Avenanthramide C Prevents Neuronal Apoptosis via PI3K/Akt/GSK3β Signaling Pathway Following Middle Cerebral Artery Occlusion



Abstract
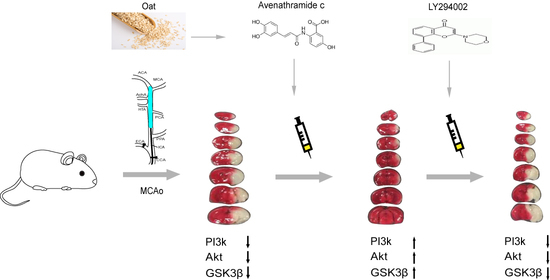
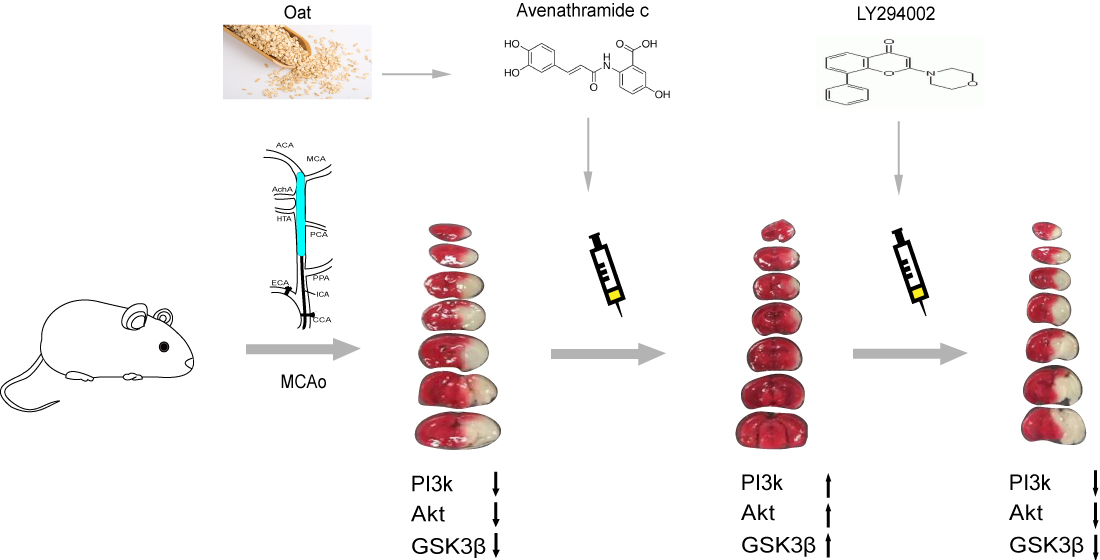{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Preparation and Study Model
2.1.1. Animals and Study Approval
2.1.2. Middle Cerebral Artery Occlusion (MCAo) Model
2.1.3. Drug Dosing and Delivery
2.2. Evaluation of Neurologic Function and Infarct Volume
2.3. Western Blot Analysis
2.4. Statistical Analysis
3. Results
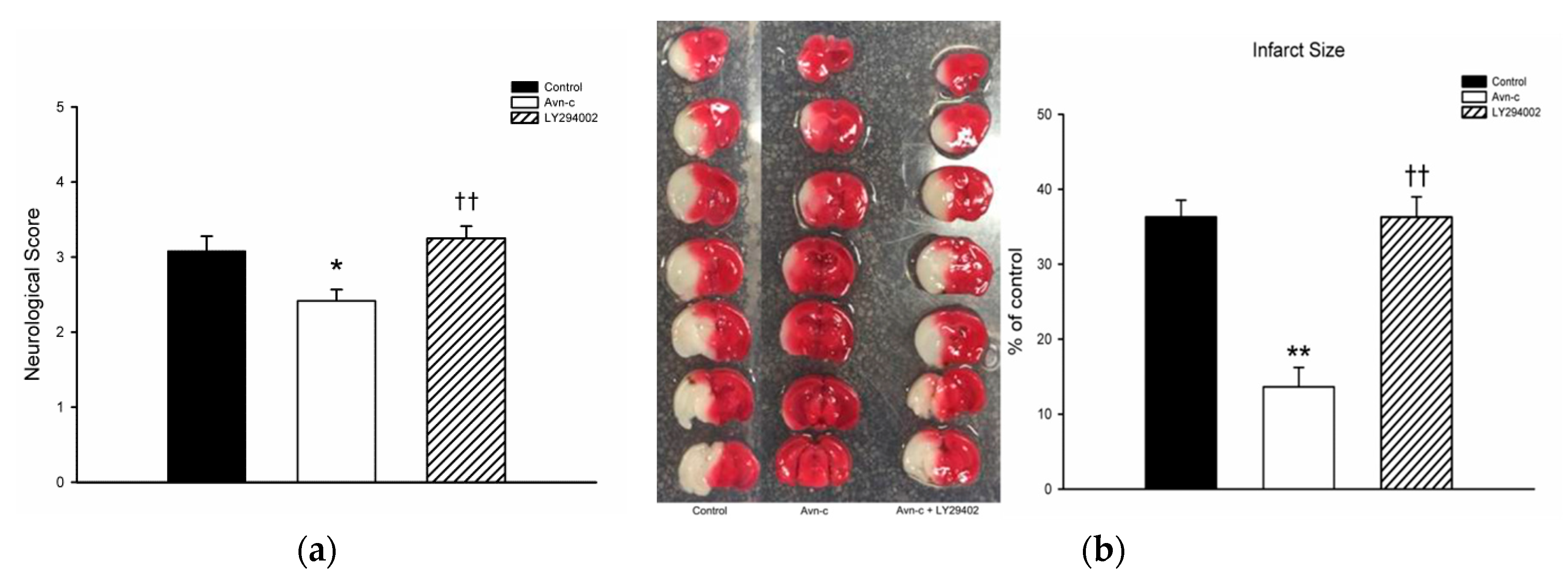
3.1. Avn-c Treatment Improved Neurologic Function and Reduced Cerebral Infarct Volume and Inhibition of PI3K Signaling Abolished the Effects of Avn-c on Cerebral Infarction
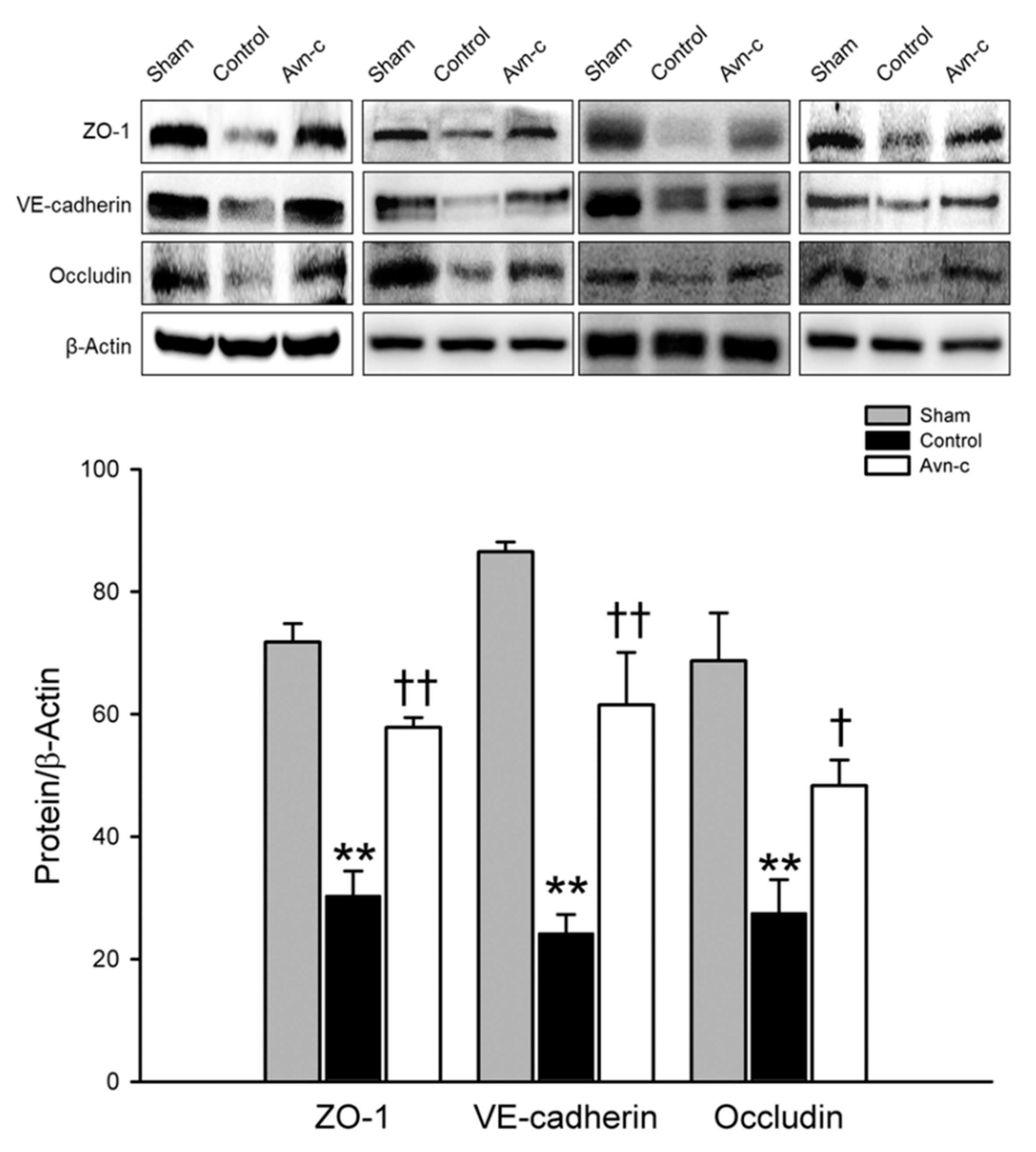
3.2. Avn-c Prevents MCAo Induced Disruption of the Blood-Brain Barrier
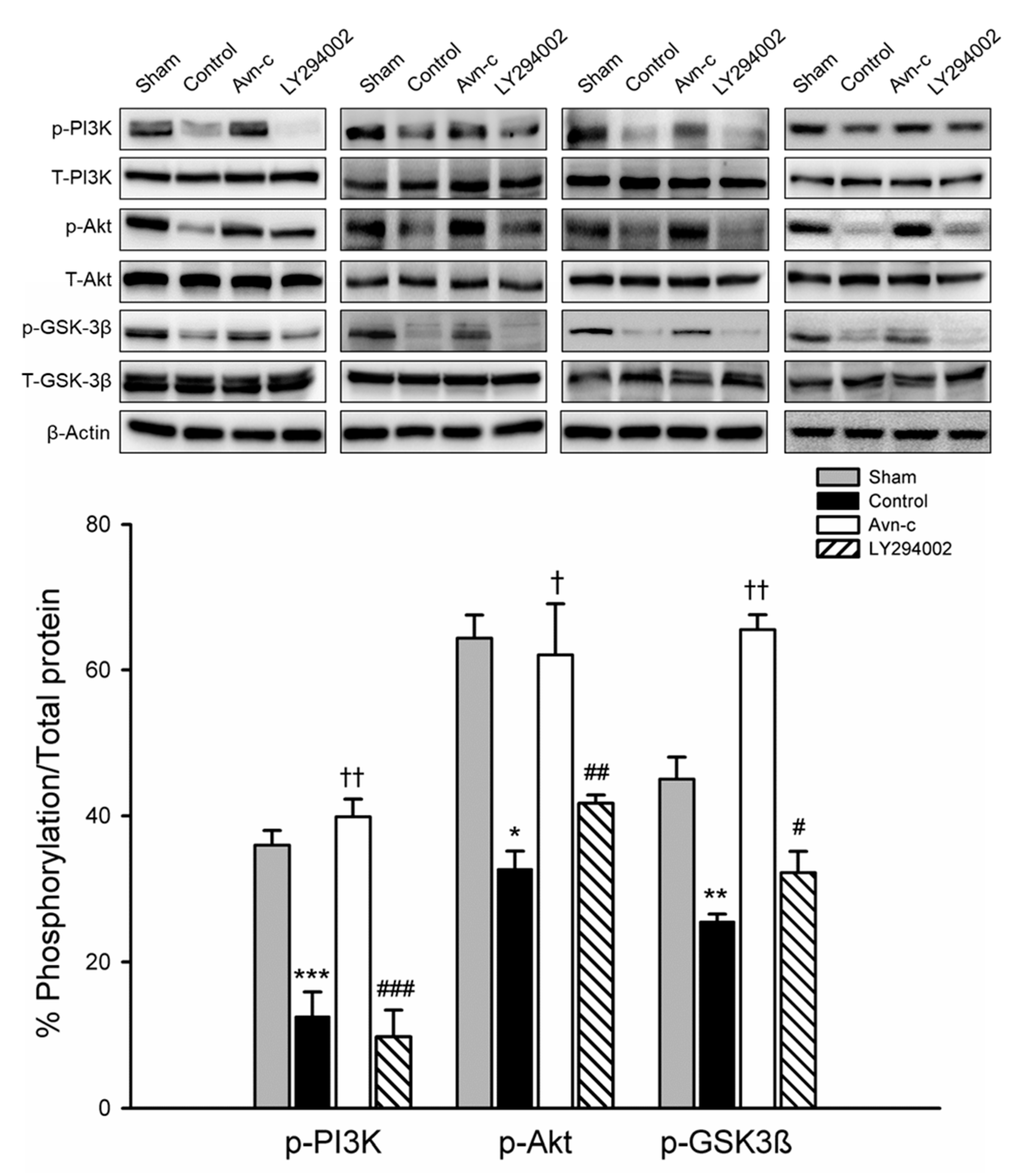
3.3. Avn-c Prevents MCAo Induced Inhibition of the PI3K/Akt Pathway
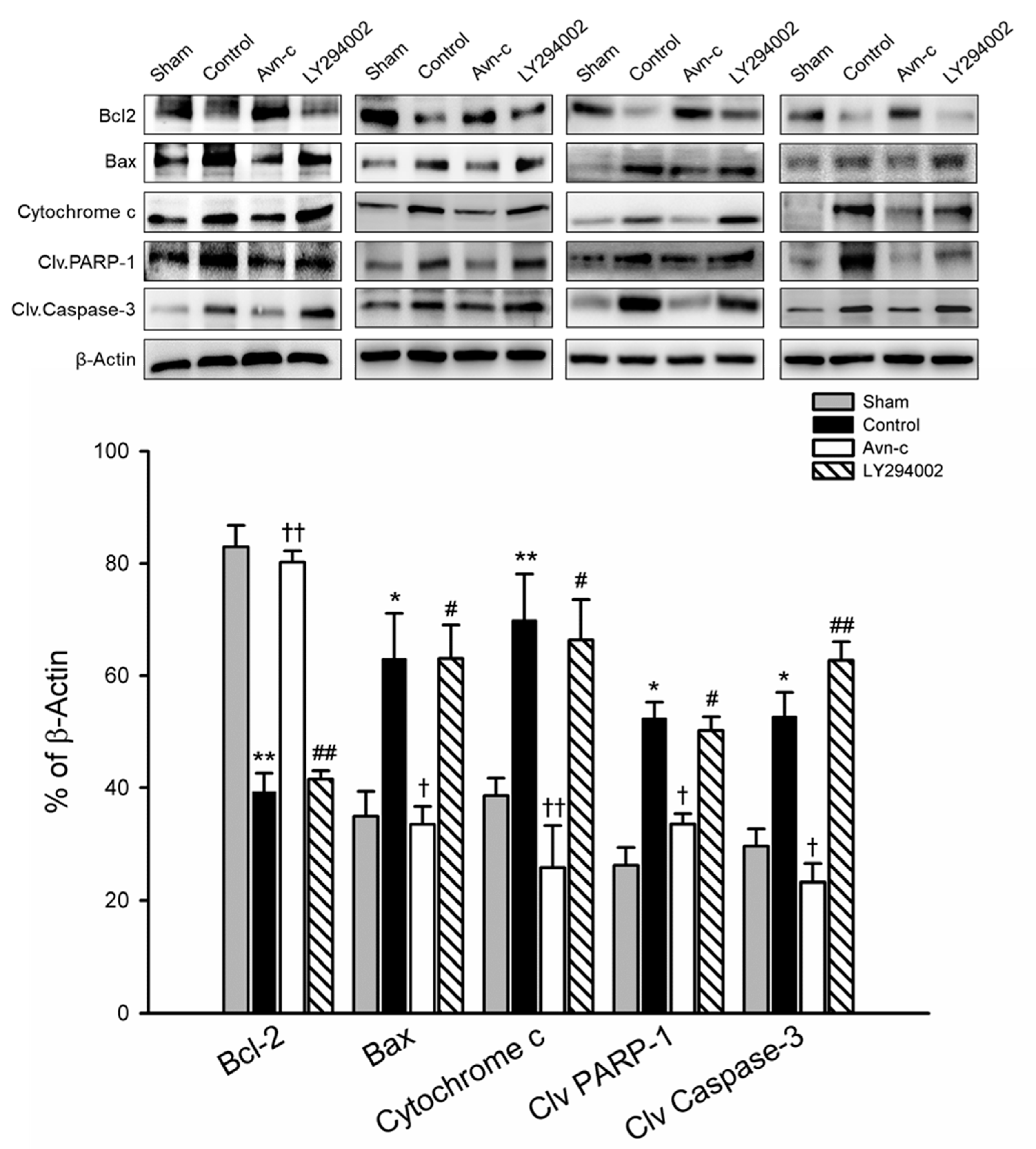
3.4. Avn-c Attenuated MCAo-Induced Changes in Apoptotic and Anti-Apoptotic Proteins and Anti-Apoptosis Markers Were Reduced Following Treatment with a PI3K Inhibitor
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Collaborators, G.B.D.S. Global, regional, and national burden of stroke, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef]
