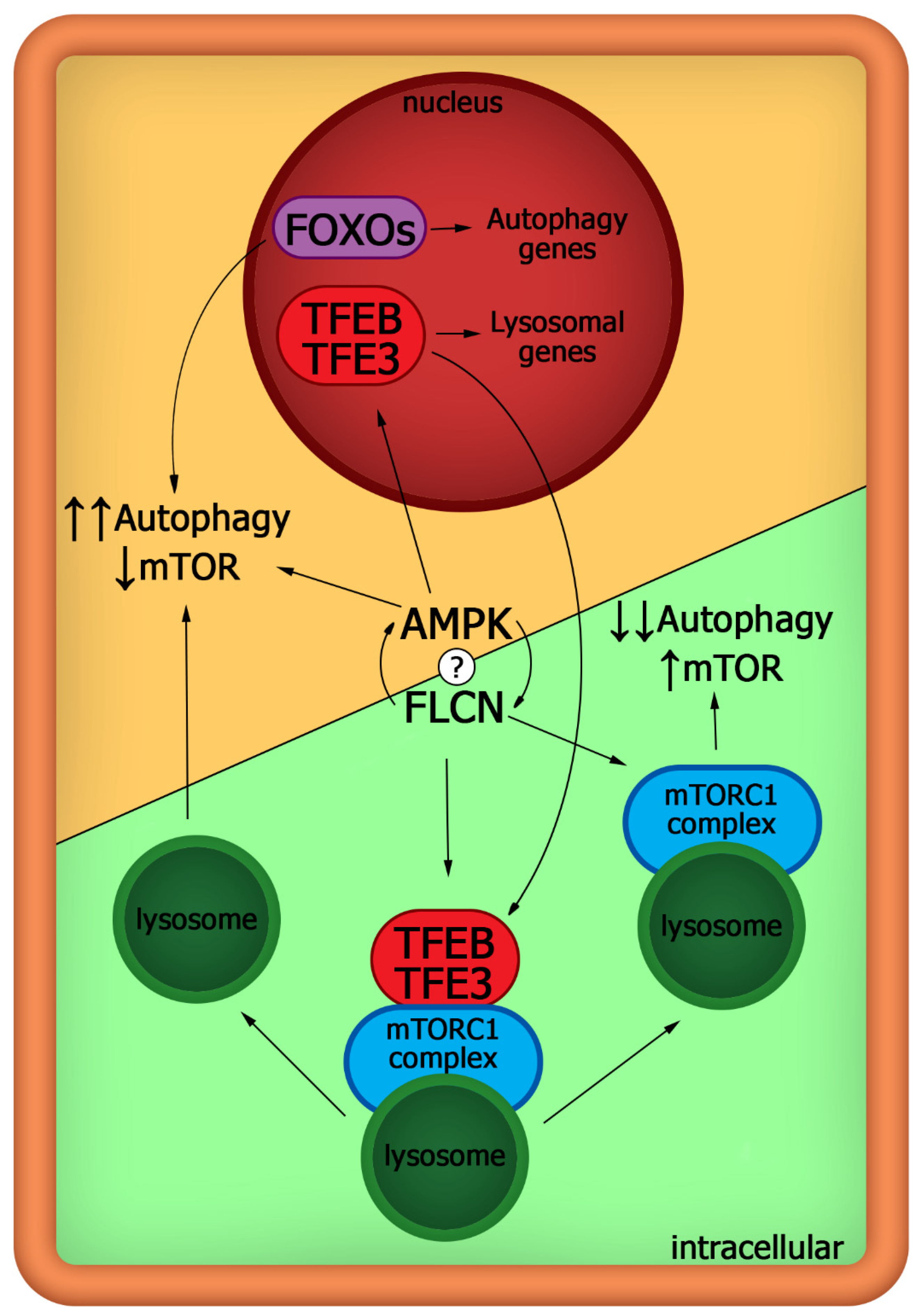
3. The mTOR Pathway versus Autophagy in Pluripotent Cells in Detail
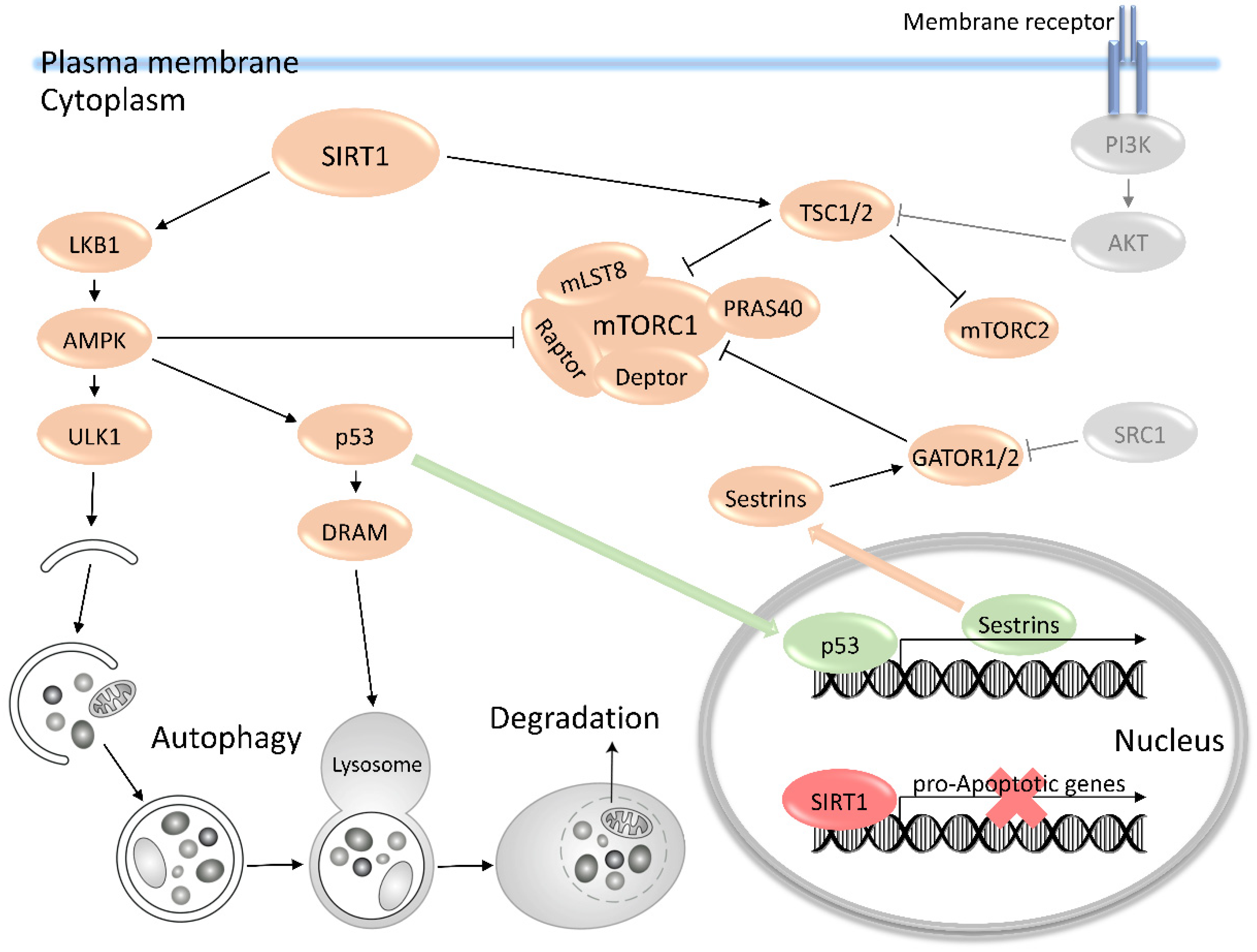
4. The Key Upstream Regulation of the mTOR Pathway and Autophagy in ESCs
5. The Role of mTOR Signaling Pathway and Autophagy in Dormant Cancer Cells
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kleffel, S.; Schatton, T. Tumor dormancy and cancer stem cells: Two sides of the same coin? Adv. Exp. Med. Biol. 2013, 734, 145–179. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Hunter, K.W. Tumor Cell Dormancy as an Adaptive Cell Stress Response Mechanism. F1000Research 2017, 6, 2134. [Google Scholar] [CrossRef] [PubMed]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; MacRae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]
- Sousa, M.I.; Correia, B.; Rodrigues, A.S.; Ramalho-Santos, J. Metabolic characterization of a paused-like pluripotent state. Biochim. Biophys. Acta Gen. Subj. 2020, 1864. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Gangloff, Y.J.; Mueller, M.; Dann, S.G.; Svoboda, P.; Sticker, M.; Spetz, J.F.; Um, S.H.; Brown, E.J.; Cereghini, S.; Thomas, G.; et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol. Cell. Biol. 2004, 24, 9508–9516. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef]
- Hentges, K.E.; Sirry, B.; Gingeras, A.C.; Sarbassov, D.; Sonenberg, N.; Sabatini, D.; Peterson, A.S. FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 13796–13801. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef]
- Shiota, C.; Woo, J.-T.; Lindner, J.; Shelton, K.D.; Magnuson, M.A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 2006, 11, 583–589. [Google Scholar] [CrossRef]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.-L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, J.; Tang, L.; Tan, C.; Zhao, Z.; Xiao, Y.; Ge, R.; Zhu, D. Involvement of Rictor/mTORC2 in cardiomyocyte differentiation of mouse embryonic stem cells in vitro. Int. J. Biol. Sci. 2017, 13, 110–121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, B.; Wang, J.; Tang, L.; Shi, J.; Zhu, D. mTORC1 and mTORC2 play different roles in regulating cardiomyocyte differentiation from embryonic stem cells. Int. J. Dev. Biol. 2017, 61, 65–72. [Google Scholar] [CrossRef]
- Zhou, J.; Su, P.; Wang, L.; Chen, J.; Zimmermann, M.; Genbacev, O.; Afonja, O.; Horne, M.C.; Tanaka, T.; Duan, E.; et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7840–7845. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-S.; Göke, J.; Ng, J.-H.; Lu, X.; Gonzales, K.A.U.; Tan, C.-P.; Tng, W.-Q.; Hong, Z.-Z.; Lim, Y.-S.; Ng, H.-H. