

Hydrogen Sulfide (H2S)-Donating Formyl Peptide Receptor 2 (FPR2) Agonists: Design, Synthesis, and Biological Evaluation in Primary Mouse Microglia Culture

 ,
,  , , , , ,
, , , , ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
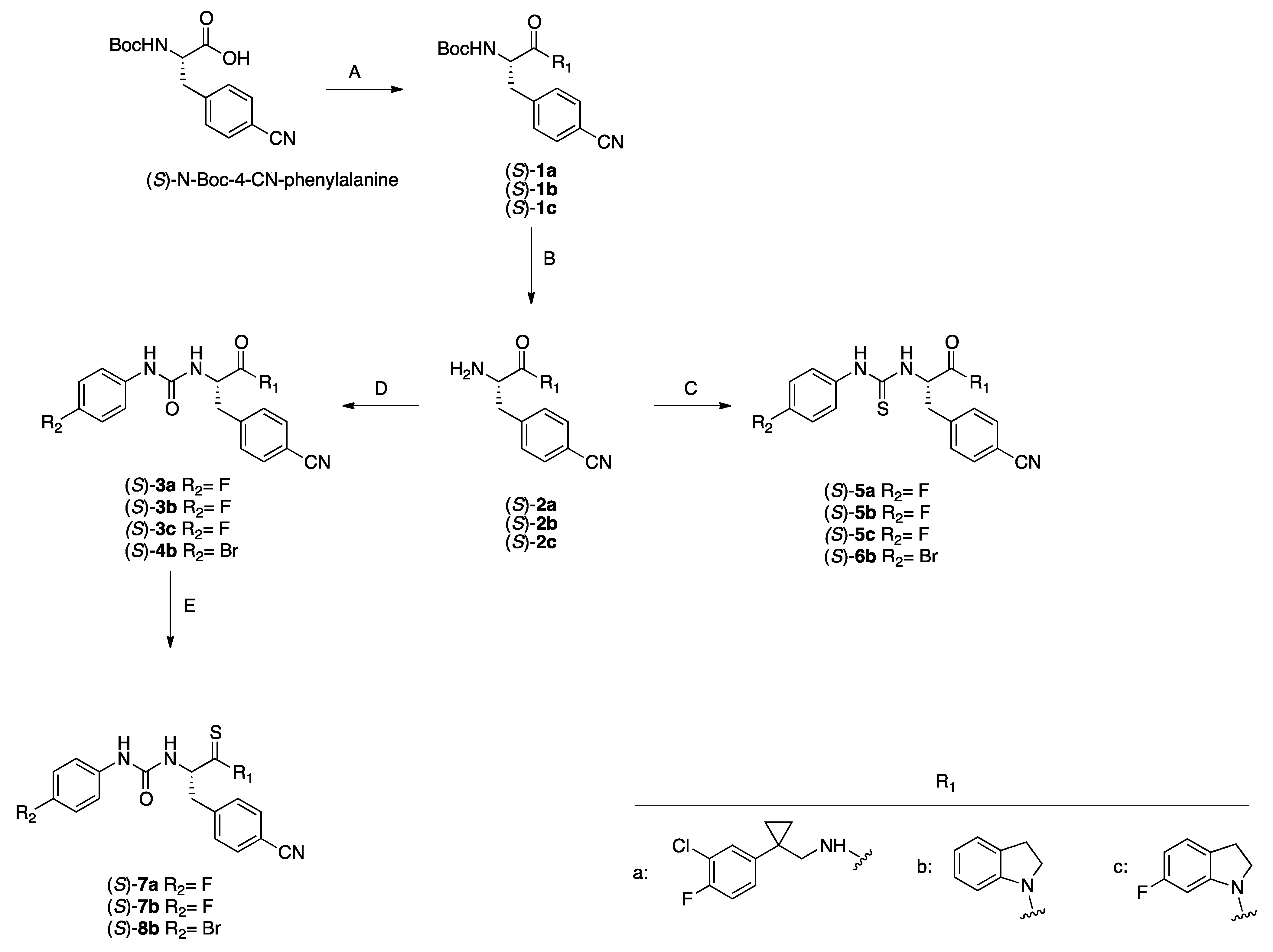
2.1.1. General Procedure for the Synthesis of Compounds 4b, 5a–c, 6b
2.1.2. General Procedure for the Synthesis of the Target Compounds 7a,b and 8b
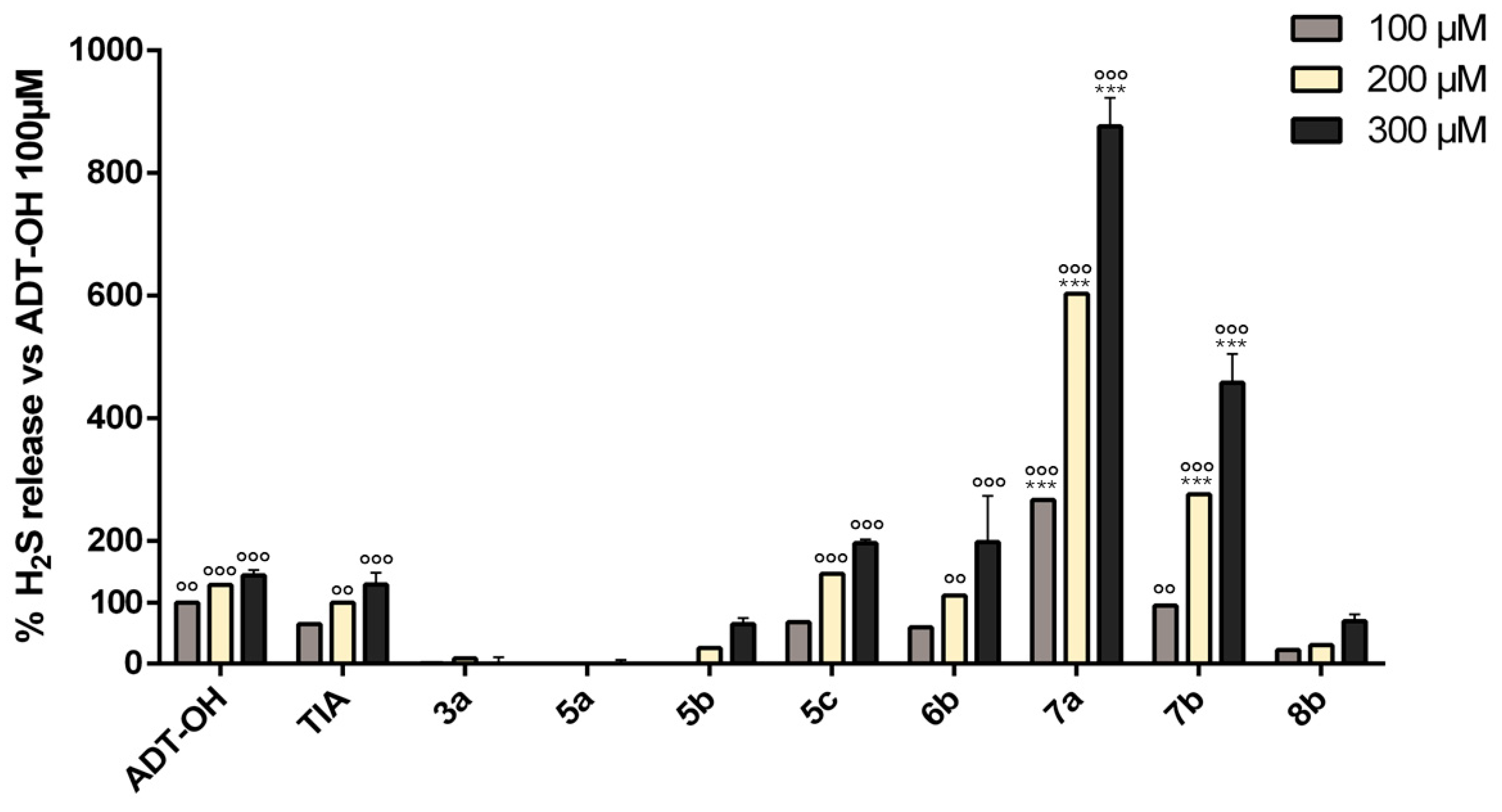
2.2. Cell-Free H2S Release Studies
2.2.1. Fluorescence Assay
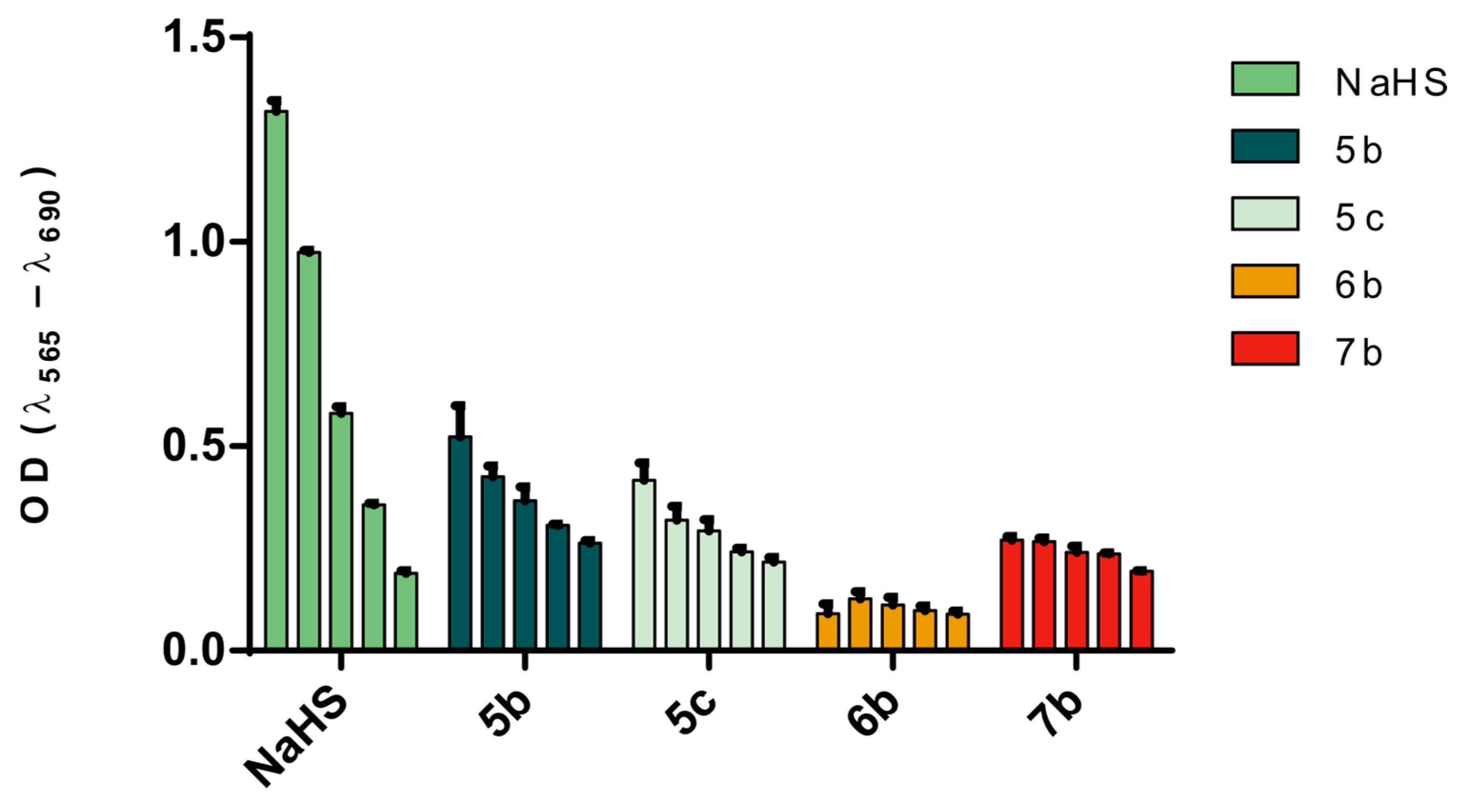
2.2.2. MTT-Based Assay
2.3. Biological Methods
2.3.1. Ca2+ Mobilization Assay
2.3.2. Cell Viability Assays
2.3.3. Intracellular H2S Release Studies
2.4. Mouse Primary Microglial Cell Cultures
2.4.1. Animals
2.4.2. Cell Culture
2.4.3. Cell Treatment
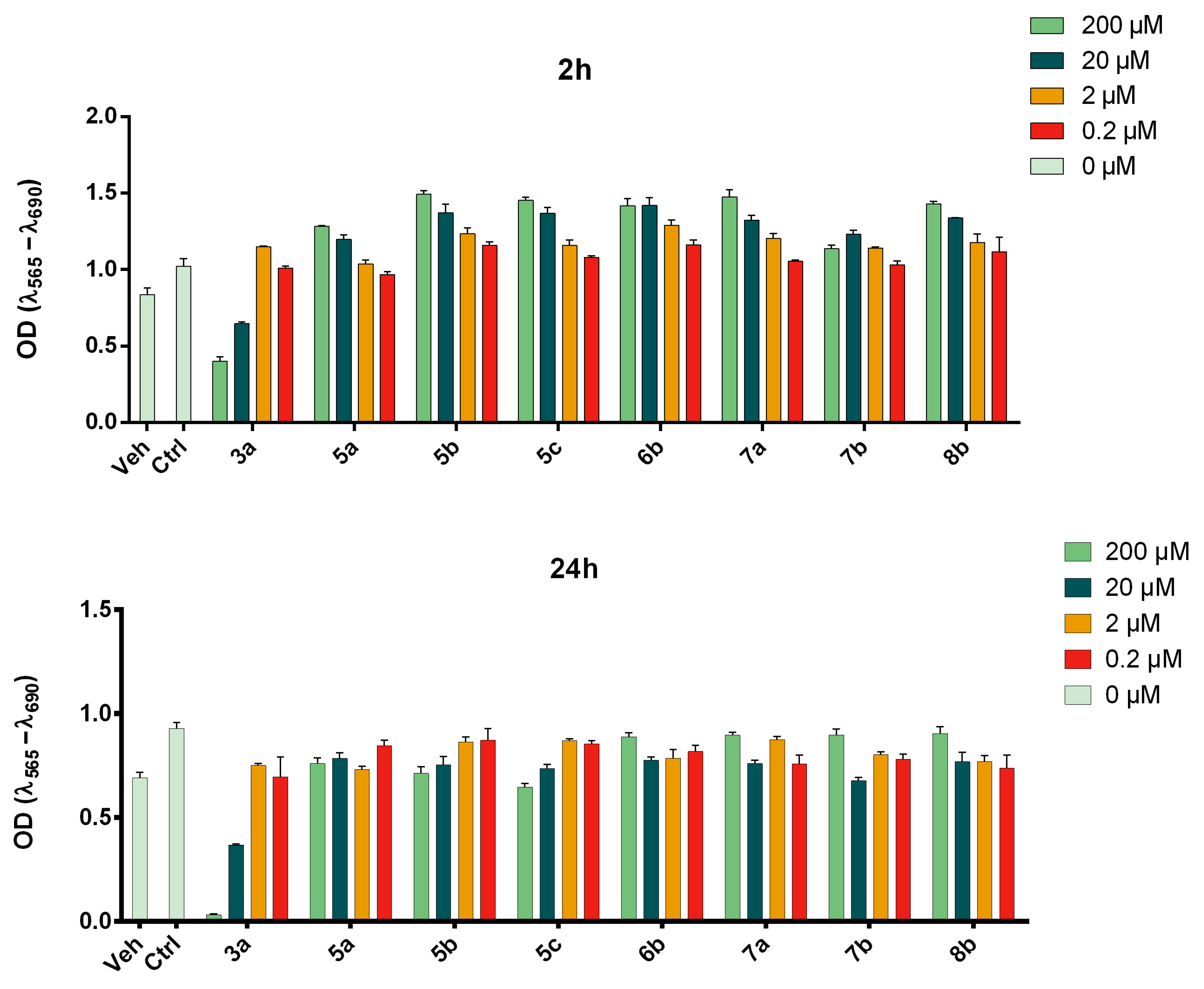
2.4.4. NO Release Assay
2.4.5. Lactate Dehydrogenase (LDH) Release Assay
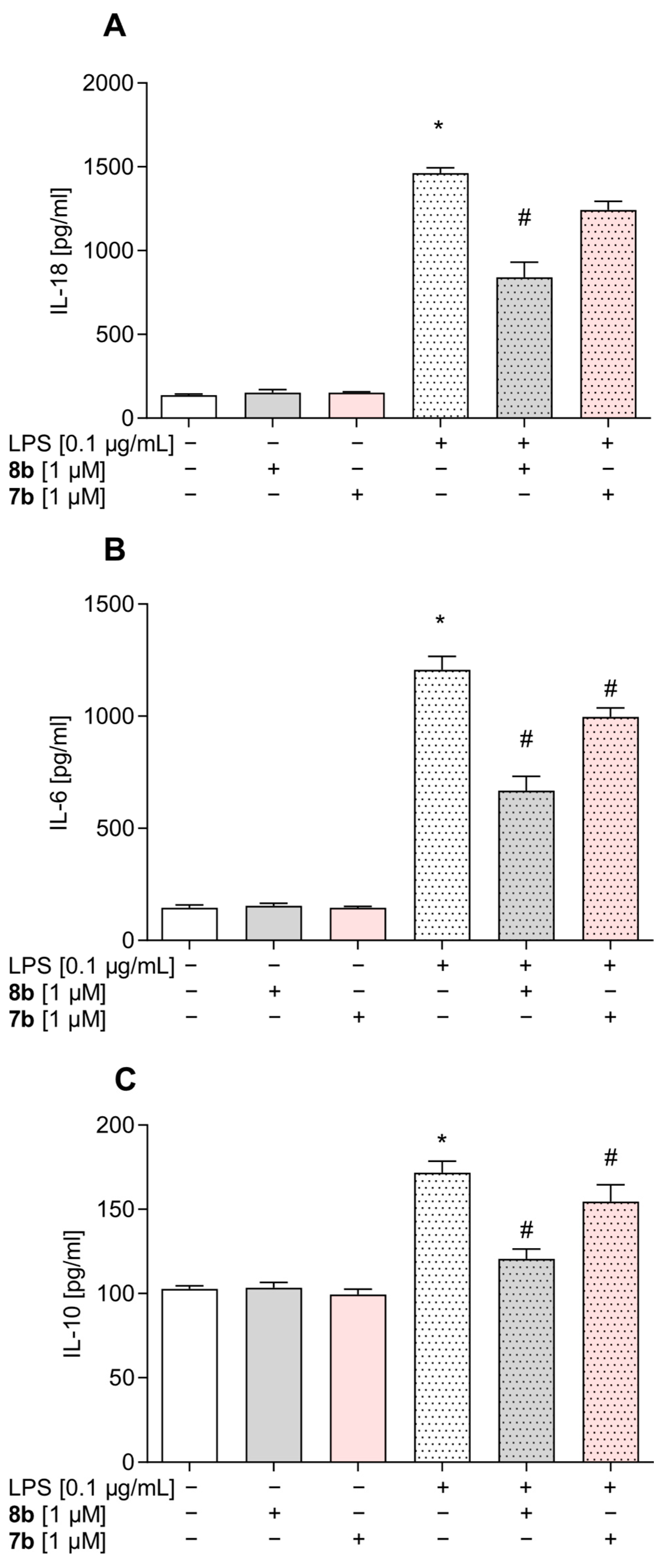
2.4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.4.7. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
3.2. Agonist Activity at FPR2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca2+ Mobilization | |||||||
|---|---|---|---|---|---|---|---|
| HL60-FPR2 | HL60-FPR1 | ||||||
| EC50, μM (Efficacy, %) | IC50, μM | EC50, μM (Efficacy, %) | IC50, μM | ||||
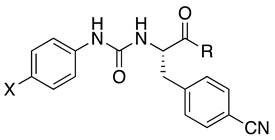 | |||||||
| Compd | X | R | |||||
| 3a | F | a | 3.9 a | N.T. b | 5.2 a | N.T. | |
| 3b | F | b | 0.026 c | 0.01 c | 0.32 c | 17.6 c | |
| 3c | F | c | 0.16 c | 0.21 c | 4.8 c | N.A. d | |
| 4b | Br | b | 0.10 ± 0.04 (120) | 0.35 | 4.8 ± 1.2 (120) | N.A. c | |
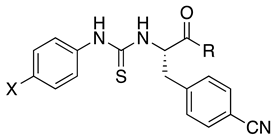 | |||||||
| 5a | F | a | N.A. | N.A. | N.A. | N.A. | |
| 5b | F | b | 2.2 ± 0.7 (65) | N.A. | N.A. | N.A. | |
| 5c | F | c | 12.0 ± 32 (105) | N.A. | N.A. | N.A. | |
| 6b | Br | b | 1.7 ± 0.7 (65) | 2.2 ± 0.4 | N.A. | N.A. | |
 | |||||||
| 7a | F | a | 2.3 ± 0.8 (145) | 0.3 ± 0.10 | N.A. | N.A. | |
| 7b | F | b | 0.6 ± 0.2 (170) | 0.06 ± 0.02 | N.A. | N.A. | |
| 8b | Br | b | 0.04 ± 0.02 (150) | 0.007 ± 0.003 | N.A. | N.A. | |
 | |||||||
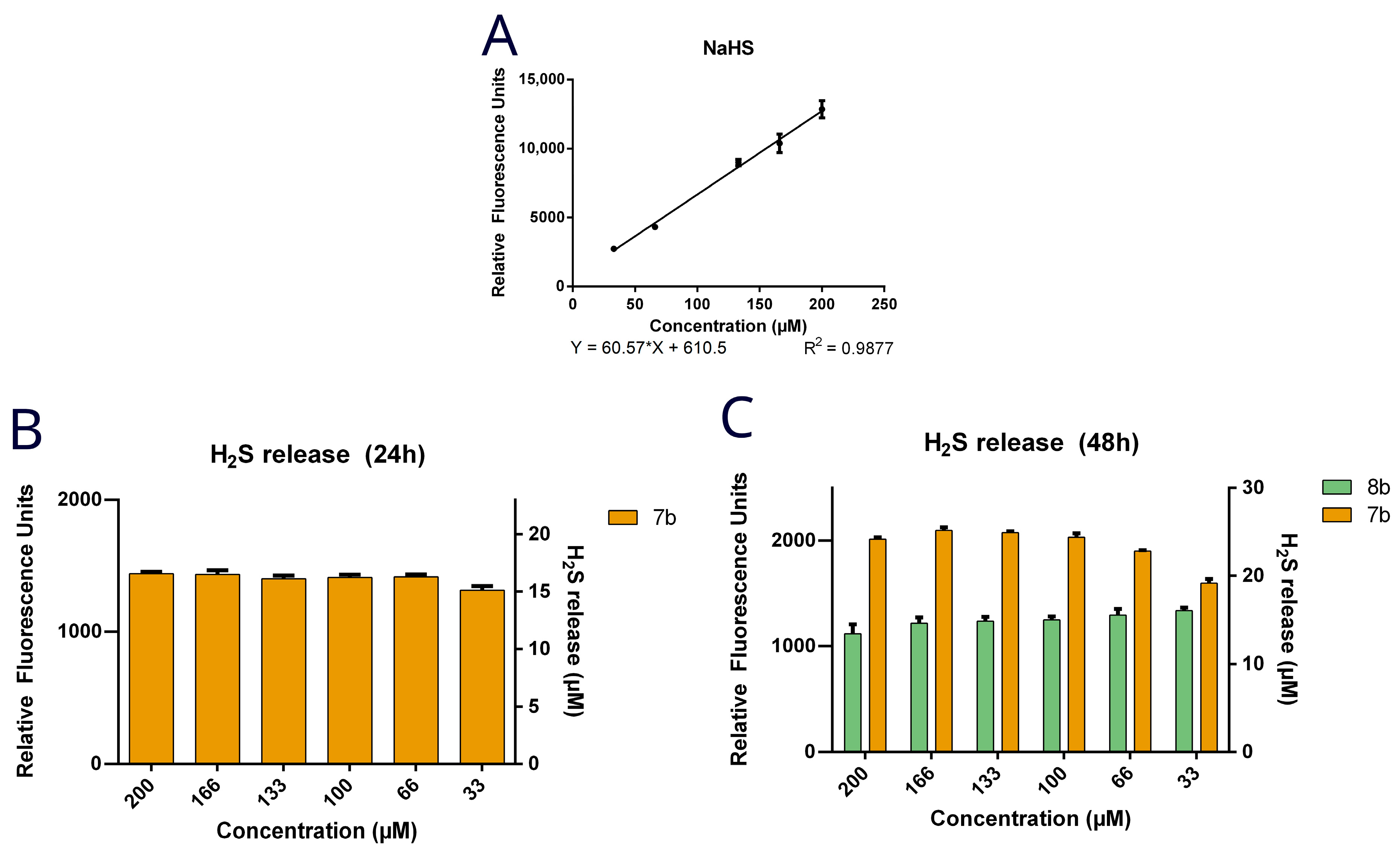
3.3. Quantification of H2S Release by Target Compounds
