
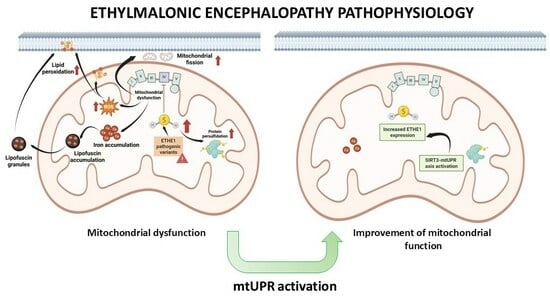
Mitochondrial Unfolded Protein Response (mtUPR) Activation Improves Pathological Alterations in Cellular Models of Ethylmalonic Encephalopathy

 , , , ,
, , , ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethical Statements
2.3. Patients and Fibroblast Culture
2.4. Immunoblotting
2.5. Quantification of Endogenous H2S
2.6. Protein Persulfidation Assay: In-Gel Persulfide Detection in Cell Lysates
2.7. Immunofluorescence Microscopy
2.8. Mitochondrial Complexes Activity
2.9. Mitochondrial Bioenergetics
2.10. Determination of L-Lactate Levels
2.11. Analysis of Mitochondrial Network and Measurement of Mitochondrial Tubular/Rounded Ratio
2.12. Measurement of Cell Membrane Lipid Peroxidation
2.13. Determination of Iron Accumulation
2.14. Determination of Lipofuscin Accumulation
2.15. Measurement of Mitochondrial Reactive Oxygen Species (ROS) Generation
2.16. TEM Analysis
2.17. Direct Reprogramming
2.18. Determination of Reduced Glutathione Levels
2.19. Determination of the Labile Iron Pool (LIP)
2.20. Transfection with the Human ETHE1 Plasmid
2.21. Cell Survival Determination After Exposure to SIRT Inhibitors
2.22. Quantitative Real-Time PCR (qPCR)
2.23. Statistical Analysis
3. Results
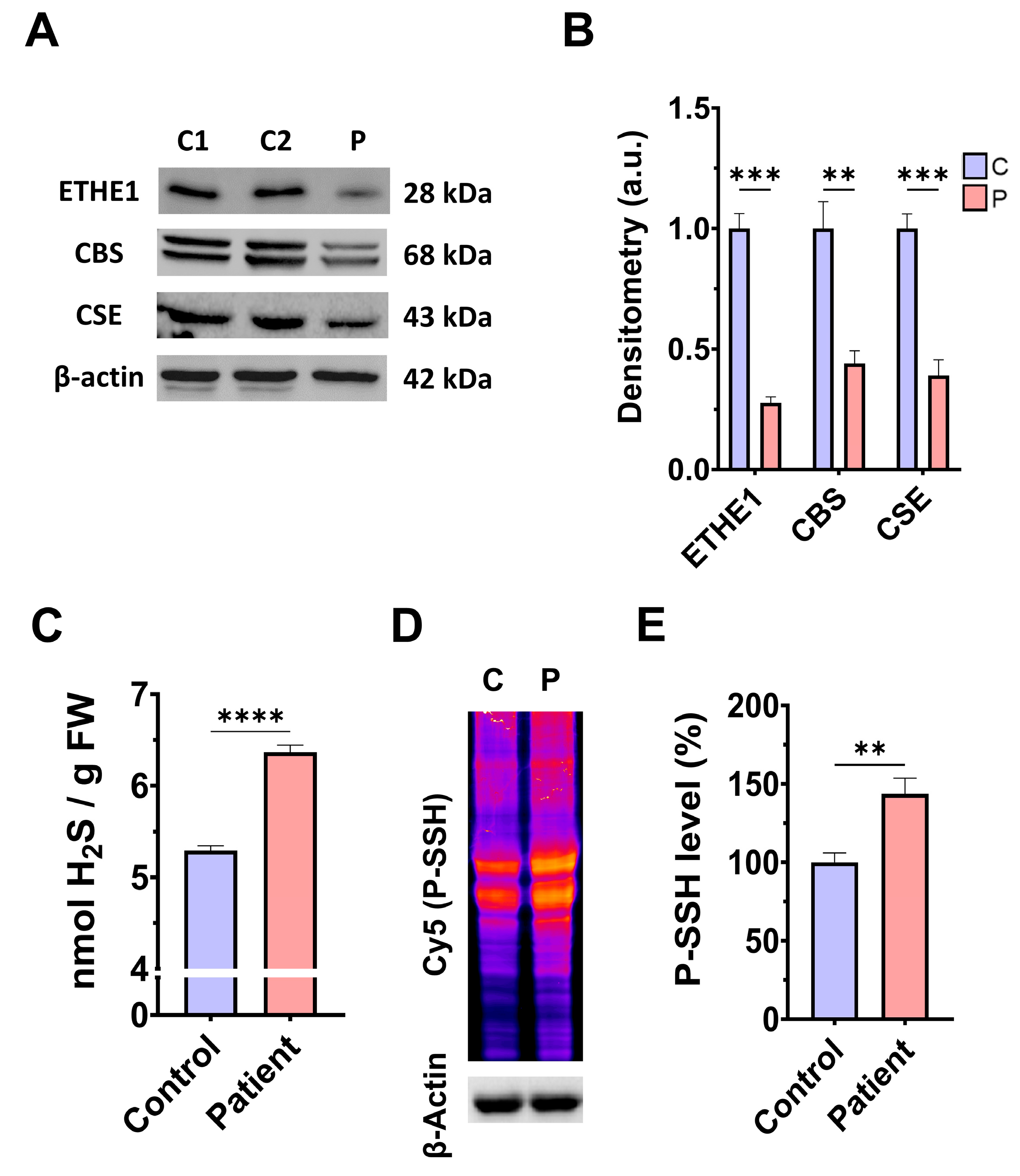
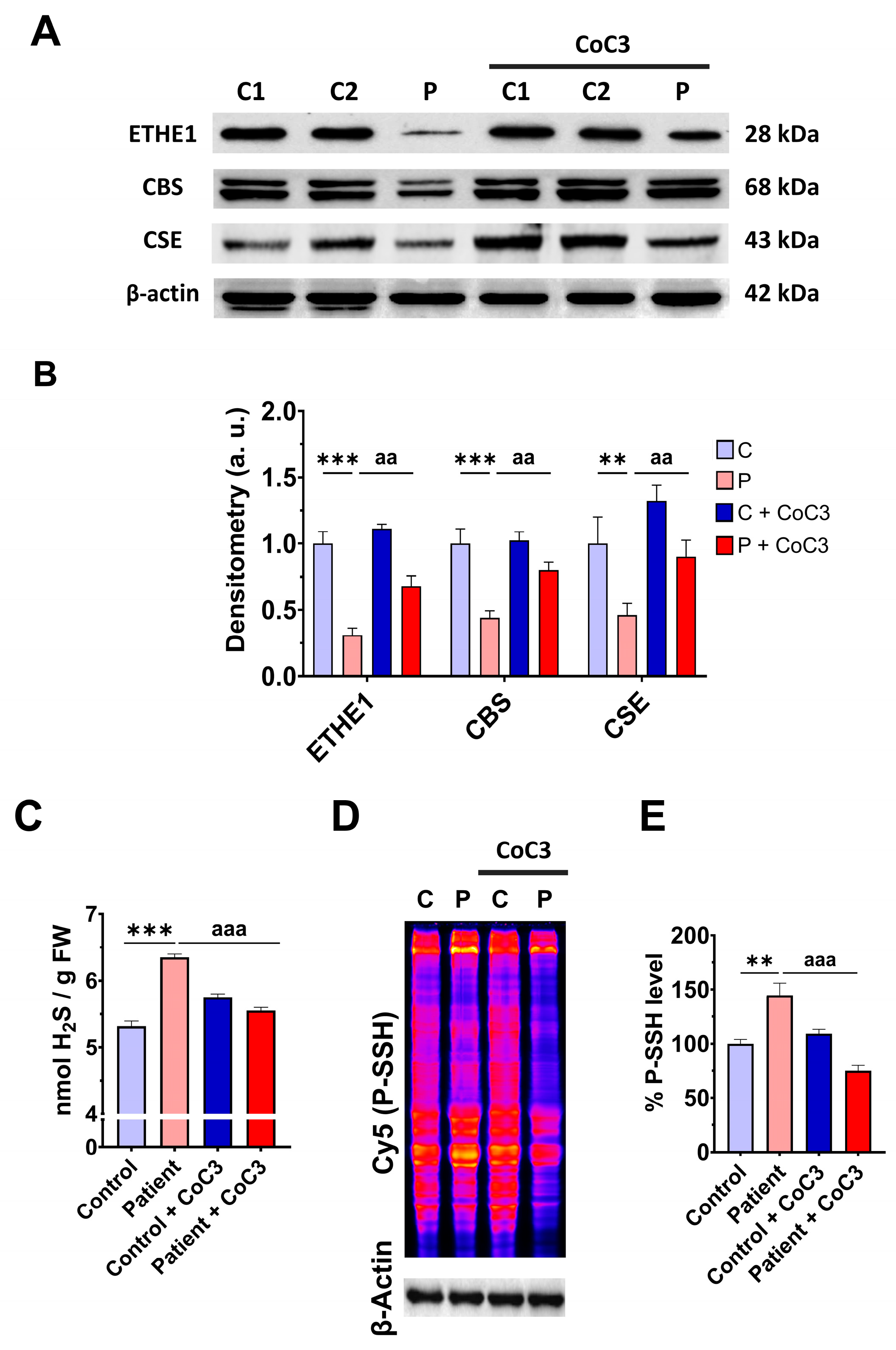
3.1. Pathological Variants of ETHE1 Cause Low Expression Levels of the ETHE1 Mutant Protein and Altered Sulfur Metabolism
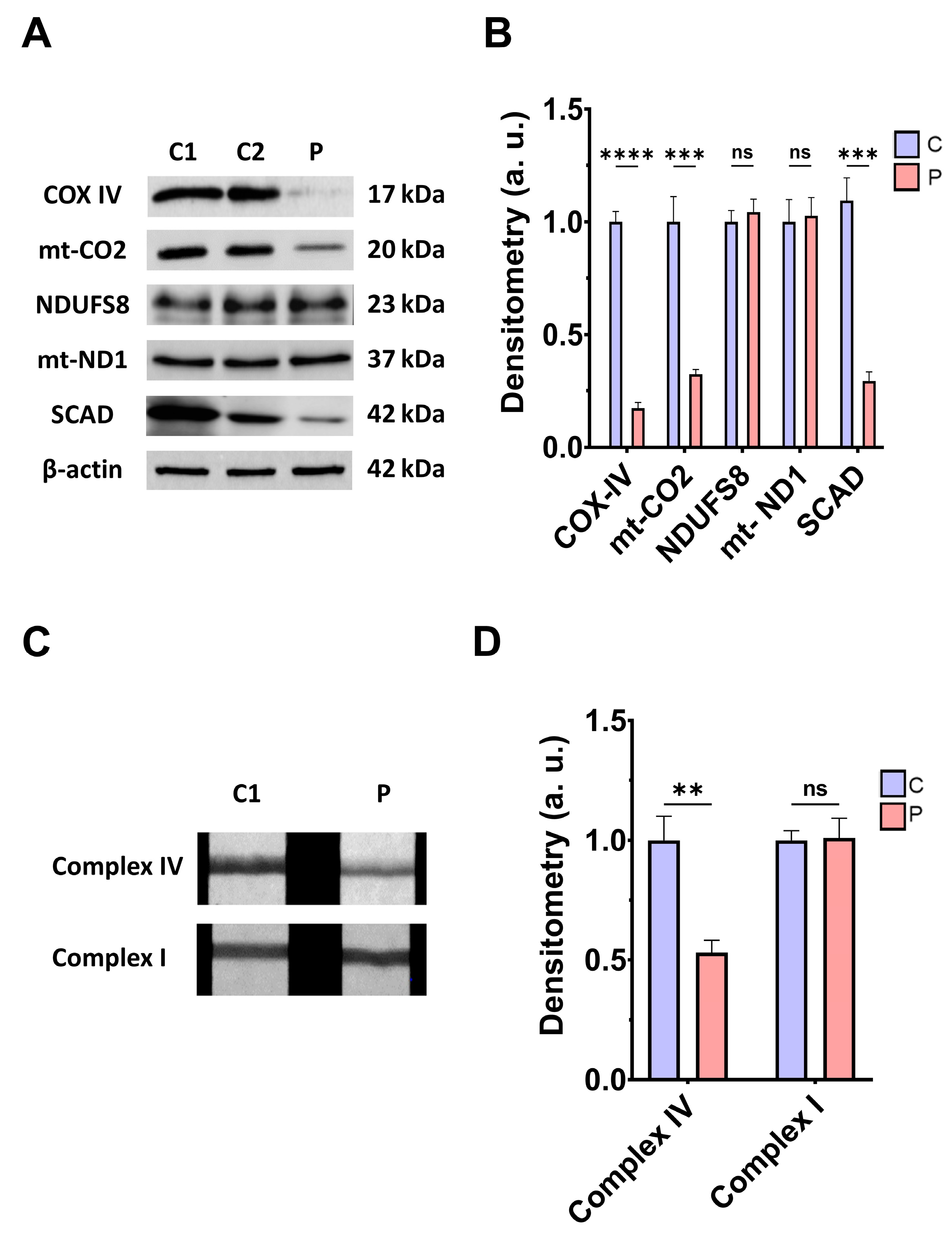
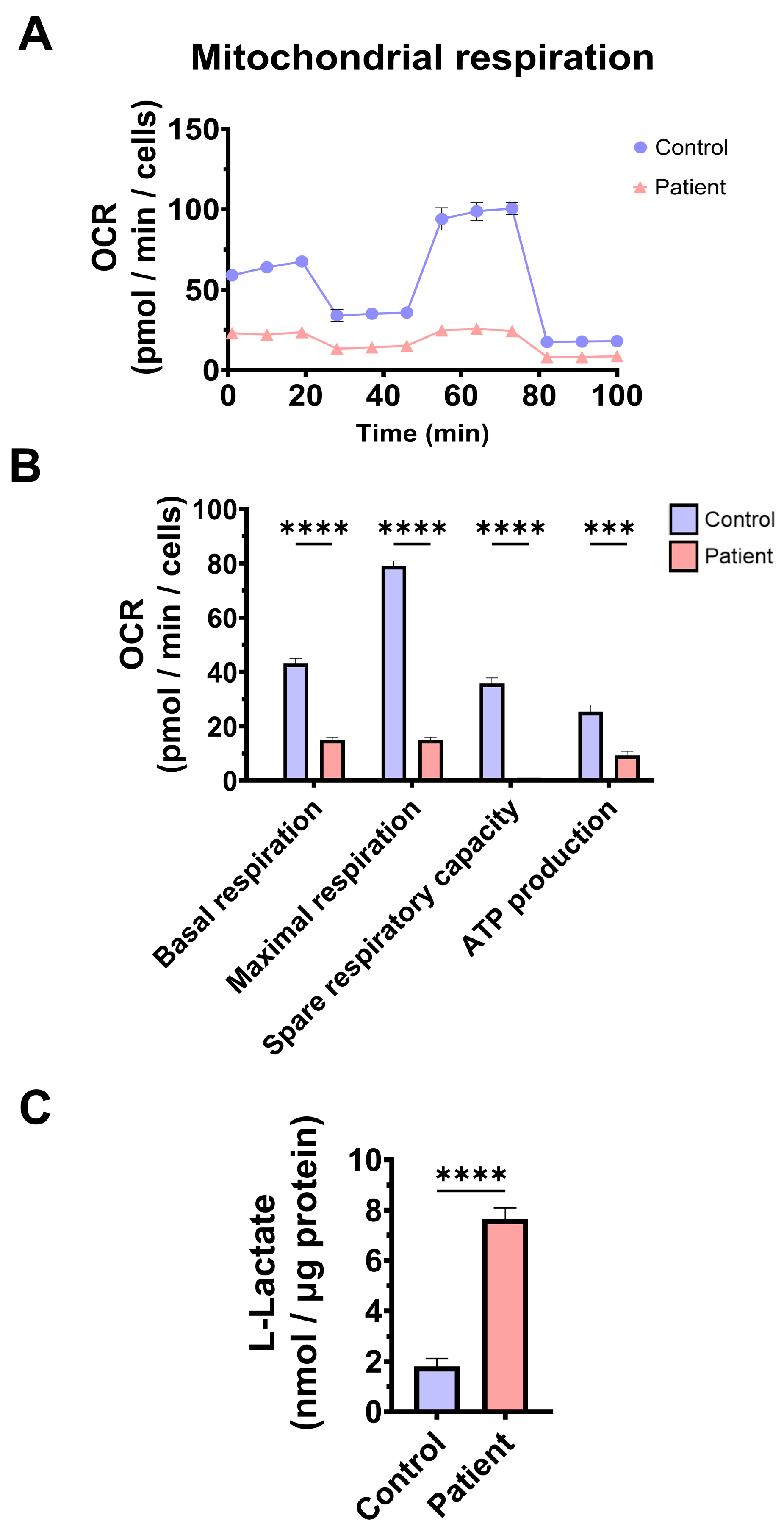
3.2. Mitochondrial Function and Bioenergetics in Fibroblasts Derived from the EE Patient
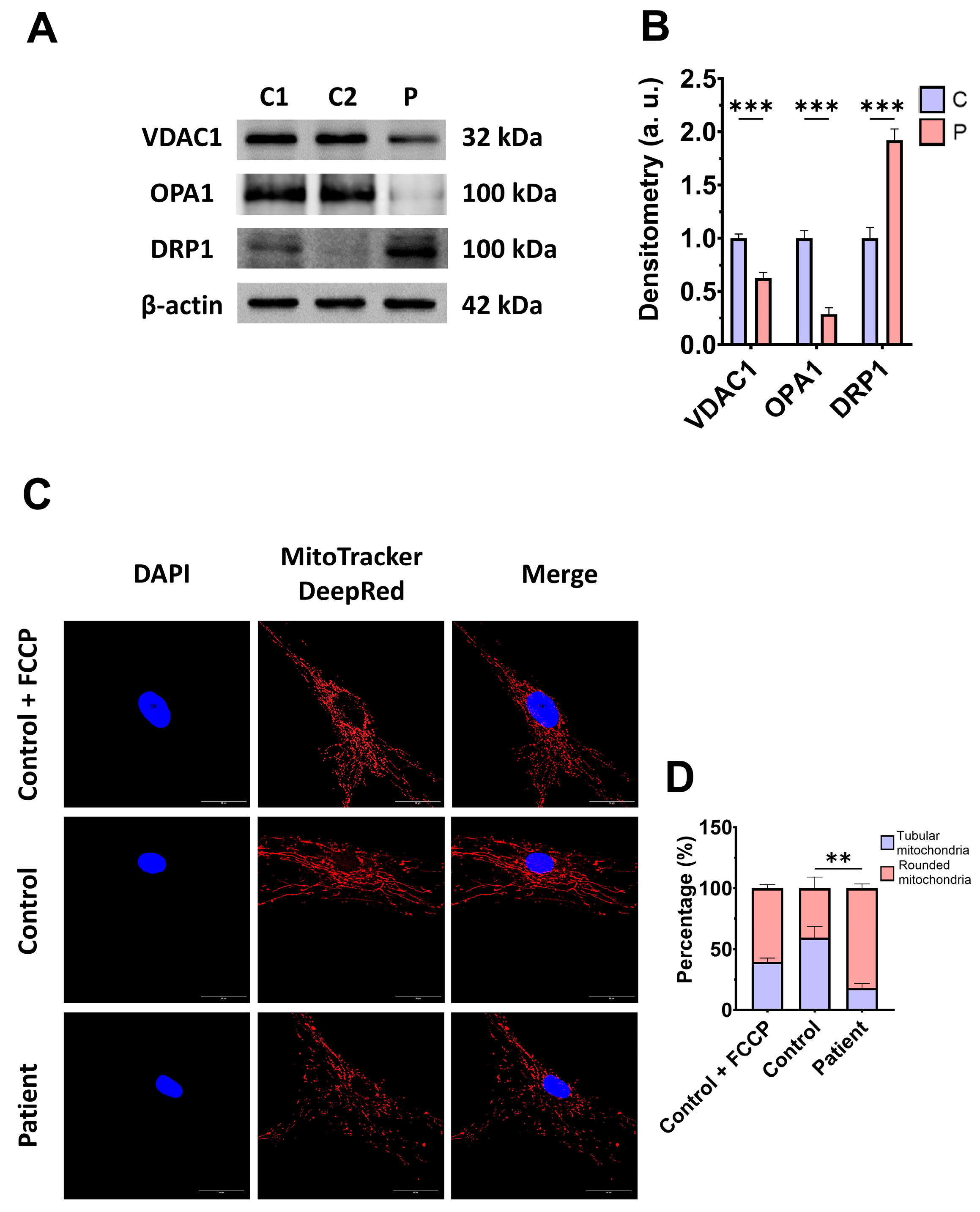
3.3. Mitochondrial Mass and Dynamics in ETHE1 Fibroblasts
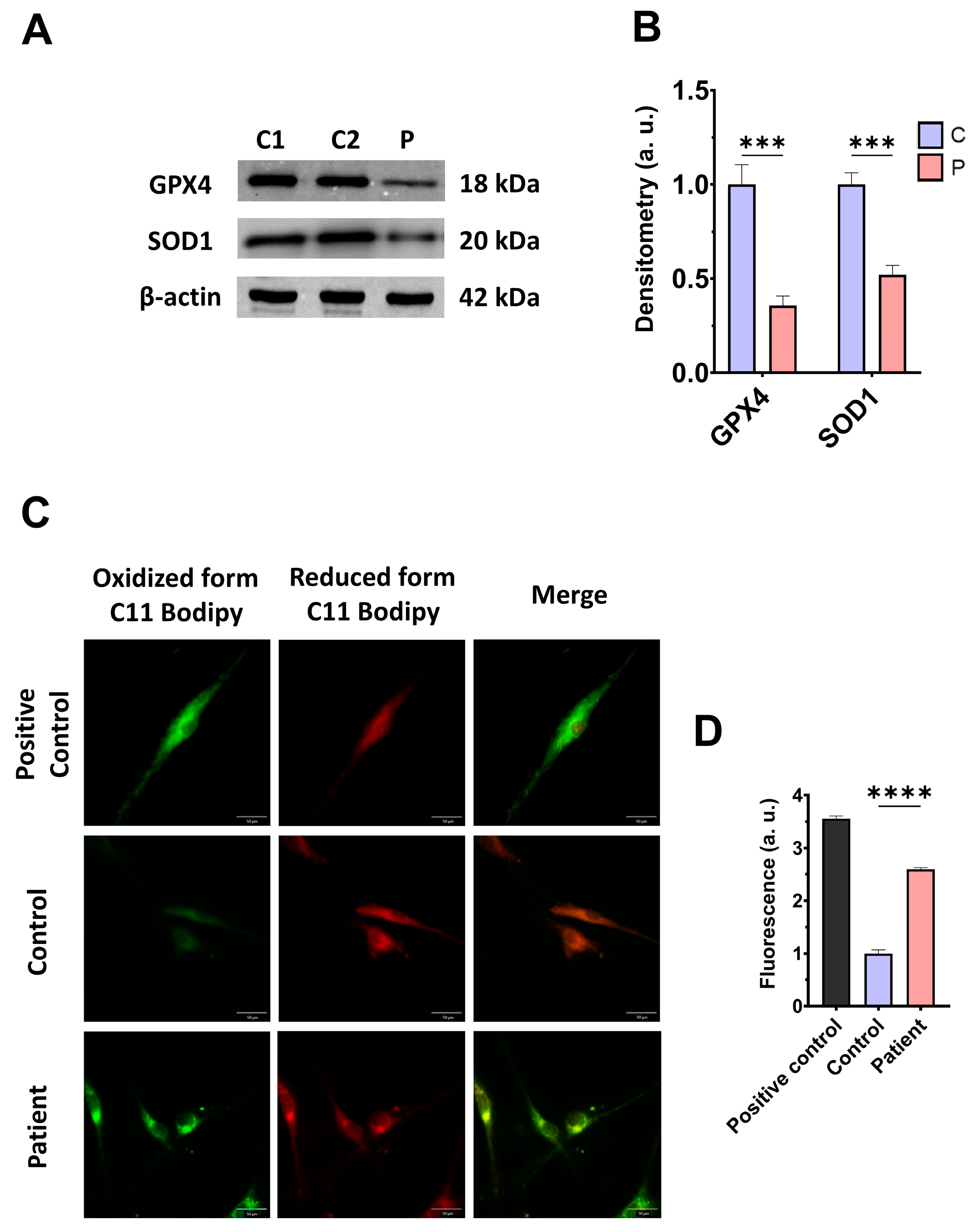
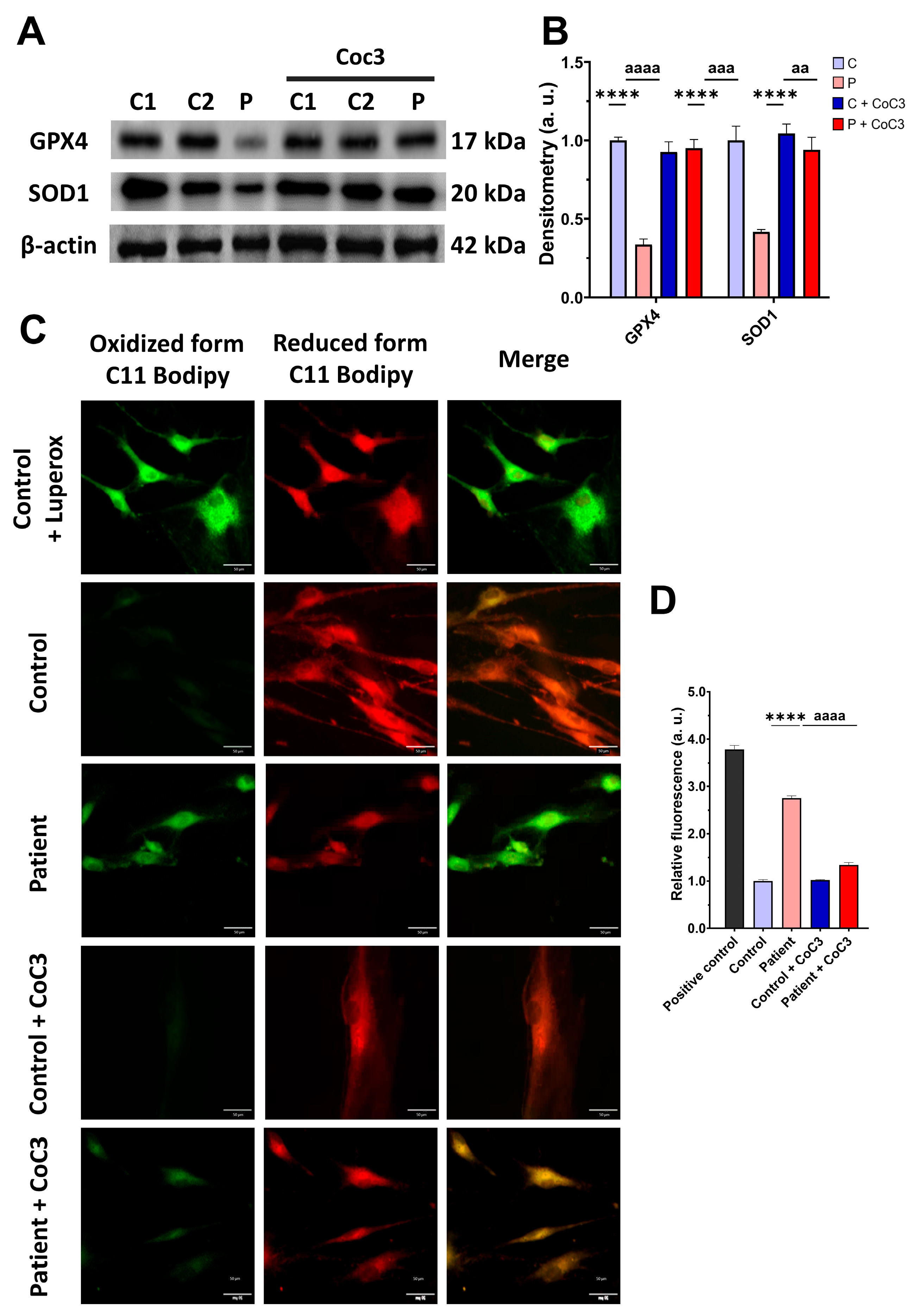
3.4. The ETHE1 Mutation Causes Oxidative Stress and Lipid Peroxidation
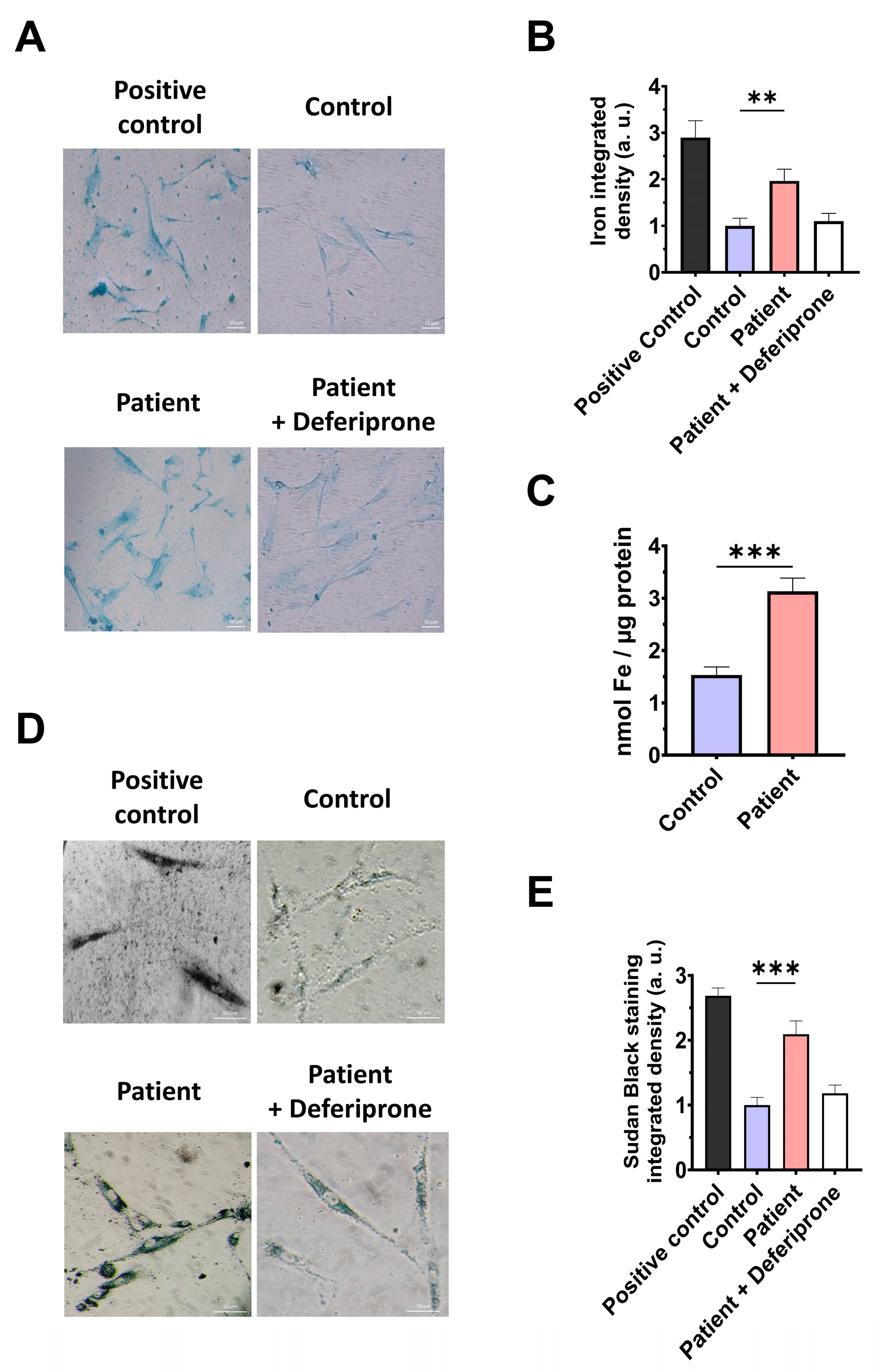
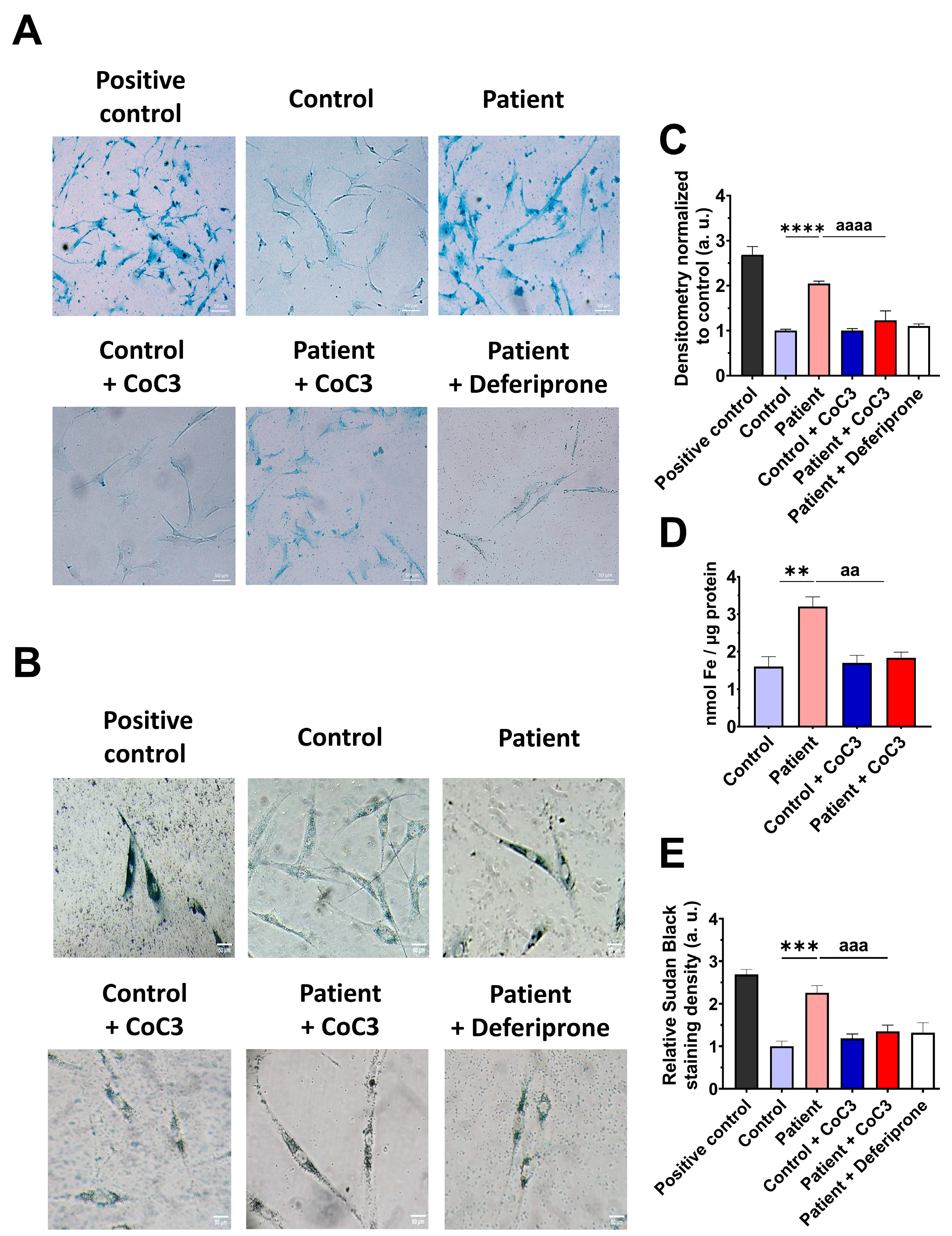
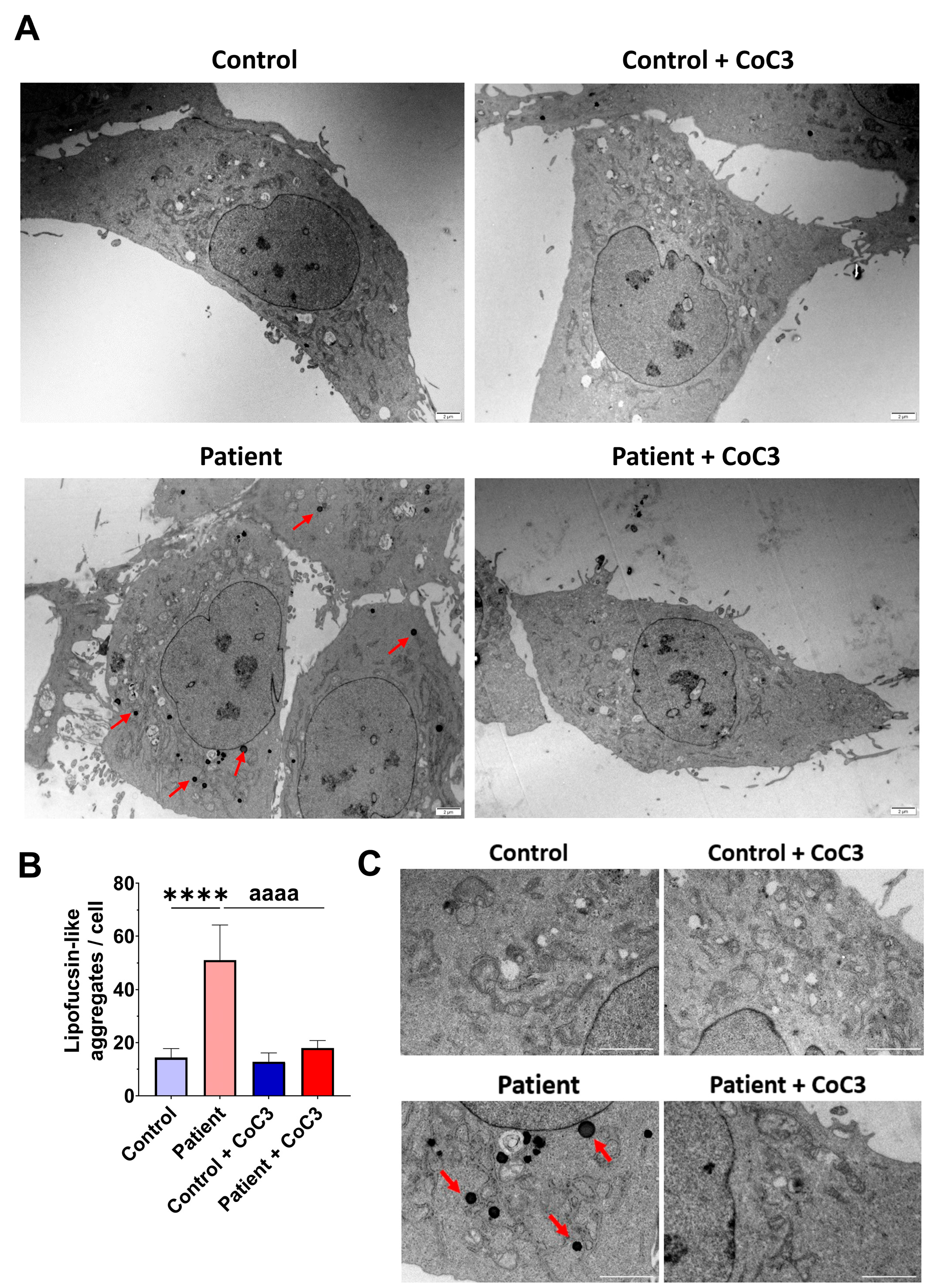
3.5. The ETHE1 Mutation Causes the Accumulation of Iron and Lipofuscin
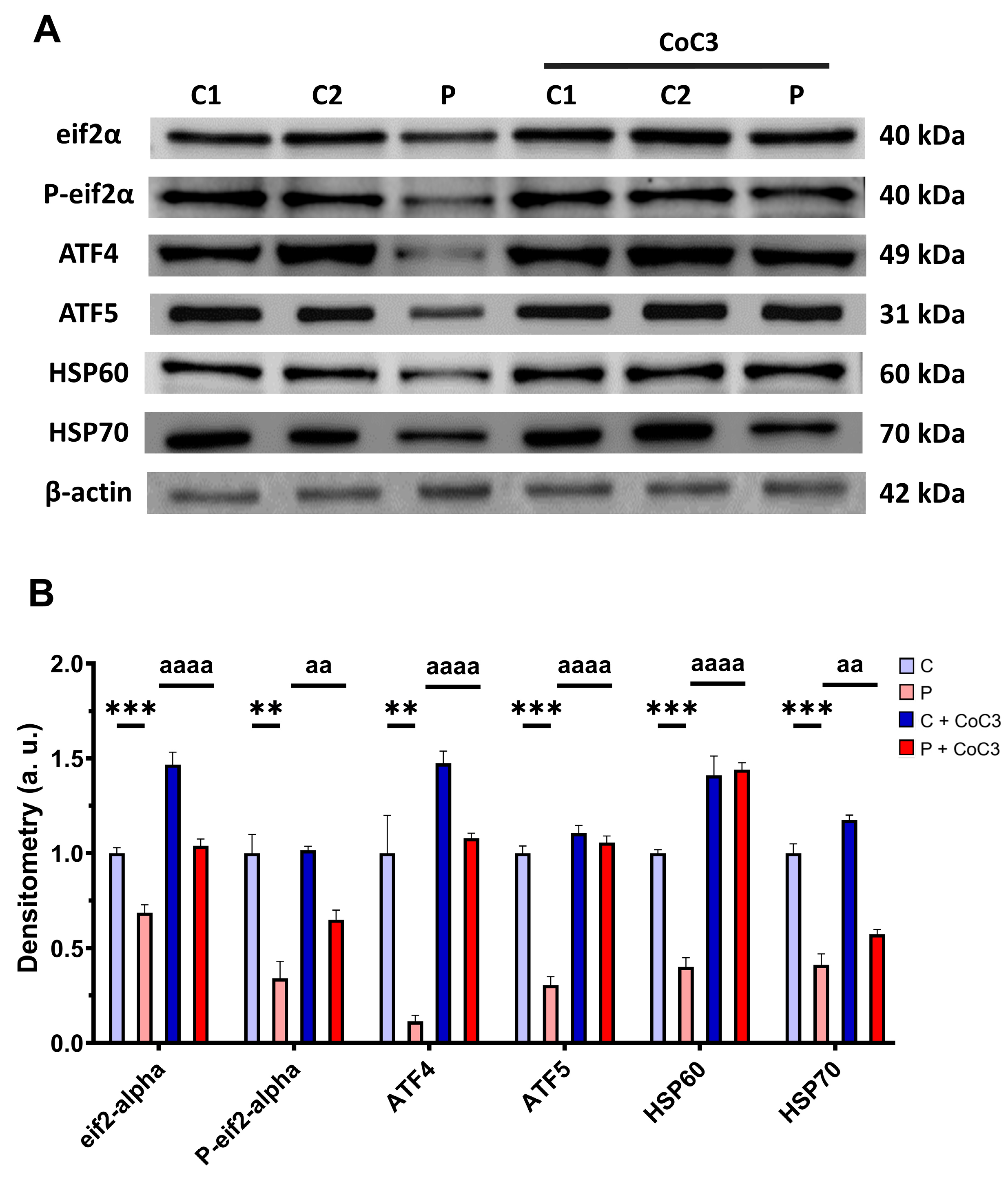
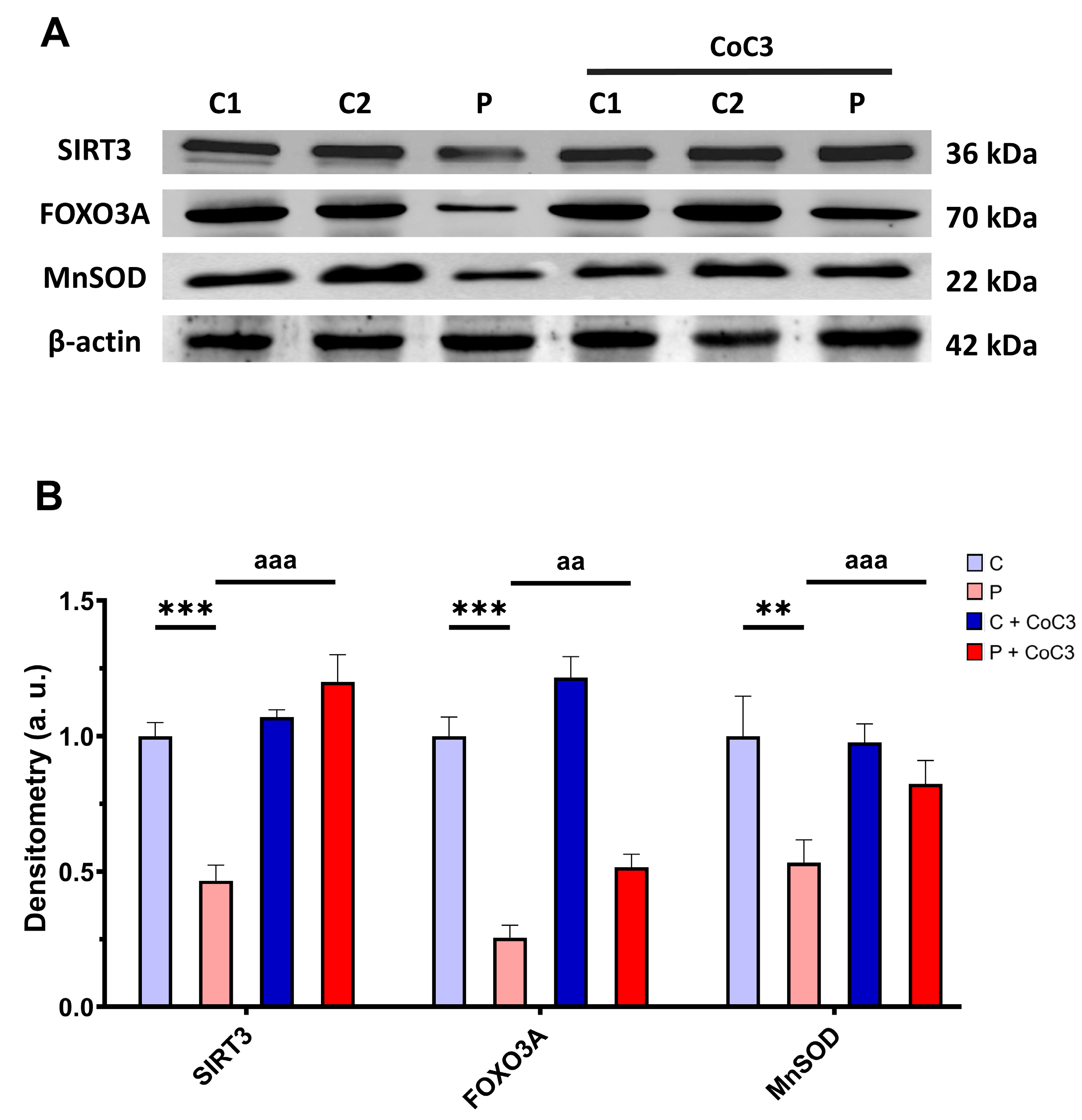
3.6. mtUPR Activation Increases the Expression Levels of the Mutant ETHE1 Protein and Normalises Sulfur Metabolism
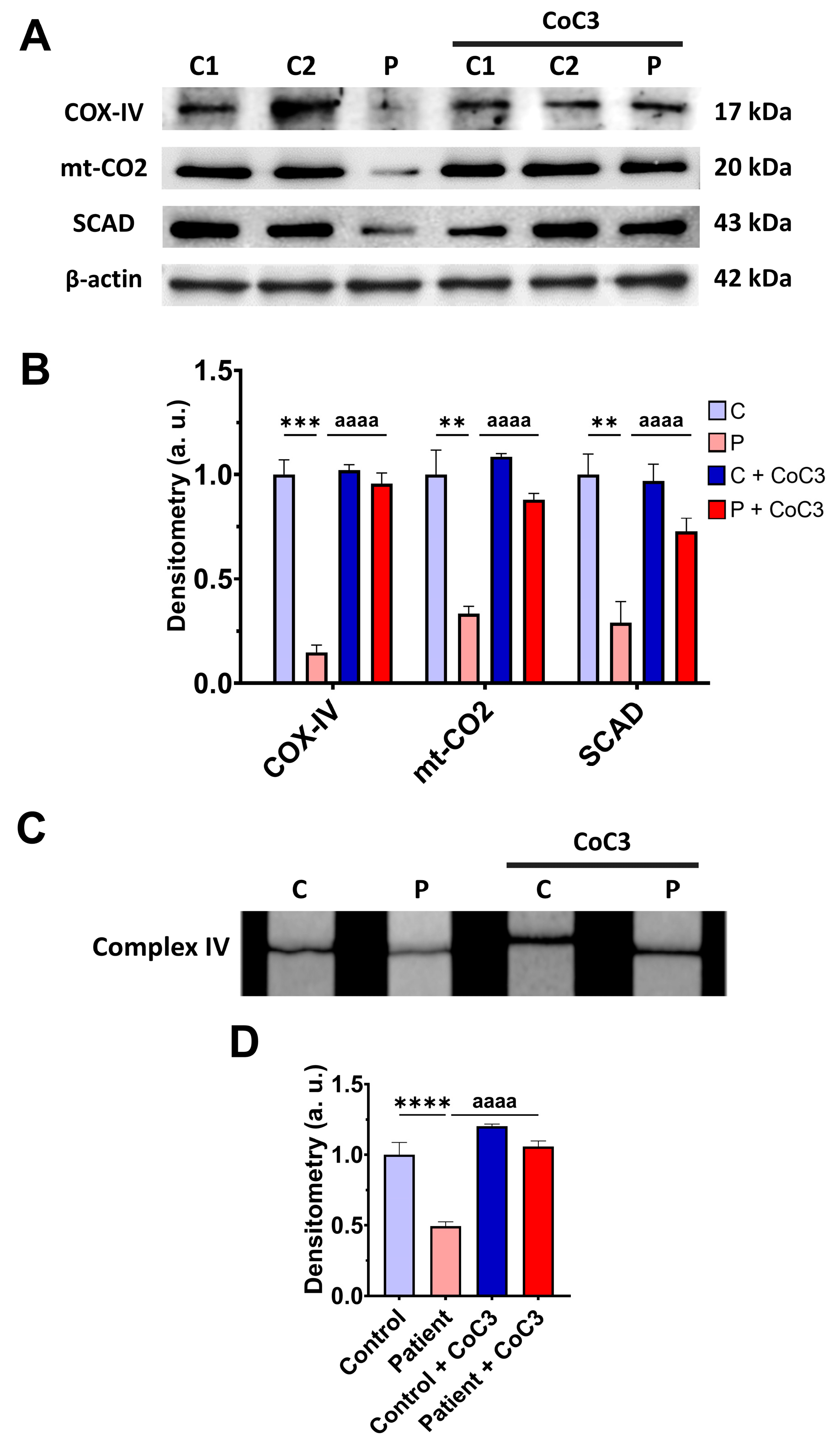
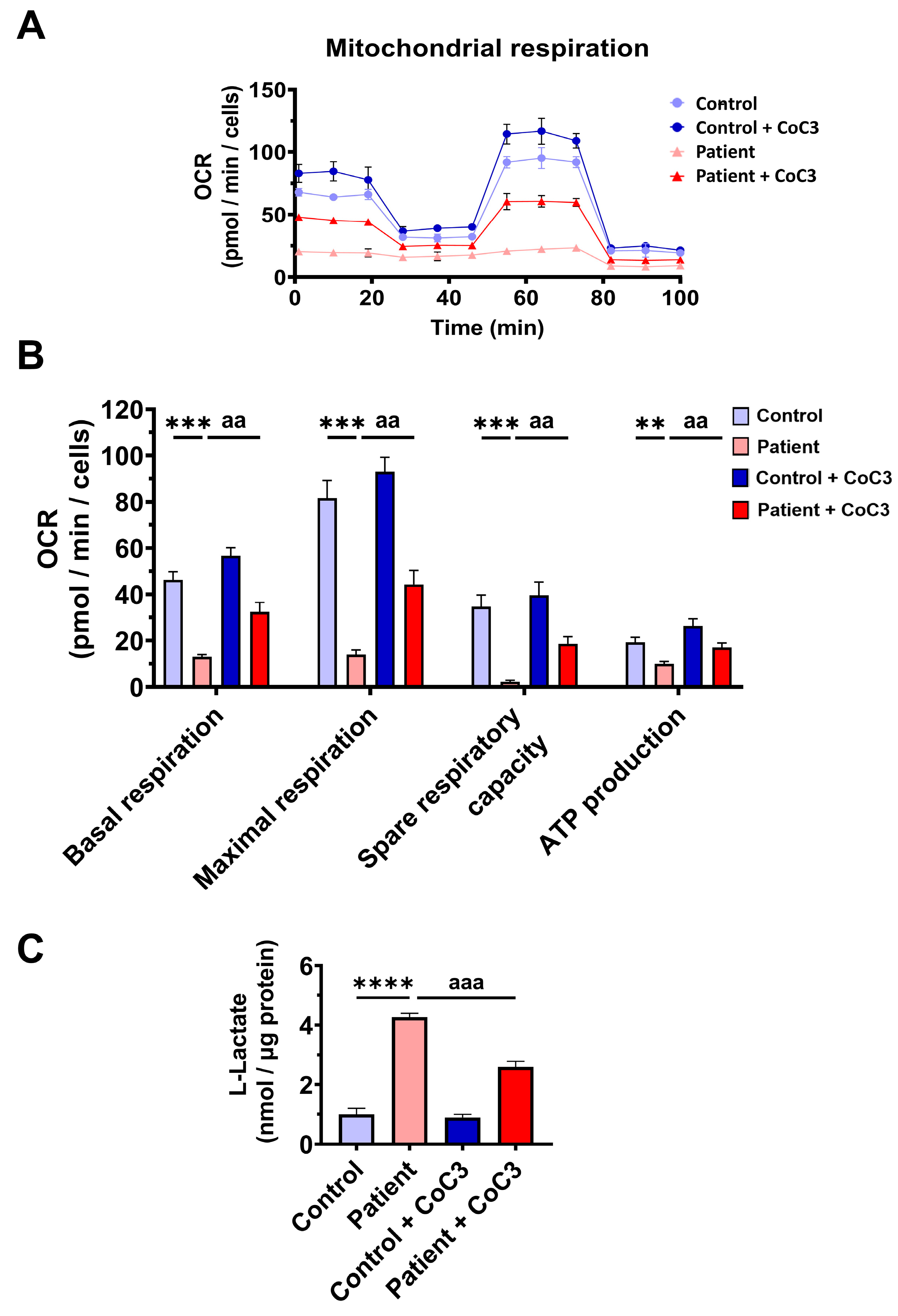
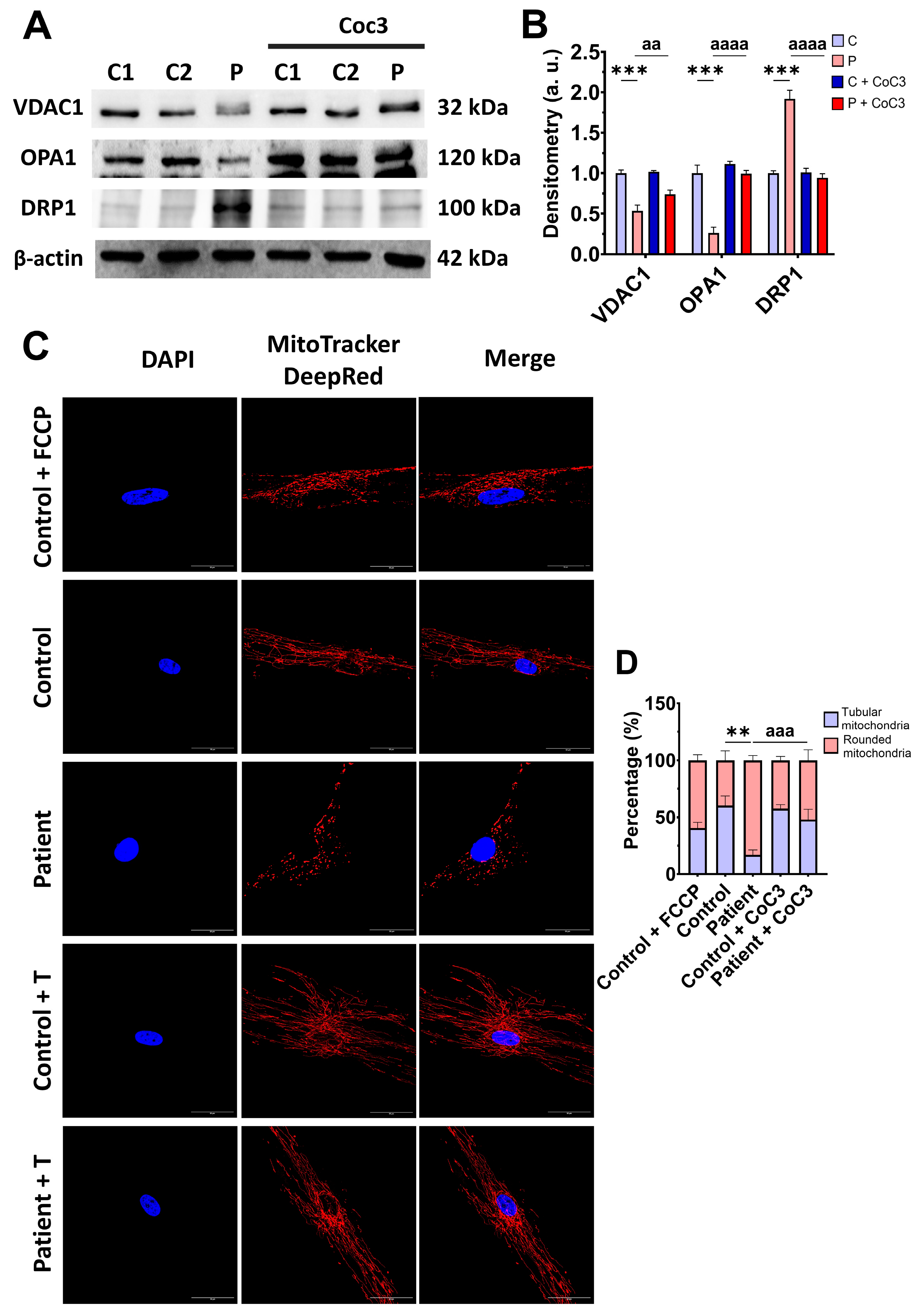
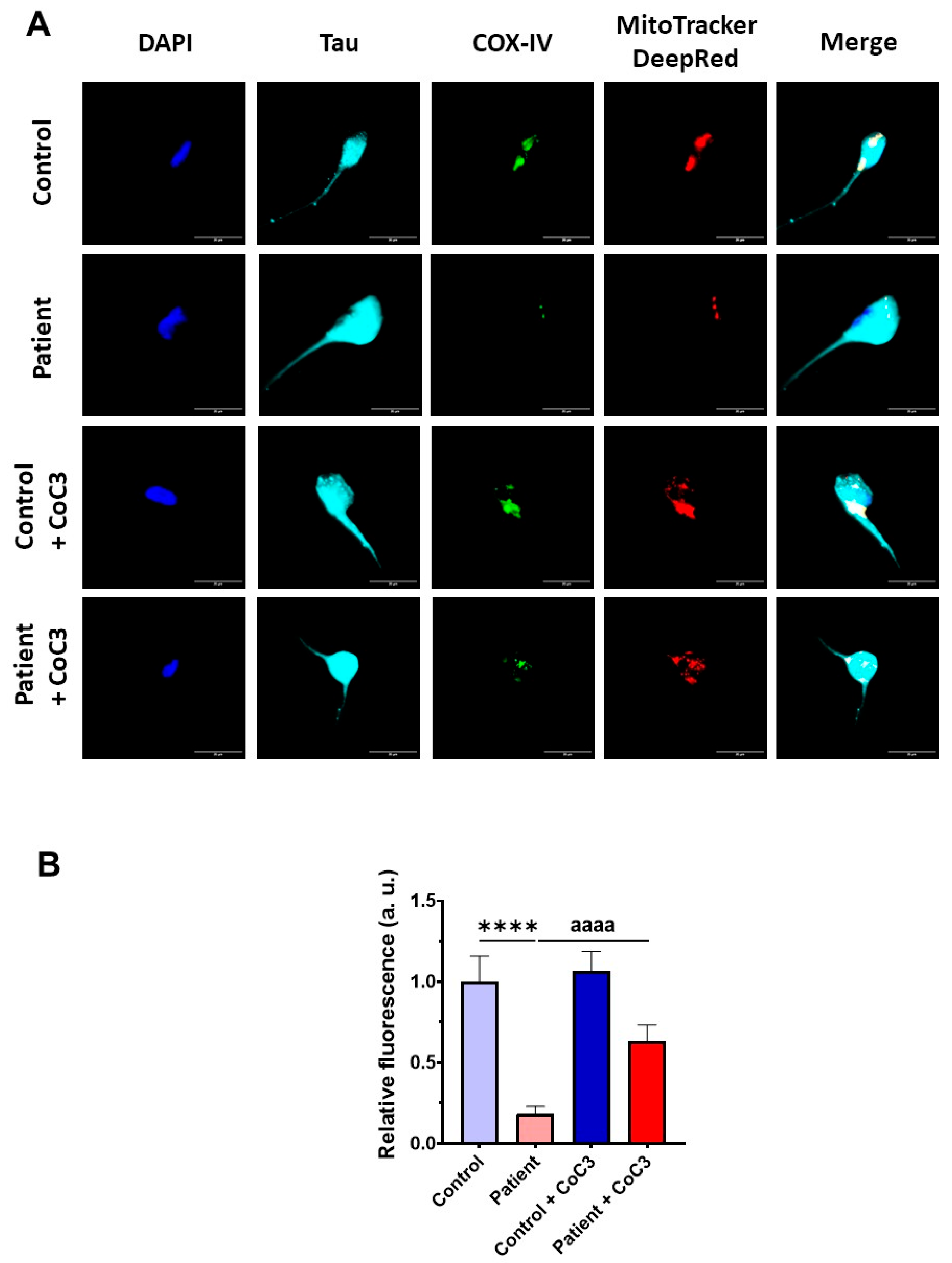
3.7. Cocktail CoC3 Treatment Increases Cytochrome C Oxidase Activity and Mitochondrial Bioenergetics and Dynamics in ETHE1 Mutant Fibroblasts
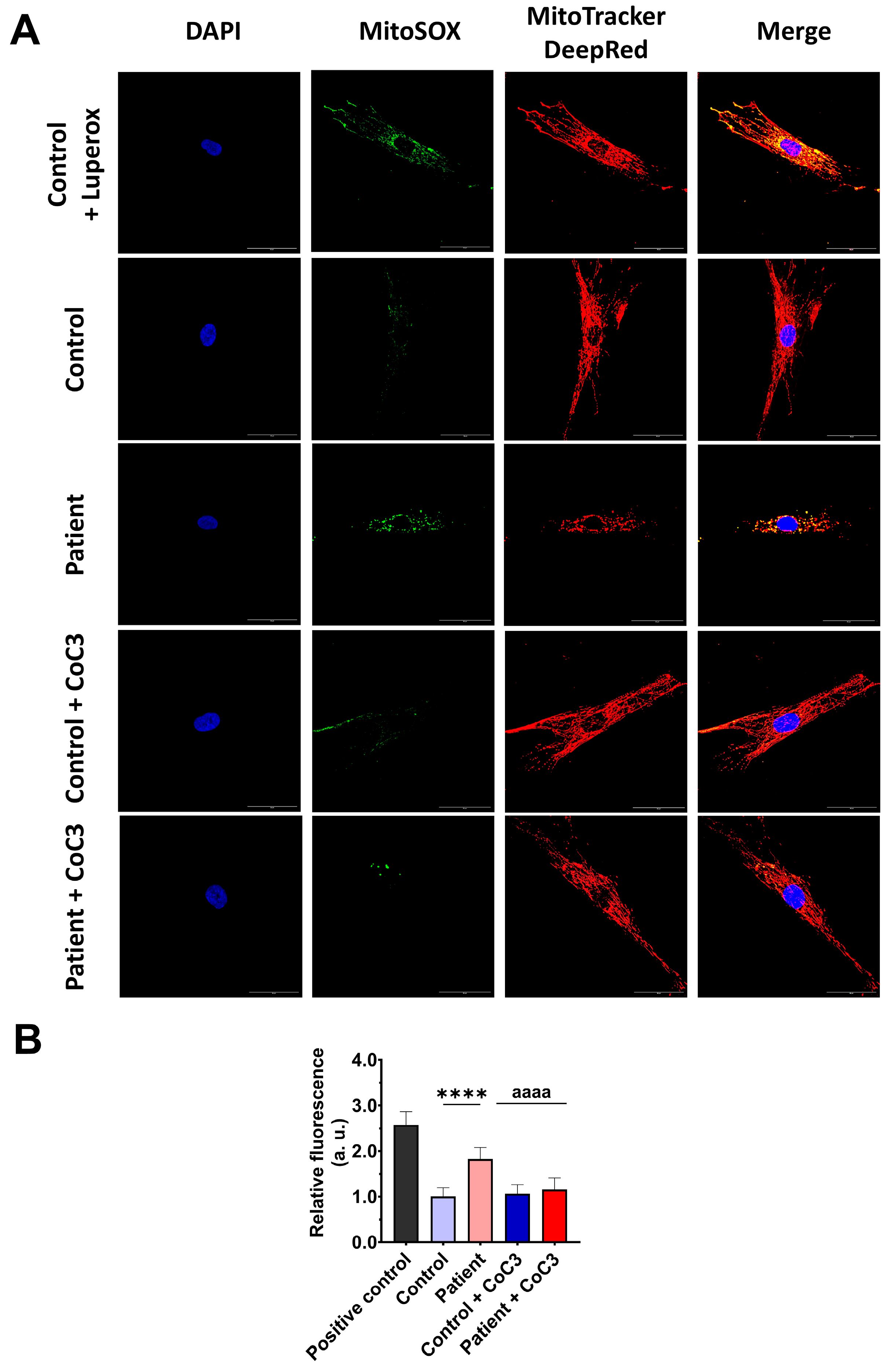
3.8. Cocktail CoC3 Treatment Improves Oxidative Stress and Lipid Peroxidation in EE Fibroblasts
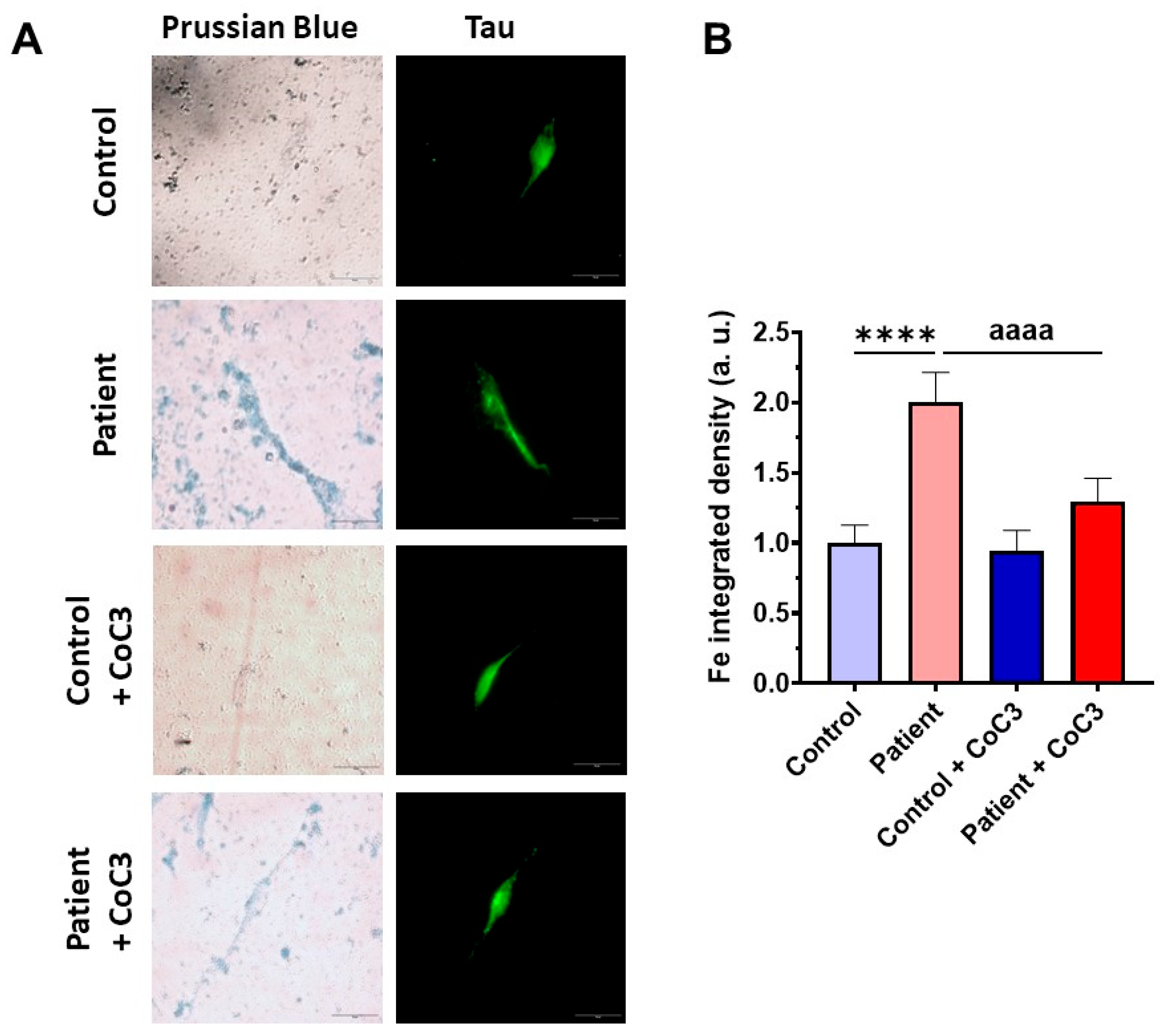
3.9. Cocktail Treatment Prevents the Accumulation of Iron and Lipofuscin in ETHE1 Mutant Fibroblasts
3.10. Functional Complementation with Wild-Type ETHE1 cDNA Reverts Pathophysiologic Characteristics in ETHE1 Mutant Fibroblasts
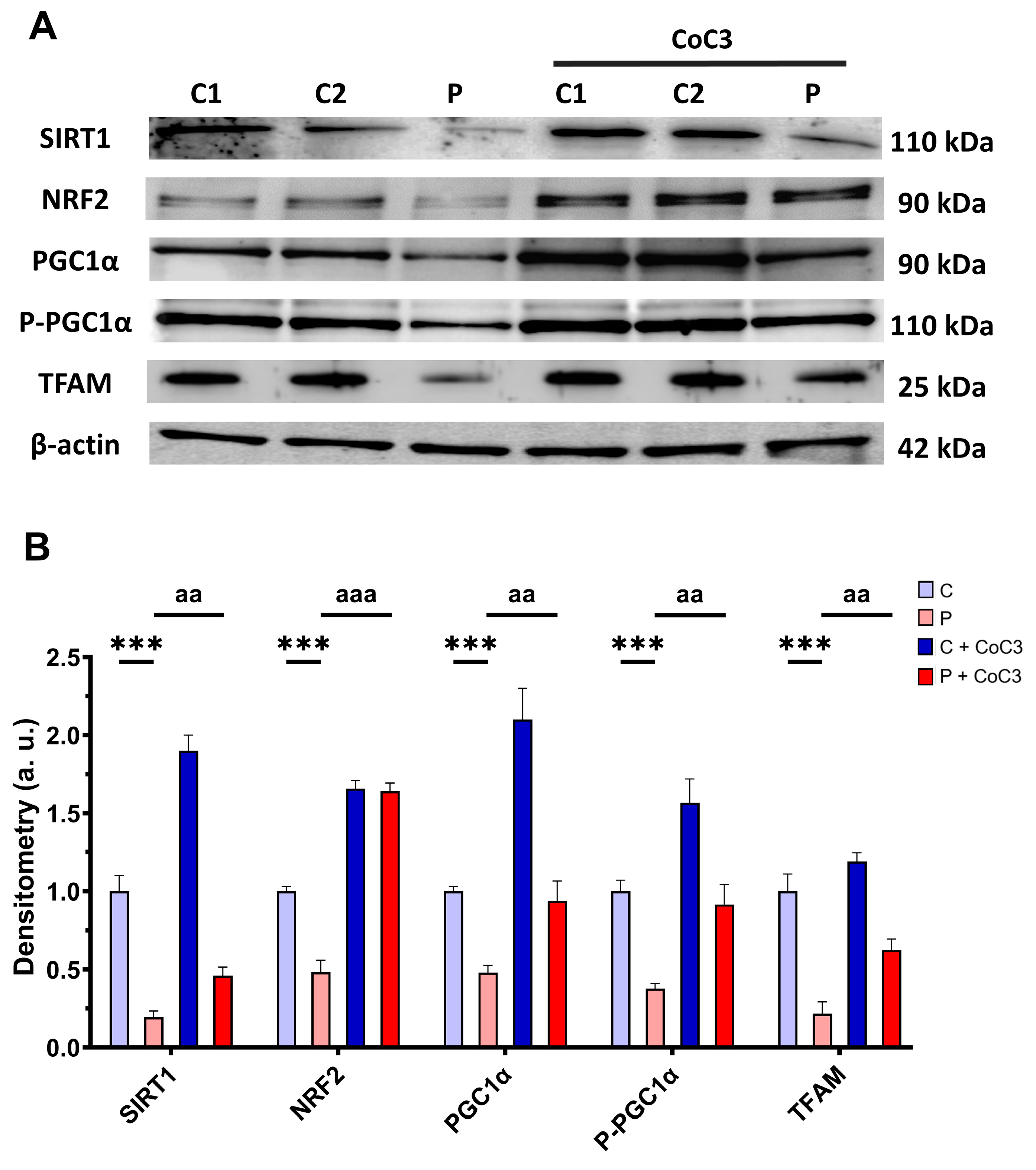
3.11. CoC3 Cocktail Treatment Activates the Canonical and SIRT3 Transcriptional Axis of mtUPR, as Well as Mitochondrial Biogenesis in ETHE1 Fibroblasts
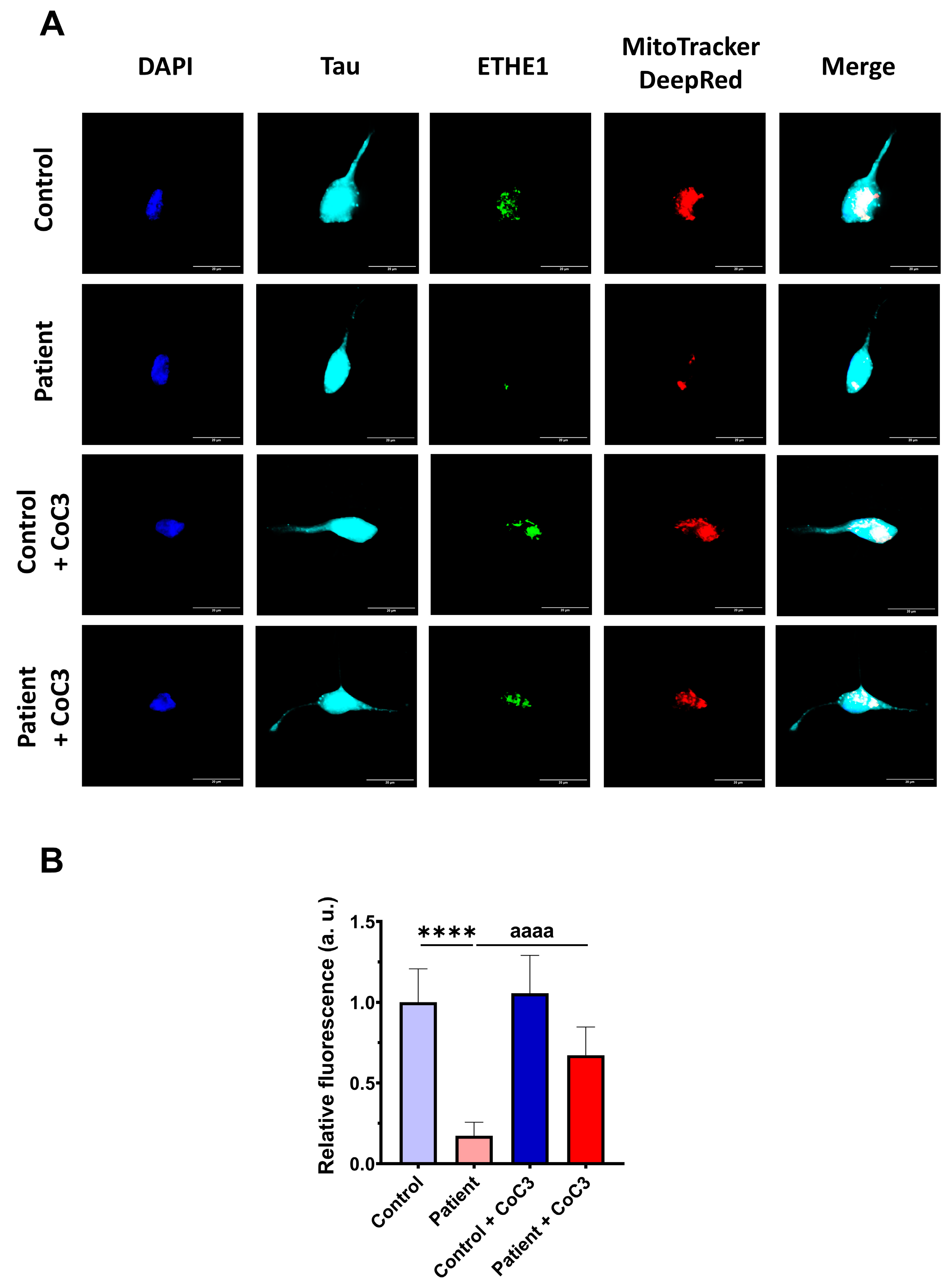