- Ge, P.; Luo, Y.; Liu, C.L.; Hu, B. Protein aggregation and proteasome dysfunction after brain ischemia. Stroke 2007, 38, 3230–3236. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef]
- Fink, K.; Zhu, J.; Namura, S.; Shimizu-Sasamata, M.; Endres, M.; Ma, J.; Dalkara, T.; Yuan, J.; Moskowitz, M.A. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J. Cereb. Blood Flow Metab. 1998, 18, 1071–1076. [Google Scholar] [CrossRef]
- Krajewski, S.; Mai, J.K.; Krajewska, M.; Sikorska, M.; Mossakowski, M.J.; Reed, J.C. Up-Regulation of Bax Protein-Levels in Neurons Following Cerebral-Ischemia. J. Neurosci. 1995, 15, 6364–6376. [Google Scholar] [CrossRef]
- Matsushita, K.; Matsuyama, T.; Kitagawa, K.; Matsumoto, M.; Yanagihara, T.; Sugita, M. Alterations of Bcl-2 family proteins precede cytoskeletal proteolysis in the penumbra, but not in infarct centres following focal cerebral ischemia in mice. Neuroscience 1998, 83, 439–448. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Noshita, N.; Lewen, A.; Sugawara, T.; Chan, P.H. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001, 21, 1442–1450. [Google Scholar] [CrossRef]