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 2013, 13, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Kobayashi, S.; Hiratani, I. Epigenetic differences between naive and primed pluripotent stem cells. Cell. Mol. Life Sci. 2018, 75, 1191. [Google Scholar] [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Manning, B.D. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat. Metab. 2019, 1, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Kuma, A.; Murakami, M.; Kishi, C.; Yamamoto, A.; Mizushima, N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008, 321, 117–120. [Google Scholar] [CrossRef]
- Schier, A.F. The maternal-zygotic transition: Death and birth of RNAs. Science 2007, 316, 406–407. [Google Scholar] [CrossRef]
- Lee, J.-E.; Oh, H.-A.; Song, H.; Jun, J.H.; Roh, C.-R.; Xie, H.; Dey, S.K.; Lim, H.J. Autophagy Regulates Embryonic Survival During Delayed Implantation. Endocrinology 2011, 152, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; González-Polo, R.-A.; Casares, N.; Perfettini, J.-L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Easley, C.A., IV; Ben-Yehudah, A.; Redinger, C.J.; Oliver, S.L.; Varum, S.T.; Eisinger, V.M.; Carlisle, D.L.; Donovan, P.J.; Schatten, G.P. mTOR-Mediated Activation of p70 S6K Induces Differentiation of Pluripotent Human Embryonic Stem Cells. Cell. Reprogram. 2010, 12, 263. [Google Scholar] [CrossRef]
- Sampath, P.; Pritchard, D.K.; Pabon, L.; Reinecke, H.; Schwartz, S.M.; Morris, D.R.; Murry, C.E. A hierarchical network controls protein translation during murine embryonic stem cell self-renewal and differentiation. Cell Stem Cell 2008, 2, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Noormohammadi, A.; Calculli, G.; Gutierrez-Garcia, R.; Khodakarami, A.; Koyuncu, S.; Vilchez, D. Mechanisms of Protein Homeostasis (Proteostasis) Maintain Stem Cell Identity in Mammalian Pluripotent Stem Cells. Cell. Mol. Life Sci. 2018, 75, 275–290. [Google Scholar] [CrossRef]
- Gong, J.; Gu, H.; Zhao, L.; Wang, L.; Liu, P.; Wang, F.; Xu, H.; Zhao, T. Phosphorylation of ULK1 by AMPK is essential for mouse embryonic stem cell self-renewal and pluripotency. Cell Death Dis. 2018, 9, 38. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Suárez-Álvarez, B.; Dawud, R.A.; Vázquez-Chantada, M.; Martínez-Chantar, M.L.; Embade, N.; López-Nieva, P.; Horrillo, A.; Hmadcha, A.; et al. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13736–13741. [Google Scholar] [CrossRef]
- Ou, X.; Chae, H.-D.; Wang, R.-H.; Shelley, W.C.; Cooper, S.; Taylor, T.; Kim, Y.-J.; Deng, C.-X.; Yoder, M.C.; Broxmeyer, H.E. SIRT1 deficiency compromises mouse embryonic stem cell hematopoietic differentiation, and embryonic and adult hematopoiesis in the mouse. Blood 2011, 117, 440–450. [Google Scholar] [CrossRef]