3.12. Induced Neurons
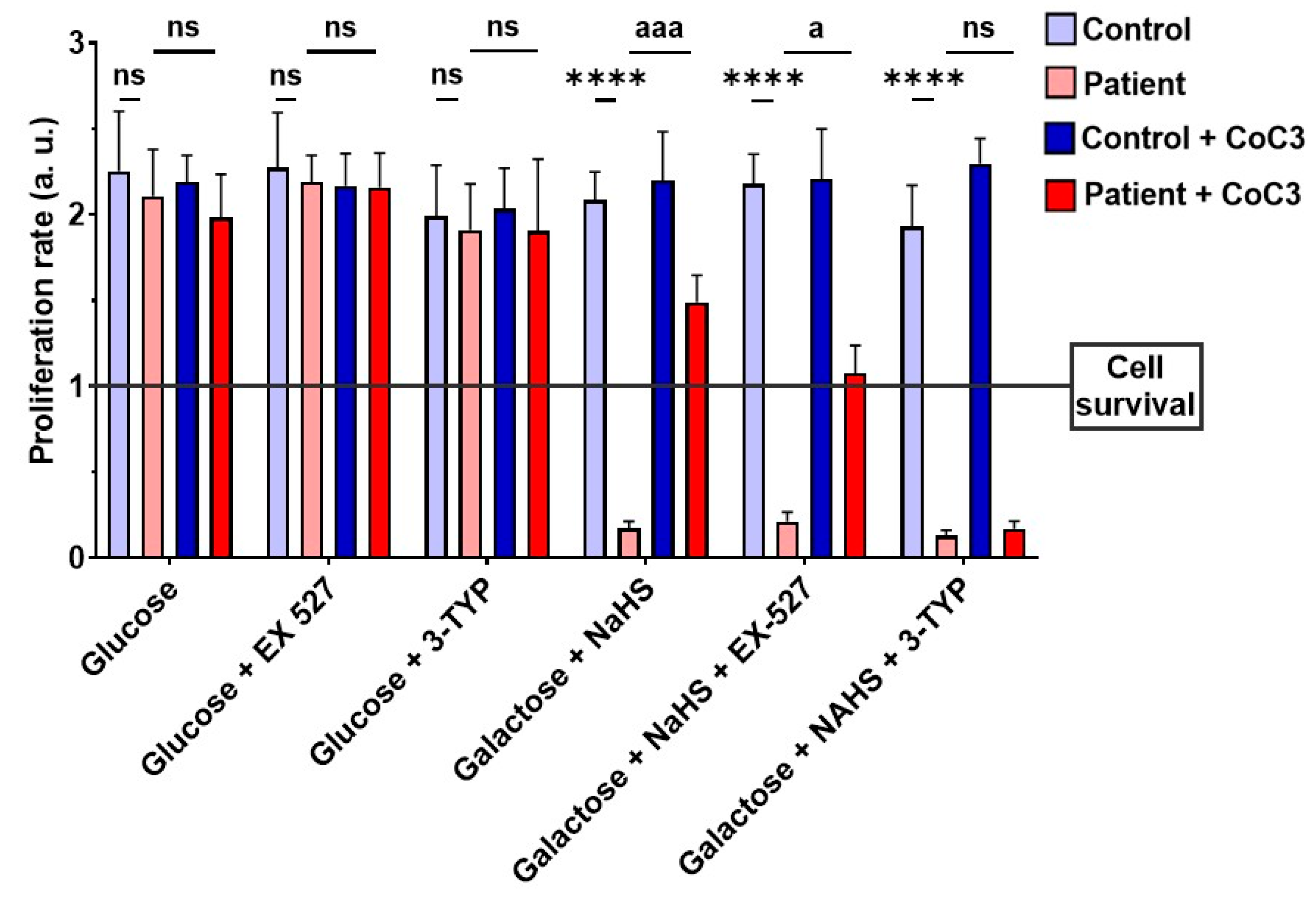
3.13. Effect of CoC3 Supplementations in Another Fibroblast Cell Line Derived from an EE Patient Recruited for This Work
4. Discussion
4.1. Consequences of H2S Accumulation
4.2. Protein Persulfidation
4.3. Mitochondrial Dysfunction, Iron/Lipofuscin Accumulation, and Lipid Peroxidation
4.4. mtUPR Activation and Improvement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-TYP | 3-(1H-1,2,3-triazol-4-yl) pyridine |
| α-LA | α-Lipoic acid |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| ATF5 | Activating transcription factor 5 |
| ATP | Adenosine triphosphate |
| BPAN | Beta-propeller protein-associated neurodegeneration |
| BSA | Bovine serum albumin |
| Calcein-AM | Calcein Acetoxymethyl ester |
| CBS | Cystathionine β-synthase |
| CO | Carbon monoxide |
| CoA | Coenzyme A |
| COX | Cytochrome c oxidase |
| COX-IV | Cytochrome c oxidase subunit IV |
| CSE | Cystathionine γ-lyase |
| Cy5 | Cyanine 5 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl solfoxide |
| DMT1 | Divalent Metal Transporter 1 |
| DNA | Desoxyrribonucleic acid |
| DRP1 | Dynamin-related protein 1 |
| EDTA | Ethylene Diamine Tetraacetic Acid |
| EE | Ethylmalonic encephalopathy |
| eif2α | Eukaryotic translation initiation factor 2 alpha |
| P-eif2α | Phosphorilated eukaryotic translation initiation factor 2 alpha |
| EMA | Ethylmalonic acid |
| ER | Endoplasmic reticulum |
| ETHE1 | Ethylmalonic encephalopathy 1 |
| FBS | Fetal bovine serum |
| FCCP | Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone |
| FM | Fluorescent marker |
| FTL | Ferritin light-chain |
| FXN | Frataxin |
| GPX4 | Glutathione Peroxidase 4 |
| GSH | Glutathione |
| GSSH | Glutathione persulfide |
| HBSS | Hanks’ Balanced Salt Solution |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| HRP | Horseradish peroxidase |
| HSP60 | Heat shock protein 60 |
| HSP70 | Heat shock protein 70 |
| IC50 | Half-maximal inhibitory concentration |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| iNs | Induced neurons |
| ISCU | Iron–sulfur cluster assembly enzyme |
| LC | Liquid chromatography |
| LIP | Labile iron pool |
| Luperox® DI | Luperox® Diisopropyl |
| LYRM4 | Lysine-rich motif containing 4 |
| MetAP2 | methionine aminopeptidase 2 |
| MDA | malondialdehyde |
| Mfrn2 | Mitoferrin 2 |
| MnSOD | Manganese superoxide dismutase |
| MOI | Multiplicity of infection |
| MRI | Magnetic resonance imaging |
| MS | Mass spectrometry |
| 3-MST | 3-mercaptopyruvate sulfotransferase |
| mt-CO2 | Mitochondrially encoded cytochrome c oxidase subunit 2 |
| mt-FTL | Mitochondrial ferritin light chain |
| mt-ND1 | Mitochondrially encoded NADH Dehydrogenase Subunit 1 |
| NAD | nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NCOA4 | Nuclear Receptor Coactivator 4 |
| NDUFS8 | NADH:Ubiquinone Oxidoreductase protein 8 |
| NFS1 | Nuclear Fe-S cluster protein 1 |
| NRF2 | Nuclear respiratory factor 2 |
| C1-NBF | 4-chloro-7-nitrobenzofurazan |
| NBIA | Neurodegeneration with Brain Iron Accumulation |
| NO | Nitric oxide |
| OCR | Oxygen consumption rate |
| OPA1 | Optic atrophy 1 |
| OR | Oxygen radicals |
| OXPHOS | Oxidative phosphorylation |