- Shibata, M.; Yamawaki, T.; Sasaki, T.; Hattori, H.; Hamada, J.; Fukuuchi, Y.; Okano, H.; Miura, M. Upregulation of Akt phosphorylation at the early stage of middle cerebral artery occlusion in mice. Brain Res. 2002, 942, 1–10. [Google Scholar] [CrossRef]
- Yao, R.J.; Cooper, G.M. Requirement for Phosphatidylinositol-3 Kinase in the Prevention of Apoptosis by Nerve Growth-Factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.A.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- KauffmanZeh, A.; RodriguezViciana, P.; Ulrich, E.; Gilbert, C.; Coffer, P.; Downward, J.; Evan, G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef]
- Nozaki, K.; Nishimura, M.; Hashimoto, N. Mitogen-activated protein kinases and cerebral ischemia. Mol. Neurobiol. 2001, 23, 1–19. [Google Scholar] [CrossRef]
- Fruman, D.A.; Cantley, L.C. Phosphoinositide 3-kinase in immunological systems. Semin. Immunol. 2002, 14, 7–18. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303, 701–704. [Google Scholar] [CrossRef]
- Meijer, L.; Flajolet, M.; Greengard, P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 2004, 25, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.W. Oat phenolics: Avenanthramides, novel substituted N-cinnamoylanthranilate alkaloids from oat groats and hulls. J. Agríe. Food Chem. 1989, 37, 60–66. [Google Scholar] [CrossRef]