3.4. Effect of Compounds 8b and 7b on NO and Cell Death in Mouse Primary Microglial Cells
3.5. Effect of Compounds 8b and 7b on the Levels of Pro- and Anti-Inflammatory Factors in Mouse Primary Microglial Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Panigrahy, D.; Gilligan, M.M.; Serhan, C.N.; Kashfi, K. Resolution of Inflammation: An Organizing Principle in Biology and Medicine. Pharmacol. Ther. 2021, 227, 107879. [Google Scholar] [CrossRef]
- Laurindo, L.F.; Dias, J.A.; Araújo, A.C.; Pomini, K.T.; Galhardi, C.M.; Detregiachi, C.R.P.; Haber, L.S.d.A.; Roque, D.D.; Bechara, M.D.; de Castro, M.V.M.; et al. Immunological Dimensions of Neuroinflammation and Microglial Activation: Exploring Innovative Immunomodulatory Approaches to Mitigate Neuroinflammatory Progression. Front. Immunol. 2024, 14, 1305933. [Google Scholar] [CrossRef]
- Gusev, E.; Sarapultsev, A. Atherosclerosis and Inflammation: Insights from the Theory of General Pathological Processes. Int. J. Mol. Sci. 2023, 24, 7910. [Google Scholar] [CrossRef]
- Goswami, S.K.; Ranjan, P.; Dutta, R.K.; Verma, S.K. Management of Inflammation in Cardiovascular Diseases. Pharmacol. Res. 2021, 173, 105912. [Google Scholar] [CrossRef]
- Khandia, R.; Munjal, A. Interplay between Inflammation and Cancer. Adv. Protein Chem. Struct. Biol. 2020, 119, 1No99–245. [Google Scholar] [CrossRef]
- Basil, M.C.; Levy, B.D. Specialized Pro-Resolving Mediators: Endogenous Regulators of Infection and Inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Julliard, W.A.; Myo, Y.P.A.; Perelas, A.; Jackson, P.D.; Thatcher, T.H.; Sime, P.J. Specialized Pro-Resolving Mediators as Modulators of Immune Responses. Semin. Immunol. 2022, 59, 101605. [Google Scholar] [CrossRef]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen Sulfide: An Endogenous Regulator of the Immune System. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Schebb, N.H.; Kühn, H.; Kahnt, A.S.; Rund, K.M.; O’Donnell, V.B.; Flamand, N.; Peters-Golden, M.; Jakobsson, P.-J.; Weylandt, K.H.; Rohwer, N.; et al. Formation, Signaling and Occurrence of Specialized Pro-Resolving Lipid Mediators-What Is the Evidence so Far? Front. Pharmacol. 2022, 13, 838782. [Google Scholar] [CrossRef]