| PB | Phosphate buffer |
| PBS | Phosphate-buffered saline |
| PDH | Pyruvate dehydrogenase |
| PFA | Paraformaldehyde |
| PGK | Phosphoglycerate kinase |
| PKAN | Pantothenate-kinase-associated neurodegeneration |
| PLAN | Phospholipase-A2-associated neurodegeneration |
| PPB | Perl’s Prussian Blue |
| REST | RE1-Silencing Transcription factor |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RS | Reactive species |
| RSH | Cysteine residues |
| RSSH | Persulfided cysteine residues |
| RT | Room temperature |
| SCAD | Short-chain acyl-CoA dehydrogenase |
| SD | Standard deviation |
| SDS | Sodium Dodecyl Sulfate |
| SDS-PAGE | Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis |
| SH | Reduced sulfur |
| SIRT | Sirtuin |
| SIRT1 | Sirtuin 1 |
| SIRT3 | Sirtuin 3 |
| SOD1 | Superoxide dismutase 1 |
| SQOR | Sulfo-quinone oxidoreductase |
| SSH | Sulfane sulfur |
| TBTA | Tris(benzyltriazolylmethyl)amine |
| TCA | Tricarboxylic acid |
| TEM | Transmission electron microscopy |
| TfR | Tranferrin receptor |
| TTBS | Tris-Buffered Saline with Tween 20 |
| UPLC | Ultra-Performance Liquid Chromatography |
| VDAC1 | Voltage-dependent anion channel 1 |
References
- Tiranti, V.; Briem, E.; Lamantea, E.; Mineri, R.; Papaleo, E.; De Gioia, L.; Forlani, F.; Rinaldo, P.; Dickson, P.; Abu-Libdeh, B.; et al. ETHE1 mutations are specific to ethylmalonic encephalopathy. J. Med. Genet. 2006, 43, 340–346. [Google Scholar] [CrossRef]
- Platt, I.; Bisgin, A.; Kilavuz, S. Ethylmalonic Encephalopathy: A literature review and two new cases of mild phenotype. Neurol. Sci. 2023, 44, 3827–3852. [Google Scholar] [CrossRef]
- Mineri, R.; Rimoldi, M.; Burlina, A.B.; Koskull, S.; Perletti, C.; Heese, B.; von Dobeln, U.; Mereghetti, P.; Di Meo, I.; Invernizzi, F.; et al. Identification of new mutations in the ETHE1 gene in a cohort of 14 patients presenting with ethylmalonic encephalopathy. J. Med. Genet. 2008, 45, 473–478. [Google Scholar] [CrossRef]
- Lim, J.; Shayota, B.J.; Lay, E.; Elsea, S.H.; Bekheirnia, M.R.; Tessier, M.E.M.; Kralik, S.F.; Rice, G.M.; Soler-Alfonso, C.; Scaglia, F. Acute Strokelike Presentation and Long-term Evolution of Diffusion Restriction Pattern in Ethylmalonic Encephalopathy. J. Child. Neurol. 2021, 36, 841–852. [Google Scholar] [CrossRef]
- Cardelo Autero, N.; Cordon Martinez, A.M.; Ramos-Fernandez, J.M. Ethylmalonic encephalopathy: Phenotype-genotype description and review of its management. Neurologia 2021, 36, 729–731. [Google Scholar] [CrossRef]
- Grings, M.; Wajner, M.; Leipnitz, G. Mitochondrial Dysfunction and Redox Homeostasis Impairment as Pathomechanisms of Brain Damage in Ethylmalonic Encephalopathy: Insights from Animal and Human Studies. Cell Mol. Neurobiol. 2022, 42, 565–575. [Google Scholar] [CrossRef]
- Tiranti, V.; Zeviani, M. Altered sulfide (H(2)S) metabolism in ethylmalonic encephalopathy. Cold Spring Harb. Perspect. Biol. 2013, 5, a011437. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef]
- Kabil, O.; Motl, N.; Banerjee, R. H2S and its role in redox signaling. Biochim. Biophys. Acta 2014, 1844, 1355–1366. [Google Scholar] [CrossRef]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010, 16, 869–871. [Google Scholar] [CrossRef]