- Mullin, W.J.; Collins, F.W. High-performance liquid chromatographic determination of avenanthramides, n-aroylanthranilic acid alkaloids from oats. J. Chromatogr. 1988, 445, 363–370. [Google Scholar] [CrossRef]
- Collins, F.W.; McLachlan, D.C.; Blackwell, B.A. Oat phenolics: Avenalumic acids, a new group of bound phenolics acids from oat groats and hulls. Cereal Chem. 1991, 68, 184–189. [Google Scholar]
- Emmonsa, C.L.; Peterson, D.M.; Hahn, M.J. Oat avenanthramides exhibit antioxidant activities in vitro. Food Chem. 2002, 79, 474–478. [Google Scholar] [CrossRef]
- Helns, A.; Kyro, C.; Andersen, I.; Lacoppidan, S.; Overvad, K.; Christensen, J.; Tjonneland, A.; Olsen, A. Intake of whole grains is associated with lower risk of myocardial infarction: The Danish Diet, Cancer and Health Cohort. Am. J. Clin. Nutr. 2016, 103, 999–1007. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef]
- Bederson, J.B.; Pitts, L.H.; Tsuji, M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke 1986, 17, 472–476. [Google Scholar] [CrossRef]
- Li, J.Y.; Boado, R.J.; Pardridge, W.M. Blood-brain barrier genomics. J. Cereb. Blood Flow Metab. 2001, 21, 61–68. [Google Scholar] [CrossRef]