- Wickstead, E.S.; Solito, E.; McArthur, S. Promiscuous Receptors and Neuroinflammation: The Formyl Peptide Class. Life 2022, 12, 2009. [Google Scholar] [CrossRef]
- Lee, H.Y.; Jeong, Y.S.; Lee, M.; Kweon, H.-S.; Huh, Y.H.; Park, J.S.; Hwang, J.E.; Kim, K.; Bae, Y.-S. Intracellular Formyl Peptide Receptor Regulates Naïve CD4 T Cell Migration. Biochem. Biophys. Res. Commun. 2018, 497, 226–232. [Google Scholar] [CrossRef]
- Qin, C.X.; Norling, L.V.; Vecchio, E.A.; Brennan, E.P.; May, L.T.; Wootten, D.; Godson, C.; Perretti, M.; Ritchie, R.H. Formylpeptide Receptor 2: Nomenclature, Structure, Signalling and Translational Perspectives: IUPHAR Review 35. Br. J. Pharmacol. 2022, 179, 4617–4639. [Google Scholar] [CrossRef]
- Zhu, J.; Li, L.; Ding, J.; Huang, J.; Shao, A.; Tang, B. The Role of Formyl Peptide Receptors in Neurological Diseases via Regulating Inflammation. Front. Cell Neurosci. 2021, 15, 753832. [Google Scholar] [CrossRef]
- Gao, J.; Su, G.; Liu, J.; Shen, M.; Zhang, Z.; Wang, M. Formyl Peptide Receptors in the Microglial Activation: New Perspectives and Therapeutic Potential for Neuroinflammation. FASEB J. 2024, 38, e70151. [Google Scholar] [CrossRef]
- Li, Q.-Q.; Ding, D.-H.; Wang, X.-Y.; Sun, Y.-Y.; Wu, J. Lipoxin A4 Regulates Microglial M1/M2 Polarization after Cerebral Ischemia-Reperfusion Injury via the Notch Signaling Pathway. Exp. Neurol. 2021, 339, 113645. [Google Scholar] [CrossRef]
- Jiang, S.; Wan, Q.; Wang, X.; Di, L.; Li, X.; Kang, R.; Li, S.; Huang, L. LXA4 Attenuates Perioperative Neurocognitive Disorders by Suppressing Neuroinflammation and Oxidative Stress. Int. Immunopharmacol. 2023, 123, 110788. [Google Scholar] [CrossRef]
- Yi, X.; Tran, E.; Odiba, J.O.; Qin, C.X.; Ritchie, R.H.; Baell, J.B. The Formyl Peptide Receptors FPR1 and FPR2 as Targets for Inflammatory Disorders: Recent Advances in the Development of Small-Molecule Agonists. Eur. J. Med. Chem. 2024, 265, 115989. [Google Scholar] [CrossRef]
- Stama, M.L.; Ślusarczyk, J.; Lacivita, E.; Kirpotina, L.N.; Schepetkin, I.A.; Chamera, K.; Riganti, C.; Perrone, R.; Quinn, M.T.; Basta-Kaim, A.; et al. Novel Ureidopropanamide Based N-Formyl Peptide Receptor 2 (FPR2) Agonists with Potential Application for Central Nervous System Disorders Characterized by Neuroinflammation. Eur. J. Med. Chem. 2017, 141, 703–720. [Google Scholar] [CrossRef]