- Ellis, M.F. Use of Bicarbonate in Patients With Metabolic Acidosis. Crit. Care Nurse 2015, 35, 73–75. [Google Scholar] [CrossRef]
- Olivieri, G.; Martinelli, D.; Longo, D.; Grimaldi, C.; Liccardo, D.; Di Meo, I.; Pietrobattista, A.; Sidorina, A.; Semeraro, M.; Dionisi-Vici, C. Ethylmalonic encephalopathy and liver transplantation: Long-term outcome of the first treated patient. Orphanet J. Rare Dis. 2021, 16, 229. [Google Scholar] [CrossRef]
- Zhou, G.P.; Qu, W.; Zhu, Z.J.; Sun, L.Y.; Wei, L.; Zeng, Z.G.; Liu, Y. Compromised therapeutic value of pediatric liver transplantation in ethylmalonic encephalopathy: A case report. World J. Gastroenterol. 2020, 26, 6295–6303. [Google Scholar] [CrossRef]
- Tao, Y.; Han, D.; Li, X.; Wang, L.; Yue, L.; Huang, C.; Lu, D.; Li, X. Identification of a novel homozygous nonsense variant in a Chinese patient with ethylmalonic encephalopathy and a genotype-phenotype spectrum review. Clin. Chim. Acta 2020, 509, 8–17. [Google Scholar] [CrossRef]
- Henriques, B.J.; Lucas, T.G.; Rodrigues, J.V.; Frederiksen, J.H.; Teixeira, M.S.; Tiranti, V.; Bross, P.; Gomes, C.M. Ethylmalonic encephalopathy ETHE1 R163W/R163Q mutations alter protein stability and redox properties of the iron centre. PLoS ONE 2014, 9, e107157. [Google Scholar] [CrossRef]
- Gomez-Fernandez, D.; Romero-Gonzalez, A.; Suarez-Rivero, J.M.; Cilleros-Holgado, P.; Alvarez-Cordoba, M.; Pinero-Perez, R.; Romero-Dominguez, J.M.; Reche-Lopez, D.; Lopez-Cabrera, A.; Ibanez-Mico, S.; et al. A Multi-Target Pharmacological Correction of a Lipoyltransferase LIPT1 Gene Mutation in Patient-Derived Cellular Models. Antioxidants 2024, 13, 1023. [Google Scholar] [CrossRef]
- Tan, B.; Jin, S.; Sun, J.; Gu, Z.; Sun, X.; Zhu, Y.; Huo, K.; Cao, Z.; Yang, P.; Xin, X.; et al. New method for quantification of gasotransmitter hydrogen sulfide in biological matrices by LC-MS/MS. Sci. Rep. 2017, 7, 46278. [Google Scholar] [CrossRef] [PubMed]
- Gallo-Oller, G.; Ordoñez, R.; Dotor, J. A new background subtraction method for Western blot densitometry band quantification through image analysis software. J. Immunol. Methods 2018, 457, 1–5. [Google Scholar] [CrossRef]
- Alcocer-Gomez, E.; Garrido-Maraver, J.; Bullon, P.; Marin-Aguilar, F.; Cotan, D.; Carrion, A.M.; Alvarez-Suarez, J.M.; Giampieri, F.; Sanchez-Alcazar, J.A.; Battino, M.; et al. Metformin and caloric restriction induce an AMPK-dependent restoration of mitochondrial dysfunction in fibroblasts from Fibromyalgia patients. Biochim. Biophys. Acta 2015, 1852, 1257–1267. [Google Scholar] [CrossRef]
- Pap, E.H.; Drummen, G.P.; Winter, V.J.; Kooij, T.W.; Rijken, P.; Wirtz, K.W.; Op den Kamp, J.A.; Hage, W.J.; Post, J.A. Ratio-fluorescence microscopy of lipid oxidation in living cells using C11-BODIPY(581/591). FEBS Lett. 1999, 453, 278–282. [Google Scholar] [CrossRef]
- Ghio, A.J.; Roggli, V.L. Perls’ Prussian Blue Stains of Lung Tissue, Bronchoalveolar Lavage, and Sputum. J. Environ. Pathol. Toxicol. Oncol. 2021, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Legault, E.M.; Drouin-Ouellet, J. Generation of Induced Dopaminergic Neurons from Human Fetal Fibroblasts. Methods Mol. Biol. 2021, 2352, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Bruzelius, A.; Kidnapillai, S.; Drouin-Ouellet, J.; Stoker, T.; Barker, R.A.; Rylander Ottosson, D. Reprogramming Human Adult Fibroblasts into GABAergic Interneurons. Cells 2021, 10, 3450. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef]