- Yoon, D.S.; Choi, Y.; Jang, Y.; Lee, M.; Choi, W.J.; Kim, S.-H.; Lee, J.W. SIRT1 directly regulates SOX2 to maintain self-renewal and multipotency in bone marrow-derived mesenchymal stem cells. Stem Cells 2014, 32, 3219–3231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-N.; Chung, S.-K.; Xu, Z.; Xu, Y. Oct4 maintains the pluripotency of human embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells 2014, 32, 157–165. [Google Scholar] [CrossRef]
- Jang, J.; Huh, Y.J.; Cho, H.-J.; Lee, B.; Park, J.; Hwang, D.-Y.; Kim, D.-W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem Cell Rep. 2017, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kawano, H.; Kanai, F.; Nakamura, E.; Tada, N.; Takai, S.; Horie, S.; Arai, H.; Kobayashi, T.; Hino, O. Establishment of Tsc2-deficient rat embryonic stem cells. Int. J. Oncol. 2015, 46, 1944–1952. [Google Scholar] [CrossRef]
- Li, M.; Yu, J.S.L.; Tilgner, K.; Ong, S.H.; Koike-Yusa, H.; Yusa, K. Genome-wide CRISPR-KO Screen Uncovers mTORC1-Mediated Gsk3 Regulation in Naive Pluripotency Maintenance and Dissolution. Cell Rep. 2018, 24, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 Complex Is Required for Proper Activation of mTOR Complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Liu, P.; Liu, K.; Gu, H.; Wang, W.; Gong, J.; Zhu, Y.; Zhao, Q.; Cao, J.; Han, C.; Gao, F.; et al. High autophagic flux guards ESC identity through coordinating autophagy machinery gene program by FOXO1. Cell Death Differ. 2017, 24, 1672–1680. [Google Scholar] [CrossRef]
- Suvorova, I.I.; Knyazeva, A.R.; Petukhov, A.V.; Aksenov, N.D.; Pospelov, V.A. Resveratrol enhances pluripotency of mouse embryonic stem cells by activating AMPK/Ulk1 pathway. Cell Death Discov. 2019, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Suvorova, I.I.; Knyazeva, A.R.; Pospelov, V.A. Resveratrol-induced p53 activation is associated with autophagy in mouse embryonic stem cells. Biochem. Biophys. Res. Commun. 2018, 503, 2180–2185. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.-L.; Karin, M.; Budanov, A.V. Sestrins Inhibit mTORC1 Kinase Activation Through the GATOR Complex. Cell Rep. 2014, 9, 1281. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ro, S.-H.; Kim, M.; Park, H.-W.; Semple, I.A.; Park, H.; Cho, U.-S.; Wang, W.; Guan, K.-L.; Karin, M.; et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef]
- Lu, J.; Temp, U.; Müller-Hartmann, A.; Esser, J.; Grönke, S.; Partridge, L. Sestrin is a key regulator of stem cell function and lifespan in response to dietary amino acids. Nat. Aging 2020, 1, 60–72. [Google Scholar] [CrossRef]
- Pal, R.; Palmieri, M.; Chaudhury, A.; Klisch, T.J.; di Ronza, A.; Neilson, J.R.; Rodney, G.G.; Sardiello, M. Src regulates amino acid-mediated mTORC1 activation by disrupting GATOR1-Rag GTPase interaction. Nat. Commun. 2018, 9, 4351. [Google Scholar] [CrossRef]