- Trojan, E.; Tylek, K.; Leśkiewicz, M.; Lasoń, W.; Brandenburg, L.-O.; Leopoldo, M.; Lacivita, E.; Basta-Kaim, A. The N-Formyl Peptide Receptor 2 (FPR2) Agonist MR-39 Exhibits Anti-Inflammatory Activity in LPS-Stimulated Organotypic Hippocampal Cultures. Cells 2021, 10, 1524. [Google Scholar] [CrossRef]
- Tylek, K.; Trojan, E.; Leśkiewicz, M.; Regulska, M.; Bryniarska, N.; Curzytek, K.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways. Cells 2021, 10, 2373. [Google Scholar] [CrossRef]
- Trojan, E.; Tylek, K.; Schröder, N.; Kahl, I.; Brandenburg, L.-O.; Mastromarino, M.; Leopoldo, M.; Basta-Kaim, A.; Lacivita, E. The N-Formyl Peptide Receptor 2 (FPR2) Agonist MR-39 Improves Ex Vivo and In Vivo Amyloid Beta (1–42)-Induced Neuroinflammation in Mouse Models of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 6203–6221. [Google Scholar] [CrossRef]
- Cristiano, C.; Volpicelli, F.; Crispino, M.; Lacivita, E.; Russo, R.; Leopoldo, M.; Calignano, A.; Perrone-Capano, C. Behavioral, Anti-Inflammatory, and Neuroprotective Effects of a Novel FPR2 Agonist in Two Mouse Models of Autism. Pharmaceuticals 2022, 15, 161. [Google Scholar] [CrossRef]
- Mastromarino, M.; Favia, M.; Schepetkin, I.A.; Kirpotina, L.N.; Trojan, E.; Niso, M.; Carrieri, A.; Leśkiewicz, M.; Regulska, M.; Darida, M.; et al. Design, Synthesis, Biological Evaluation, and Computational Studies of Novel Ureidopropanamides as Formyl Peptide Receptor 2 (FPR2) Agonists to Target the Resolution of Inflammation in Central Nervous System Disorders. J. Med. Chem. 2022, 65, 5004–5028. [Google Scholar] [CrossRef]
- Tylek, K.; Trojan, E.; Leśkiewicz, M.; Francavilla, F.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Stimulation of Formyl Peptide Receptor-2 by the New Agonist CMC23 Protects against Endotoxin-Induced Neuroinflammatory Response: A Study in Organotypic Hippocampal Cultures. ACS Chem. Neurosci. 2023, 14, 3869–3882. [Google Scholar] [CrossRef]
- Tylek, K.; Trojan, E.; Leśkiewicz, M.; Ghafir El Idrissi, I.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Microglia Depletion Attenuates the Pro-Resolving Activity of the Formyl Peptide Receptor 2 Agonist AMS21 Related to Inhibition of Inflammasome NLRP3 Signalling Pathway: A Study of Organotypic Hippocampal Cultures. Cells 2023, 12, 2570. [Google Scholar] [CrossRef]