- Kamalian, L.; Douglas, O.; Jolly, C.E.; Snoeys, J.; Simic, D.; Monshouwer, M.; Williams, D.P.; Park, B.K.; Chadwick, A.E. Acute Metabolic Switch Assay Using Glucose/Galactose Medium in HepaRG Cells to Detect Mitochondrial Toxicity. Curr. Protoc. Toxicol. 2019, 80, e76. [Google Scholar] [CrossRef]
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef]
- He, B.; Zhang, Z.; Huang, Z.; Duan, X.; Wang, Y.; Cao, J.; Li, L.; He, K.; Nice, E.C.; He, W.; et al. Protein persulfidation: Rewiring the hydrogen sulfide signaling in cell stress response. Biochem. Pharmacol. 2023, 209, 115444. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-Gonzalez, A.; Gomez-Fernandez, D.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination With Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharmacol. 2022, 13, 862085. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gomez-Fernandez, D.; Pinero-Perez, R.; Reche-Lopez, D.; Alvarez-Cordoba, M.; Munuera-Cabeza, M.; Talaveron-Rey, M.; Povea-Cabello, S.; Suarez-Carrillo, A.; Romero-Gonzalez, A.; et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines 2022, 10, 1611. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.Y.; Mitoma, H.; Fatemi, A. Metabolic ataxias. Handb. Clin. Neurol. 2018, 155, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155. [Google Scholar] [CrossRef]
- Ersoy, M.; Tiranti, V.; Zeviani, M. Ethylmalonic encephalopathy: Clinical course and therapy response in an uncommon mild case with a severe ETHE1 mutation. Mol. Genet. Metab. Rep. 2020, 25, 100641. [Google Scholar] [CrossRef]
- Tiranti, V.; D’Adamo, P.; Briem, E.; Ferrari, G.; Mineri, R.; Lamantea, E.; Mandel, H.; Balestri, P.; Garcia-Silva, M.T.; Vollmer, B.; et al. Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am. J. Hum. Genet. 2004, 74, 239–252. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Augoustides-Savvopoulou, P.; Haas, D.; Smet, J.; Triantafyllou, P.; Vargiami, E.; Tamiolaki, M.; Gombakis, N.; van Coster, R.; Sewell, A.C.; et al. Ethylmalonic encephalopathy: Clinical and biochemical observations. Neuropediatrics 2007, 38, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, Z.H.; Ren, Z.; Qu, S.L.; Liu, M.H.; Liu, L.S.; Jiang, Z.S. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol. Cell Biol. 2013, 33, 1104–1113. [Google Scholar] [CrossRef]
- Wang, R. Gasotransmitters: Growing pains and joys. Trends Biochem. Sci. 2014, 39, 227–232. [Google Scholar] [CrossRef]
- Di Meo, I.; Lamperti, C.; Tiranti, V. Mitochondrial diseases caused by toxic compound accumulation: From etiopathology to therapeutic approaches. EMBO Mol. Med. 2015, 7, 1257–1266. [Google Scholar] [CrossRef]
- Petrovic, D.; Kouroussis, E.; Vignane, T.; Filipovic, M.R. The Role of Protein Persulfidation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2021, 13, 674135. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. H2S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H2S: A Novel Gasotransmitter that Signals by Sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef]