- Meyn, M.A.; Schreiner, S.J.; Dumitrescu, T.P.; Nau, G.J.; Smithgall, T.E. SRC family kinase activity is required for murine embryonic stem cell growth and differentiation. Mol. Pharmacol. 2005, 68, 1320–1330. [Google Scholar] [CrossRef]
- Zhang, X.; Simerly, C.; Hartnett, C.; Schatten, G.; Smithgall, T.E. Src-family tyrosine kinase activities are essential for differentiation of human embryonic stem cells. Stem Cell Res. 2014, 13, 379–389. [Google Scholar] [CrossRef]
- Villegas, F.; Lehalle, D.; Mayer, D.; Rittirsch, M.; Stadler, M.B.; Zinner, M.; Olivieri, D.; Vabres, P.; Duplomb-Jego, L.; De Bont, E.S.J.M.; et al. Lysosomal Signaling Licenses Embryonic Stem Cell Differentiation via Inactivation of Tfe3. Cell Stem Cell 2019, 24, 257–270.e8. [Google Scholar] [CrossRef]
- Hasumi, Y.; Baba, M.; Ajima, R.; Hasumi, H.; Valera, V.A.; Klein, M.E.; Haines, D.C.; Merino, M.J.; Hong, S.-B.; Yamaguchi, T.P.; et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. USA 2009, 106, 18722–18727. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-B.; Oh, H.; Valera, V.A.; Stull, J.; Ngo, D.-T.; Baba, M.; Merino, M.J.; Linehan, W.M.; Schmidt, L.S. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-β signaling. Mol. Cancer 2010, 9, 160. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, D.; Rubera, I.; Futami, K.; Wang, P.; Zickert, P.; Khoo, S.-K.; Dykema, K.; Zhao, P.; Petillo, D.; et al. Disruption of tubular Flcn expression as a mouse model for renal tumor induction. Kidney Int. 2015, 88, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Si, S.; Li, Y.; Schoen, S.; Xiao, G.-Q.; Li, X.; Teh, B.T.; Wu, G.; Chen, J. Flcn-deficient renal cells are tumorigenic and sensitive to mTOR suppression. Oncotarget 2015, 6, 32761. [Google Scholar] [CrossRef]
- Betschinger, J.; Nichols, J.; Dietmann, S.; Corrin, P.D.; Paddison, P.J.; Smith, A. Exit from Pluripotency Is Gated by Intracellular Redistribution of the bHLH Transcription Factor Tfe3. Cell 2013, 153, 335. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Detraux, D.; Kuppers, D.; Wang, Y.; Cavanaugh, C.; Sidhu, S.; Levy, S.; Robitaille, A.M.; Ferreccio, A.; Bottorff, T.; et al. Folliculin regulates mTORC1/2 and WNT pathways in early human pluripotency. Nat. Commun. 2019, 10, 632. [Google Scholar] [CrossRef]
- Collodet, C.; Foretz, M.; Deak, M.; Bultot, L.; Metairon, S.; Viollet, B.; Lefebvre, G.; Raymond, F.; Parisi, A.; Civiletto, G.; et al. AMPK promotes induction of the tumor suppressor FLCN through activation of TFEB independently of mTOR. FASEB J. 2019, 33, 12374. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; C Zirden, L.; Puustinen, P.; Blanchette, P.; Jeong, H.; Dejgaard, K.; Siegel, P.M.; Pause, A. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 2021, 1–19. [Google Scholar] [CrossRef]