- Dey, A.; Pramanik, P.K.; Dwivedi, S.K.D.; Neizer-Ashun, F.; Kiss, T.; Ganguly, A.; Rice, H.; Mukherjee, P.; Xu, C.; Ahmad, M.; et al. A Role for the Cystathionine-β-Synthase/H2S Axis in Astrocyte Dysfunction in the Aging Brain. Redox Biol. 2023, 68, 102958. [Google Scholar] [CrossRef]
- Morikawa, T.; Kajimura, M.; Nakamura, T.; Hishiki, T.; Nakanishi, T.; Yukutake, Y.; Nagahata, Y.; Ishikawa, M.; Hattori, K.; Takenouchi, T.; et al. Hypoxic Regulation of the Cerebral Microcirculation Is Mediated by a Carbon Monoxide-Sensitive Hydrogen Sulfide Pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 1293–1298. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen Sulfide Is Neuroprotective in Alzheimer’s Disease by Sulfhydrating GSK3β and Inhibiting Tau Hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Zhang, M.; Shan, H.; Chang, P.; Ma, L.; Chu, Y.; Shen, X.; Wu, Q.; Wang, Z.; Luo, C.; Wang, T.; et al. Upregulation of 3-MST Relates to Neuronal Autophagy After Traumatic Brain Injury in Mice. Cell Mol. Neurobiol. 2017, 37, 291–302. [Google Scholar] [CrossRef]
- Zhao, H.; Chan, S.-J.; Ng, Y.-K.; Wong, P.T.-H. Brain 3-Mercaptopyruvate Sulfurtransferase (3MST): Cellular Localization and Downregulation after Acute Stroke. PLoS ONE 2013, 8, e67322. [Google Scholar] [CrossRef]
- Nagahara, N.; Ito, T.; Kitamura, H.; Nishino, T. Tissue and Subcellular Distribution of Mercaptopyruvate Sulfurtransferase in the Rat: Confocal Laser Fluorescence and Immunoelectron Microscopic Studies Combined with Biochemical Analysis. Histochemistry 1998, 110, 243–250. [Google Scholar] [CrossRef]
- Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L.; Zanardo, R.C.O.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; et al. Hydrogen Sulfide Is an Endogenous Modulator of Leukocyte-Mediated inflammation Hydrogen Sulfide Is an Endogenous Modulator of Leukocyte-Mediated Inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid. Redox Signal. 2019, 30, 184–197. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, M.; Zhu, R.; Zhou, J.; Ni, L.; Wang, Z.; Liu, N.; Zhu, F.; Shi, T.; Deng, Z.; et al. Hydrogen Sulfide Protects against Particle-Induced Inflammatory Response and Osteolysis via SIRT1 Pathway in Prosthesis Loosening. FASEB J. 2020, 34, 3743–3754. [Google Scholar] [CrossRef]