- Luo, S.; Kong, C.; Ye, D.; Liu, X.; Wang, Y.; Meng, G.; Han, Y.; Xie, L.; Ji, Y. Protein Persulfidation: Recent Progress and Future Directions. Antioxid. Redox Signal. 2023, 39, 829–852. [Google Scholar] [CrossRef]
- Barschak, A.G.; Ferreira Gda, C.; Andre, K.R.; Schuck, P.F.; Viegas, C.M.; Tonin, A.; Dutra Filho, C.S.; Wyse, A.T.; Wannmacher, C.M.; Vargas, C.R.; et al. Inhibition of the electron transport chain and creatine kinase activity by ethylmalonic acid in human skeletal muscle. Metab. Brain Dis. 2006, 21, 11–19. [Google Scholar] [CrossRef]
- Nicholls, P.; Marshall, D.C.; Cooper, C.E.; Wilson, M.T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 2013, 41, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Rockfield, S.; Chhabra, R.; Robertson, M.; Rehman, N.; Bisht, R.; Nanjundan, M. Links Between Iron and Lipids: Implications in Some Major Human Diseases. Pharmaceuticals 2018, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Bilska-Wilkosz, A.; Iciek, M.; Gorny, M. Chemistry and Biochemistry Aspects of the 4-Hydroxy-2,3-trans-nonenal. Biomolecules 2022, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Guo, X.; Sun, Y. Iron Accumulation and Lipid Peroxidation in the Aging Retina: Implication of Ferroptosis in Age-Related Macular Degeneration. Aging Dis. 2021, 12, 529–551. [Google Scholar] [CrossRef]
- Villalón-García, I.; Álvarez-Córdoba, M.; Povea-Cabello, S.; Talaverón-Rey, M.; Villanueva-Paz, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinós, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [CrossRef]
- Arber, C.E.; Li, A.; Houlden, H.; Wray, S. Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathol. Appl. Neurobiol. 2016, 42, 220–241. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef]
- Beck, G.; Shinzawa, K.; Hayakawa, H.; Baba, K.; Yasuda, T.; Sumi-Akamaru, H.; Tsujimoto, Y.; Mochizuki, H. Deficiency of Calcium-Independent Phospholipase A2 Beta Induces Brain Iron Accumulation through Upregulation of Divalent Metal Transporter 1. PLoS ONE 2016, 10, e0141629. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Wani, W.Y.; Redmann, M.; Benavides, G.A.; Johnson, M.S.; Ouyang, X.; Cofield, S.S.; Mitra, K.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy 2017, 13, 1828–1840. [Google Scholar] [CrossRef]
- Frolova, M.S.; Surin, A.M.; Braslavski, A.V.; Vekshin, N.L. Degradation of Mitochondria to Lipofuscin upon Heating and Illumination. Biofizika 2015, 60, 1125–1131. [Google Scholar] [CrossRef]
- Ilie, O.-D.; Ciobica, A.; Riga, S.; Dhunna, N.; McKenna, J.; Mavroudis, I.; Doroftei, B.; Ciobanu, A.-M.; Riga, D. Mini-Review on Lipofuscin and Aging: Focusing on The Molecular Interface, The Biological Recycling Mechanism, Oxidative Stress, and The Gut-Brain Axis Functionality. Medicina 2020, 56, 626. [Google Scholar] [CrossRef]