- Greer, E.L.; Banko, M.R.; Brunet, A. AMP-activated Protein Kinase and FoxO Transcription Factors in Dietary Restriction–induced Longevity. Ann. N. Y. Acad. Sci. 2009, 1170, 688. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liao, W.; Yang, J.; Ma, K.; Li, X.; Wang, Y.; Wang, D.; Wang, L.; Zhang, Y.; Yin, Y.; et al. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012, 8, 1712–1723. [Google Scholar] [CrossRef]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Toyoshima, F. Dormant Pluripotent Cells Emerge during Neural Differentiation of Embryonic Stem Cells in a FoxO3-Dependent Manner. Mol. Cell. Biol. 2017, 37, e00417-16. [Google Scholar] [CrossRef]
- Delpuech, O.; Griffiths, B.; East, P.; Essafi, A.; Lam, E.W.-F.; Burgering, B.; Downward, J.; Schulze, A. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol. Cell. Biol. 2007, 27, 4917–4930. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Cannell, I.G.; Brenkman, A.B.; Samans, B.; Gaestel, M.; Roepman, P.; Burgering, B.M.; Bushell, M.; Rosenwald, A.; Eilers, M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol. Cell 2011, 41, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, Á.M.; Baumgärtner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S.; et al. Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef]
- Shimamoto, S.; Nishimura, Y.; Nagamatsu, G.; Hamada, N.; Kita, H.; Hikabe, O.; Hamazaki, N.; Hayashi, K. Hypoxia induces the dormant state in oocytes through expression of Foxo3. Proc. Natl. Acad. Sci. USA 2019, 116, 12321–12326. [Google Scholar] [CrossRef] [PubMed]
- Touil, Y.; Segard, P.; Ostyn, P.; Begard, S.; Aspord, C.; El Machhour, R.; Masselot, B.; Vandomme, J.; Flamenco, P.; Idziorek, T.; et al. Melanoma dormancy in a mouse model is linked to GILZ/FOXO3A-dependent quiescence of disseminated stem-like cells. Sci. Rep. 2016, 6, 30405. [Google Scholar] [CrossRef] [PubMed]
- Possik, E.; Jalali, Z.; Nouët, Y.; Yan, M.; Gingras, M.-C.; Schmeisser, K.; Panaite, L.; Dupuy, F.; Kharitidi, D.; Chotard, L.; et al. Folliculin Regulates Ampk-Dependent Autophagy and Metabolic Stress Survival. PLoS Genet. 2014, 10, e1004273. [Google Scholar] [CrossRef]
- Ramirez Reyes, J.M.J.; Cuesta, R.; Pause, A. Folliculin: A Regulator of Transcription Through AMPK and MTOR Signaling Pathways. Front. Cell Dev. Biol. 2021, 9, 961. [Google Scholar] [CrossRef]