- Li, J.Y.; Boado, R.J.; Pardridge, W.M. Rat blood-brain barrier genomics. II. J. Cereb. Blood Flow Metab. 2002, 22, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Mark, K.S.; Davis, T.P. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1485–H1494. [Google Scholar] [CrossRef] [PubMed]
- Pulsinelli, W.A. Selective neuronal vulnerability: Morphological and molecular characteristics. Prog. Brain Res. 1985, 63, 29–37. [Google Scholar] [CrossRef]
- Heron, A.; Pollard, H.; Dessi, F.; Moreau, J.; Lasbennes, F.; Ben-Ari, Y.; Charriaut-Marlangue, C. Regional variability in DNA fragmentation after global ischemia evidenced by combined histological and gel electrophoresis observations in the rat brain. J. Neurochem. 1993, 61, 1973–1976. [Google Scholar] [CrossRef]
- Li, Y.; Chopp, M.; Jiang, N.; Yao, F.; Zaloga, C. Temporal Profile of in-Situ DNA Fragmentation after Transient Middle Cerebral-Artery Occlusion in the Rat. J. Cerebr. Blood Flow Metab. 1995, 15, 389–397. [Google Scholar] [CrossRef]
- Guegan, C.; Boutin, H.; Boudry, C.; MacKenzie, E.T.; Sola, B. Apoptotic death in cortical neurons of mice subjected to focal ischemia. Cr. Acad. Sci. III-Vie 1996, 319, 879–885. [Google Scholar]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Spittaels, K.; Van den Haute, C.; Van Dorpe, J.; Terwel, D.; Vandezande, K.; Lasrado, R.; Bruynseels, K.; Irizarry, M.; Verhoye, M.; Van Lint, J.; et al. Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 2002, 113, 797–808. [Google Scholar] [CrossRef]