- Xiao, Q.; Ying, J.; Qiao, Z.; Yang, Y.; Dai, X.; Xu, Z.; Zhang, C.; Xiang, L. Exogenous Hydrogen Sulfide Inhibits Human Melanoma Cell Development via Suppression of the PI3K/AKT/ mTOR Pathway. J. Dermatol. Sci. 2020, 98, 26–34. [Google Scholar] [CrossRef]
- Zheng, J.; Zhao, T.; Yuan, Y.; Hu, N.; Tang, X. Hydrogen Sulfide (H2S) Attenuates Uranium-Induced Acute Nephrotoxicity through Oxidative Stress and Inflammatory Response via Nrf2-NF-κB Pathways. Chem. Biol. Interact. 2015, 242, 353–362. [Google Scholar] [CrossRef]
- Duan, H.; Li, L.; Shen, S.; Ma, Y.; Yin, X.; Liu, Z.; Yuan, C.; Wang, Y.; Zhang, J. Hydrogen Sulfide Reduces Cognitive Impairment in Rats After Subarachnoid Hemorrhage by Ameliorating Neuroinflammation Mediated by the TLR4/NF-κB Pathway in Microglia. Front. Cell Neurosci. 2020, 14, 210. [Google Scholar] [CrossRef]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen Sulfide Alleviates Diabetic Nephropathy in a Streptozotocin-Induced Diabetic Rat Model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef]
- Bourque, C.; Zhang, Y.; Fu, M.; Racine, M.; Greasley, A.; Pei, Y.; Wu, L.; Wang, R.; Yang, G. H2S Protects Lipopolysaccharide-Induced Inflammation by Blocking NFκB Transactivation in Endothelial Cells. Toxicol. Appl. Pharmacol. 2018, 338, 20–29. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.-I.; Kimura, H. Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen Sulfide Protects Neurons from Oxidative Stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef]
- Rafaiee, R.; Khastar, H.; Garmabi, B.; Taleb, M.; Norouzi, P.; Khaksari, M. Hydrogen Sulfide Protects Hippocampal CA1 Neurons against Lead Mediated Neuronal Damage via Reduction Oxidative Stress in Male Rats. J. Chem. Neuroanat. 2021, 112, 101917. [Google Scholar] [CrossRef]
- Tripathi, S.J.; Chakraborty, S.; Miller, E.; Pieper, A.A.; Paul, B.D. Hydrogen Sulfide Signalling in Neurodegenerative Diseases. Br. J. Pharmacol. 2023, 1–18. [Google Scholar] [CrossRef]