- Kabraji, S.; Solé, X.; Huang, Y.; Bango, C.; Bowden, M.; Bardia, A.; Sgroi, D.; Loda, M.; Ramaswamy, S. AKT1low Quiescent Cancer Cells Persist after Neoadjuvant Chemotherapy in Triple Negative Breast Cancer. Breast Cancer Res. 2017, 19, 88. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Adamski, V.; Hattermann, K.; Kubelt, C.; Cohrs, G.; Lucius, R.; Synowitz, M.; Sebens, S.; Held-Feindt, J. Entry and Exit of Chemotherapeutically-Promoted Cellular Dormancy in Glioblastoma Cells Is Differentially Affected by the Chemokines CXCL12, CXCL16, and CX3CL1. Oncogene 2020, 39, 4421–4435. [Google Scholar] [CrossRef]
- Lin, W.; Rajbhandari, N.; Liu, C.; Sakamoto, K.; Zhang, Q.; Triplett, A.A.; Batra, S.K.; Opavsky, R.; Felsher, D.W.; DiMaio, D.J.; et al. Dormant Cancer Cells Contribute to Residual Disease in a Model of Reversible Pancreatic Cancer. Cancer Res. 2013, 73, 1821–1830. [Google Scholar] [CrossRef]
- Zeuner, A.; Francescangeli, F.; Contavalli, P.; Zapparelli, G.; Apuzzo, T.; Eramo, A.; Baiocchi, M.; De Angelis, M.L.; Biffoni, M.; Sette, G.; et al. Elimination of Quiescent/Slow-Proliferating Cancer Stem Cells by Bcl-XL Inhibition in Non-Small Cell Lung Cancer. Cell Death Differ. 2014, 21, 1877–1888. [Google Scholar] [CrossRef]
- Lei, X.; He, Q.; Li, Z.; Zou, Q.; Xu, P.; Yu, H.; Ding, Y.; Zhu, W. Cancer Stem Cells in Colorectal Cancer and the Association with Chemotherapy Resistance. Med. Oncol. 2021, 38, 43. [Google Scholar] [CrossRef] [PubMed]
- Buczacki, S.J.A.; Popova, S.; Biggs, E.; Koukorava, C.; Buzzelli, J.; Vermeulen, L.; Hazelwood, L.; Francies, H.; Garnett, M.J.; Winton, D.J. Itraconazole Targets Cell Cycle Heterogeneity in Colorectal Cancer. J. Exp. Med. 2018, 215, 1891–1912. [Google Scholar] [CrossRef]
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.-L.; Bast, R.C., Jr. DIRAS3 Regulates the Autophagosome Initiation Complex in Dormant Ovarian Cancer Cells. Autophagy 2014, 10, 1071–1092. [Google Scholar] [CrossRef]
- Ebinger, S.; Özdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant Tumour Cells, Their Niches and the Influence of Immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Santos-de-Frutos, K.; Djouder, N. When Dormancy Fuels Tumour Relapse. Commun. Biol. 2021, 4, 747. [Google Scholar] [CrossRef]
- De Angelis, M.L.; Francescangeli, F.; La Torre, F.; Zeuner, A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/MTOR Signaling Pathway as a Therapeutic Target for Ovarian Cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Raphael, J.; Desautels, D.; Pritchard, K.I.; Petkova, E.; Shah, P.S. Phosphoinositide 3-Kinase Inhibitors in Advanced Breast Cancer: A Systematic Review and Meta-Analysis. Eur. J. Cancer 2018, 91, 38–46. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-MTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/MTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, A. PI3K/ Akt/ MTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/Akt/MTOR Pathway in Non-Small Cell Lung Cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, M.; Song, J.; Zheng, X.; Xu, G.; Bao, Y.; Lan, J.; Luo, D.; Hu, J.; Li, J.J.; et al. Integrin-Src-YAP1 Signaling Mediates the Melanoma Acquired Resistance to MAPK and PI3K/MTOR Dual Targeted Therapy. Mol. Biomed. 2020, 1, 12. [Google Scholar] [CrossRef]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 Regulates Prostate Cancer Dormancy through MTOR Inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of Disseminated Cancer Cell Dormancy: An Awakening Field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.J.M.; Peart, T.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. Modulation of AKT Activity Is Associated with Reversible Dormancy in Ascites-Derived Epithelial Ovarian Cancer Spheroids. Carcinogenesis 2012, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Carcereri de Prati, A.; Butturini, E.; Rigo, A.; Oppici, E.; Rossin, M.; Boriero, D.; Mariotto, S. Metastatic Breast Cancer Cells Enter Into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia. J. Cell. Biochem. 2017, 118, 3237–3248. [Google Scholar] [CrossRef]
- Gupta, A.; Roy, S.; Lazar, A.J.F.; Wang, W.-L.; McAuliffe, J.C.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy Inhibition and Antimalarials Promote Cell Death in Gastrointestinal Stromal Tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hoque, A.; Luo, R.Z.; Yuan, J.; Lu, Z.; Nishimoto, A.; Liu, J.; Sahin, A.A.; Lippman, S.M.; Bast, R.C.J.; et al. Loss of the Expression of the Tumor Suppressor Gene ARHI Is Associated with Progression of Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 3660–3666. [Google Scholar]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The Tumor Suppressor Gene ARHI Regulates Autophagy and Tumor Dormancy in Human Ovarian Cancer Cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Peart, T.; Ramos Valdes, Y.; Correa, R.J.M.; Fazio, E.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Intact LKB1 Activity Is Required for Survival of Dormant Ovarian Cancer Spheroids. Oncotarget 2015, 6, 22424–22438. [Google Scholar] [CrossRef] [PubMed]
- Trapp, E.K.; Majunke, L.; Zill, B.; Sommer, H.; Andergassen, U.; Koch, J.; Harbeck, N.; Mahner, S.; Friedl, T.W.P.; Janni, W.; et al. LKB1 Pro-Oncogenic Activity Triggers Cell Survival in Circulating Tumor Cells. Mol. Oncol. 2017, 11, 1508–1526. [Google Scholar] [CrossRef] [PubMed]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy Promotes the Survival of Dormant Breast Cancer Cells and Metastatic Tumour Recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef]