- Milward, E.; Acikyol, B.; Bassett, B.; Williams, E.; Graham, R.; Delima, R.; Trinder, D.; Olynyk, J.; Johnstone, D. Brain changes in iron loading disorders. In Metal Ions in Neurological Systems; Linert, W., Kozlowski, H., Eds.; Springer-Verlag: Vienna, Austria, 2012; pp. 17–29. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, G.M.F.; Pletsch, J.T.; Parmeggiani, B.; Grings, M.; Glanzel, N.M.; Bobermin, L.D.; Amaral, A.U.; Wajner, M.; Leipnitz, G. Bioenergetics dysfunction, mitochondrial permeability transition pore opening and lipid peroxidation induced by hydrogen sulfide as relevant pathomechanisms underlying the neurological dysfunction characteristic of ethylmalonic encephalopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2192–2201. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, A.; Ali, S.; Saha, A.; Haushalter, K.J.; Lam, W.L.; Glasheen, M.; Parker, J.; Brenner, M.; Mahon, S.B.; et al. Hydrogen Sulfide--Mechanisms of Toxicity and Development of an Antidote. Sci. Rep. 2016, 6, 20831. [Google Scholar] [CrossRef]
- Höhn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef]
- Villalon-Garcia, I.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Talaveron-Rey, M.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Pinero-Perez, R.; et al. Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration. Neural Regen. Res. 2023, 18, 1196–1202. [Google Scholar] [CrossRef]
- Sitte, N.; Merker, K.; Grune, T.; Zglinicki, T.v. Lipofuscin accumulation in proliferating fibroblasts in vitro: An indicator of oxidative stress. Exp. Gerontol. 2001, 36, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.N.; Crane, J.S. Histology, Lipofuscin; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Jolly, R.D.; Douglas, B.V.; Davey, P.M.; Roiri, J.E. Lipofuscin in bovine muscle and brain: A model for studying age pigment. Gerontology 1995, 41 (Suppl. S2), 283–295. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: Role in senescent cells. Free Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef]
- Kakimoto, Y.; Okada, C.; Kawabe, N.; Sasaki, A.; Tsukamoto, H.; Nagao, R.; Osawa, M. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci. Rep. 2019, 9, 3304. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current Questions. Expert. Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef]
- Bindoli, A. Lipid peroxidation in mitochondria. Free Radic. Biol. Med. 1988, 5, 247–261. [Google Scholar] [CrossRef]
- Alvarez-Cordoba, M.; Talaveron-Rey, M.; Povea-Cabello, S.; Cilleros-Holgado, P.; Gomez-Fernandez, D.; Pinero-Perez, R.; Reche-Lopez, D.; Munuera-Cabeza, M.; Suarez-Carrillo, A.; Romero-Gonzalez, A.; et al. Patient-Derived Cellular Models for Polytarget Precision Medicine in Pantothenate Kinase-Associated Neurodegeneration. Pharmaceuticals 2023, 16, 1359. [Google Scholar] [CrossRef]
- Suarez-Carrillo, A.; Alvarez-Cordoba, M.; Romero-Gonzalez, A.; Talaveron-Rey, M.; Povea-Cabello, S.; Cilleros-Holgado, P.; Pinero-Perez, R.; Reche-Lopez, D.; Gomez-Fernandez, D.; Romero-Dominguez, J.M.; et al. Antioxidants Prevent Iron Accumulation and Lipid Peroxidation, but Do Not Correct Autophagy Dysfunction or Mitochondrial Bioenergetics in Cellular Models of BPAN. Int. J. Mol. Sci. 2023, 24, 14576. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef]