- Kim, W.Y.; Wang, X.S.; Wu, Y.H.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Pap, M.; Cooper, G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, B.; Kim, H.; Choi, J.-I.; Bae, H.-B.; Jeong, S. Avenanthramide C Prevents Neuronal Apoptosis via PI3K/Akt/GSK3β Signaling Pathway Following Middle Cerebral Artery Occlusion. Brain Sci. 2020, 10, 878. https://doi.org/10.3390/brainsci10110878
Jin B, Kim H, Choi J-I, Bae H-B, Jeong S. Avenanthramide C Prevents Neuronal Apoptosis via PI3K/Akt/GSK3β Signaling Pathway Following Middle Cerebral Artery Occlusion. Brain Sciences. 2020; 10(11):878. https://doi.org/10.3390/brainsci10110878
Chicago/Turabian StyleJin, Baoyuan, Hyehyun Kim, Jeong-Il Choi, Hong-Beom Bae, and Seongtae Jeong. 2020. "Avenanthramide C Prevents Neuronal Apoptosis via PI3K/Akt/GSK3β Signaling Pathway Following Middle Cerebral Artery Occlusion" Brain Sciences 10, no. 11: 878. https://doi.org/10.3390/brainsci10110878
APA StyleJin, B., Kim, H., Choi, J.-I., Bae, H.-B., & Jeong, S. (2020). Avenanthramide C Prevents Neuronal Apoptosis via PI3K/Akt/GSK3β Signaling Pathway Following Middle Cerebral Artery Occlusion. Brain Sciences, 10(11), 878. https://doi.org/10.3390/brainsci10110878