- Paul, B.D.; Pieper, A.A. Neuroprotective Signaling by Hydrogen Sulfide and Its Dysregulation in Alzheimer’s Disease. Curr. Opin. Chem. Biol. 2024, 82, 102511. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A Review of Hydrogen Sulfide (H2S) Donors: Chemistry and Potential Therapeutic Applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Xu, S.; Shieh, M.; Paul, B.D.; Xian, M. Hydrogen Sulfide: Recent Development of Its Dual Donors and Hybrid Drugs. Br. J. Pharmacol. 2023, 1–13. [Google Scholar] [CrossRef]
- Zaorska, E.; Hutsch, T.; Gawryś-Kopczyńska, M.; Ostaszewski, R.; Ufnal, M.; Koszelewski, D. Evaluation of Thioamides, Thiolactams and Thioureas as Hydrogen Sulfide (H2S) Donors for Lowering Blood Pressure. Bioorg. Chem. 2019, 88, 102941. [Google Scholar] [CrossRef]
- Sato, K.; Sugimoto, H.; Rikimaru, K.; Imoto, H.; Kamaura, M.; Negoro, N.; Tsujihata, Y.; Miyashita, H.; Odani, T.; Murata, T. Discovery of a Novel Series of Indoline Carbamate and Indolinylpyrimidine Derivatives as Potent GPR119 Agonists. Bioorg. Med. Chem. 2014, 22, 1649–1666. [Google Scholar] [CrossRef]
- Giordano, F.; Corvino, A.; Scognamiglio, A.; Citi, V.; Gorica, E.; Fattorusso, C.; Persico, M.; Caliendo, G.; Fiorino, F.; Magli, E.; et al. Hybrids between H2S-Donors and Betamethasone 17-Valerate or Triamcinolone Acetonide Inhibit Mast Cell Degranulation and Promote Hyperpolarization of Bronchial Smooth Muscle Cells. Eur. J. Med. Chem. 2021, 221, 113517. [Google Scholar] [CrossRef]
- Peng, B.; Chen, W.; Liu, C.; Rosser, E.W.; Pacheco, A.; Zhao, Y.; Aguilar, H.C.; Xian, M. Fluorescent Probes Based on Nucleophilic Substitution-Cyclization for Hydrogen Sulfide Detection and Bioimaging. Chemistry 2014, 20, 1010–1016. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Jutila, M.A.; Quinn, M.T. Gastrin-Releasing Peptide/Neuromedin B Receptor Antagonists PD176252, PD168368, and Related Analogs Are Potent Agonists of Human Formyl-Peptide Receptors. Mol. Pharmacol. 2011, 79, 77–90. [Google Scholar] [CrossRef]
- Francavilla, F.; Vitone, D.; Schepetkin, I.A.; Kirpotina, L.N.; Carrieri, A.; Brunetti, L.; Ghafir El Idrissi, I.; Frydrych, J.K.; Trojan, E.; Quinn, M.T.; et al. Design, Synthesis, and Biological Evaluation of Novel Heteroaryl, Squaramide, and Indolcarboxamide Derivatives as Formyl Peptide Receptor 2 (FPR2) Agonists to Target Neuroinflammation. ACS Chem. Neurosci. 2025, submitted.