- Meng, H.; Yan, W.Y.; Lei, Y.H.; Wan, Z.; Hou, Y.Y.; Sun, L.K.; Zhou, J.P. SIRT3 Regulation of Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Aging Neurosci. 2019, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Huang, Y.; Huang, R.Q.; Huang, C.G.; Wang, W.H.; Gu, J.M.; Dong, Y. SIRT3 Expression Decreases with Reactive Oxygen Species Generation in Rat Cortical Neurons during Early Brain Injury Induced by Experimental Subarachnoid Hemorrhage. BioMed Res. Int. 2016, 2016, 8263926. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.; Santoro, A.; Salvioli, S. Mitochondria and mitochondria-induced signalling molecules as longevity determinants. Mech. Ageing Dev. 2017, 165, 115–128. [Google Scholar] [CrossRef]
- Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response-synchronizing genomes. Curr. Opin. Cell Biol. 2015, 33, 74–81. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. The NAD+/PARP1/SIRT1 Axis in Aging. Rejuvenation Res. 2017, 20, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Zhang, X.; Xin, Z.; Xu, B.; Jin, Z.; Wu, J.; Hu, W.; Yang, Y. Does perturbation in the mitochondrial protein folding pave the way for neurodegeneration diseases? Ageing Res. Rev. 2020, 57, 100997. [Google Scholar] [CrossRef]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Chen, Y.; Aponte, A.M.; Battaglia, V.; Gucek, M.; Sack, M.N. Prolonged fasting identifies heat shock protein 10 as a Sirtuin 3 substrate: Elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J. Biol. Chem. 2015, 290, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Domínguez, J.M.; Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Romero-González, A.; Álvarez-Córdoba, M.; López-Cabrera, A.; De Oliveira, M.C.; Rodríguez-Sacristán, A.; et al. Mitochondrial Unfolded Protein Response (mtUPR) Activation Improves Pathological Alterations in Cellular Models of Ethylmalonic Encephalopathy. Antioxidants 2025, 14, 741. https://doi.org/10.3390/antiox14060741
Romero-Domínguez JM, Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, Reche-López D, Romero-González A, Álvarez-Córdoba M, López-Cabrera A, De Oliveira MC, Rodríguez-Sacristán A, et al. Mitochondrial Unfolded Protein Response (mtUPR) Activation Improves Pathological Alterations in Cellular Models of Ethylmalonic Encephalopathy. Antioxidants. 2025; 14(6):741. https://doi.org/10.3390/antiox14060741
Chicago/Turabian StyleRomero-Domínguez, José Manuel, Paula Cilleros-Holgado, David Gómez-Fernández, Rocío Piñero-Pérez, Diana Reche-López, Ana Romero-González, Mónica Álvarez-Córdoba, Alejandra López-Cabrera, Marta Castro De Oliveira, Andrés Rodríguez-Sacristán, and et al. 2025. "Mitochondrial Unfolded Protein Response (mtUPR) Activation Improves Pathological Alterations in Cellular Models of Ethylmalonic Encephalopathy" Antioxidants 14, no. 6: 741. https://doi.org/10.3390/antiox14060741
APA StyleRomero-Domínguez, J. M., Cilleros-Holgado, P., Gómez-Fernández, D., Piñero-Pérez, R., Reche-López, D., Romero-González, A., Álvarez-Córdoba, M., López-Cabrera, A., De Oliveira, M. C., Rodríguez-Sacristán, A., González-Granero, S., García-Verdugo, J. M., Aroca, A., & Sánchez-Alcázar, J. A. (2025). Mitochondrial Unfolded Protein Response (mtUPR) Activation Improves Pathological Alterations in Cellular Models of Ethylmalonic Encephalopathy. Antioxidants, 14(6), 741. https://doi.org/10.3390/antiox14060741