- Washington, M.N.; Suh, G.; Orozco, A.F.; Sutton, M.N.; Yang, H.; Wang, Y.; Mao, W.; Millward, S.; Ornelas, A.; Atkinson, N.; et al. ARHI (DIRAS3)-Mediated Autophagy-Associated Cell Death Enhances Chemosensitivity to Cisplatin in Ovarian Cancer Cell Lines and Xenografts. Cell Death Dis. 2015, 6, e1836. [Google Scholar] [CrossRef]
- Münst, B.; Thier, M.C.; Winnemöller, D.; Helfen, M.; Thummer, R.P.; Edenhofer, F. Nanog induces suppression of senescence through downregulation of p27KIP1 expression. J. Cell Sci. 2016, 129, 912–920. [Google Scholar] [CrossRef]
- Hepburn, A.C.; Steele, R.E.; Veeratterapillay, R.; Wilson, L.; Kounatidou, E.E.; Barnard, A.; Berry, P.; Cassidy, J.R.; Moad, M.; El-Sherif, A.; et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 2019, 38, 4412–4424. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Urbán, N.; Cheung, T.H. Stem cell quiescence: The challenging path to activation. Development 2021, 148, dev165084. [Google Scholar] [CrossRef]
- Lanzkron, S.M.; Collector, M.I.; Sharkis, S.J. Homing of long-term and short-term engrafting cells in vivo. Ann. N. Y. Acad. Sci. 1999, 872, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Fibbe, W.E.; Mark, J.; Zijlmans, J.M.; Willemze, R. Stem cells with short-term and long-term repopulating ability in the mouse. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1996, 7 (Suppl. 2), 15–18. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, M.; Tang, Y.; Liang, X. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. OncoTargets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, K.; Yao, G. Cell dormancy plasticity: Quiescence deepens into senescence through a dimmer switch. Physiol. Genom. 2020, 52, 558–562. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhasan, B.A.; Gordeev, S.A.; Knyazeva, A.R.; Aleksandrova, K.V.; Margulis, B.A.; Guzhova, I.V.; Suvorova, I.I. The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy. Membranes 2021, 11, 858. https://doi.org/10.3390/membranes11110858
Alhasan BA, Gordeev SA, Knyazeva AR, Aleksandrova KV, Margulis BA, Guzhova IV, Suvorova II. The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy. Membranes. 2021; 11(11):858. https://doi.org/10.3390/membranes11110858
Chicago/Turabian StyleAlhasan, Bashar A., Sergei A. Gordeev, Aleksandra R. Knyazeva, Kseniia V. Aleksandrova, Boris A. Margulis, Irina V. Guzhova, and Irina I. Suvorova. 2021. "The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy" Membranes 11, no. 11: 858. https://doi.org/10.3390/membranes11110858
APA StyleAlhasan, B. A., Gordeev, S. A., Knyazeva, A. R., Aleksandrova, K. V., Margulis, B. A., Guzhova, I. V., & Suvorova, I. I. (2021). The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy. Membranes, 11(11), 858. https://doi.org/10.3390/membranes11110858