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef]
- Smith, H.M.; Pluth, M.D. Advances and Opportunities in H2S Measurement in Chemical Biology. JACS Au 2023, 3, 2677–2691. [Google Scholar] [CrossRef]
- Gerő, D.; Torregrossa, R.; Perry, A.; Waters, A.; Le-Trionnaire, S.; Whatmore, J.L.; Wood, M.; Whiteman, M. The Novel Mitochondria-Targeted Hydrogen Sulfide (H2S) Donors AP123 and AP39 Protect against Hyperglycemic Injury in Microvascular Endothelial Cells in Vitro. Pharmacol. Res. 2016, 113, 186–198. [Google Scholar] [CrossRef]
- Dash, U.C.; Bhol, N.K.; Swain, S.K.; Samal, R.R.; Nayak, P.K.; Raina, V.; Panda, S.K.; Kerry, R.G.; Duttaroy, A.K.; Jena, A.B. Oxidative Stress and Inflammation in the Pathogenesis of Neurological Disorders: Mechanisms and Implications. Acta Pharm. Sin. B 2025, 15, 15–34. [Google Scholar] [CrossRef]
- Wang, R. Two’s Company, Three’s a Crowd: Can H2S Be the Third Endogenous Gaseous Transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Oh, G.-S.; Pae, H.-O.; Lee, B.-S.; Kim, B.-N.; Kim, J.-M.; Kim, H.-R.; Jeon, S.B.; Jeon, W.K.; Chae, H.-J.; Chung, H.-T. Hydrogen Sulfide Inhibits Nitric Oxide Production and Nuclear Factor-kappaB via Heme Oxygenase-1 Expression in RAW264.7 Macrophages Stimulated with Lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef]
- Suk, K.; Kim, S.Y.; Kim, H. Regulation of IL-18 production by IFN gamma and PGE2 in mouse microglial cells: Involvement of NF-kB pathway in the regulatory processes. Immunol. Lett. 2001, 77, 79–85. [Google Scholar] [CrossRef]
- Hanamsagar, R.; Torres, V.; Kielian, T. Inflammasome activation and IL-1β/IL-18 processing are influenced by distinct pathways in microglia. J. Neurochem. 2011, 119, 736–748. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Dufton, N.; Natividad, J.; Verdu, E.F.; Wallace, J.L. Hydrogen Sulfide and Resolution of Acute Inflammation: A Comparative Study Utilizing a Novel Fluorescent Probe. Sci. Rep. 2012, 2, 499. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Fu, X.; Wang, K.; Yang, J.; Zhang, X.; Wang, H. The Role of Hydrogen Sulfide Regulation of Pyroptosis in Different Pathological Processes. Eur. J. Med. Chem. 2024, 268, 116254. [Google Scholar] [CrossRef]
- Medeiros, R.; Kitazawa, M.; Passos, G.F.; Baglietto-Vargas, D.; Cheng, D.; Cribbs, D.H.; LaFerla, F.M. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am. J. Pathol. 2013, 182, 1780–1789. [Google Scholar] [CrossRef]
- Chen, J.J.; Chen, J.; Jiang, Z.X.; Zhou, Z.; Zhou, C.N. Resolvin D1 alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 signaling pathway. J. Biol. Regul. Homeost. Agents 2020, 34. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, L.; Francavilla, F.; Niso, M.; Frydrych, J.K.; Trojan, E.; Schepetkin, I.A.; Kirpotina, L.N.; Grygier, B.; Łukowicz, K.; Quinn, M.T.; et al. Hydrogen Sulfide (H2S)-Donating Formyl Peptide Receptor 2 (FPR2) Agonists: Design, Synthesis, and Biological Evaluation in Primary Mouse Microglia Culture. Antioxidants 2025, 14, 827. https://doi.org/10.3390/antiox14070827
Brunetti L, Francavilla F, Niso M, Frydrych JK, Trojan E, Schepetkin IA, Kirpotina LN, Grygier B, Łukowicz K, Quinn MT, et al. Hydrogen Sulfide (H2S)-Donating Formyl Peptide Receptor 2 (FPR2) Agonists: Design, Synthesis, and Biological Evaluation in Primary Mouse Microglia Culture. Antioxidants. 2025; 14(7):827. https://doi.org/10.3390/antiox14070827
Chicago/Turabian StyleBrunetti, Leonardo, Fabio Francavilla, Mauro Niso, Jakub Kosma Frydrych, Ewa Trojan, Igor A. Schepetkin, Liliya N. Kirpotina, Beata Grygier, Krzysztof Łukowicz, Mark T. Quinn, and et al. 2025. "Hydrogen Sulfide (H2S)-Donating Formyl Peptide Receptor 2 (FPR2) Agonists: Design, Synthesis, and Biological Evaluation in Primary Mouse Microglia Culture" Antioxidants 14, no. 7: 827. https://doi.org/10.3390/antiox14070827
APA StyleBrunetti, L., Francavilla, F., Niso, M., Frydrych, J. K., Trojan, E., Schepetkin, I. A., Kirpotina, L. N., Grygier, B., Łukowicz, K., Quinn, M. T., Basta-Kaim, A., Lacivita, E., & Leopoldo, M. (2025). Hydrogen Sulfide (H2S)-Donating Formyl Peptide Receptor 2 (FPR2) Agonists: Design, Synthesis, and Biological Evaluation in Primary Mouse Microglia Culture. Antioxidants, 14(7), 827. https://doi.org/10.3390/antiox14070827