Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis



Abstract
:1. Introduction
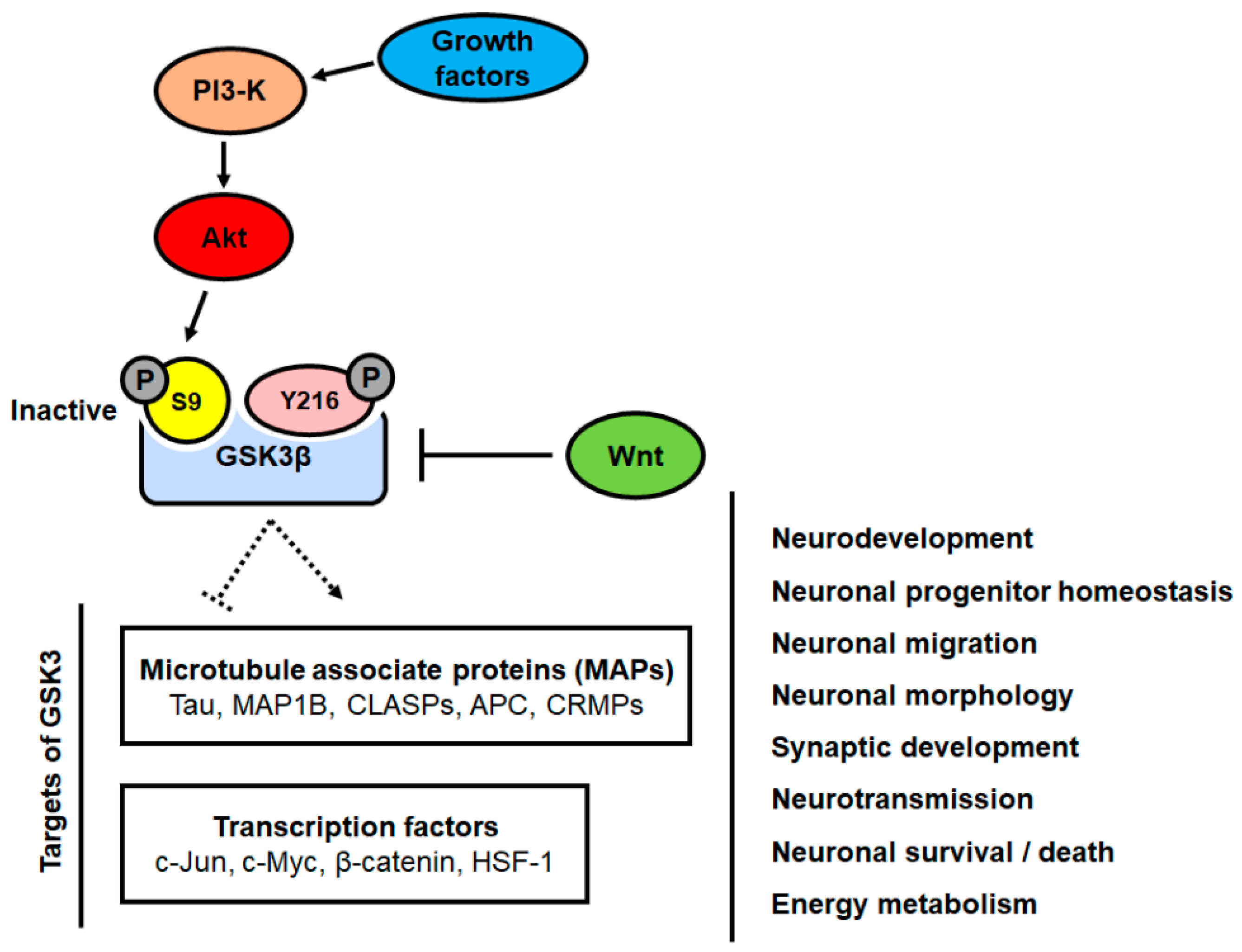
2. Role of GSK3β Signaling in Neurons
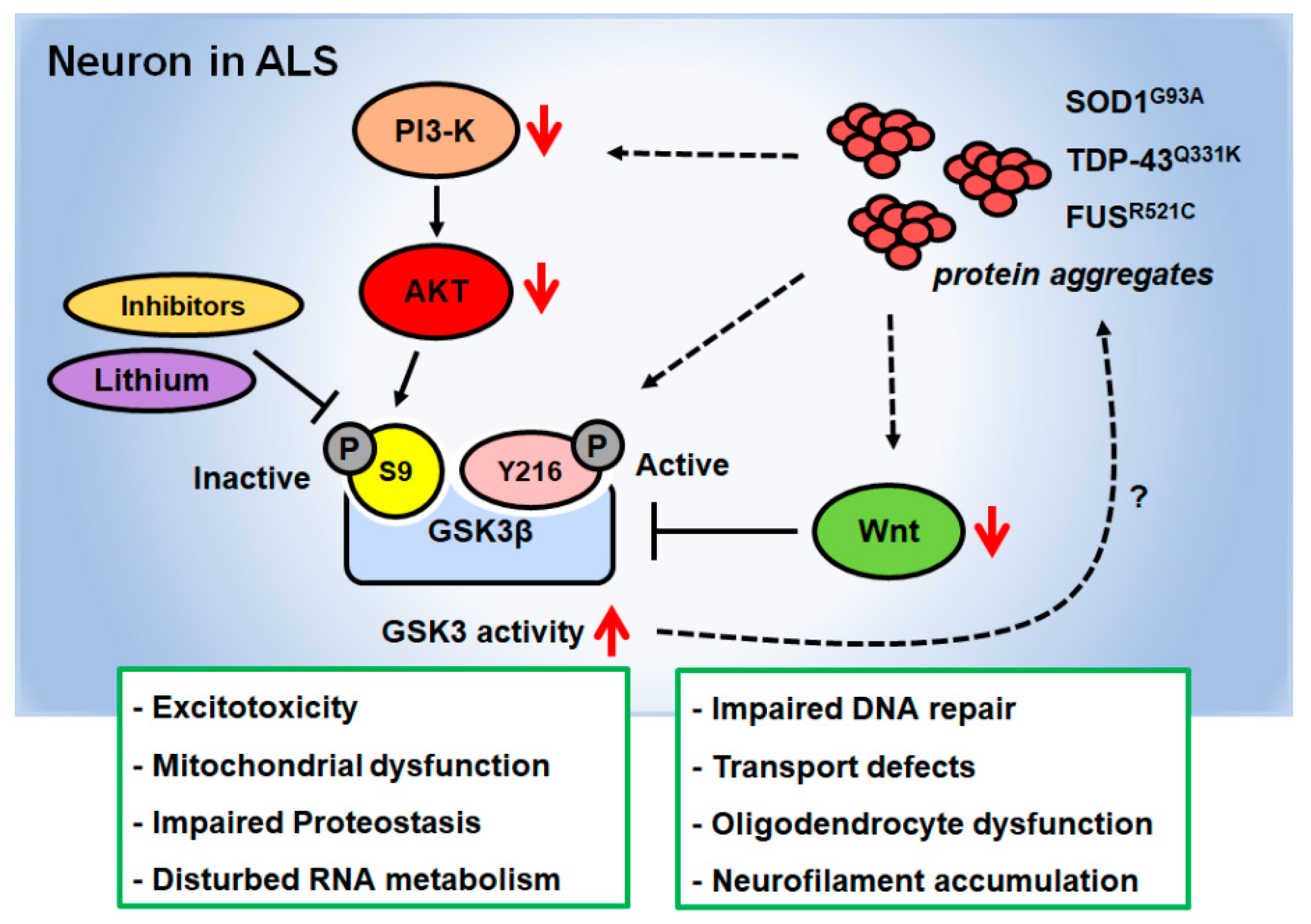
3. Role of GSK3β in Neurodegenerative Diseases
4. Studies Exploring the Role of GSK3β in In Vivo Models of ALS
5. Studies Exploring the Role of GSK3β in In Vitro Models of ALS
6. Inhibitors of GSK3β in ALS Clinical Trials
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Alonso, A.; Logroscino, G.; Jick, S.S.; Hernán, M.A. Incidence and lifetime risk of motor neuron disease in the United Kingdom: A population-based study. Eur. J. Neurol. 2009, 16, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.; Leclerc, A.; Tamrazian, E.; Vanderburg, C.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.; Munsat, T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle: Separation from Cyclic-AMP-Dependent Protein Kinase and Phosphorylase Kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Rylatt, D.B.; Aitken, A.; Bilham, T.; Condon, G.D.; Embi, N.; Cohen, P. Glycogen synthase from rabbit skeletal muscle: Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur. J. Biochem. 1980, 107, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Jack, S.L.; Vijayaraghavan, S. Changes in sperm glycogen synthase kinase-3 serine phosphorylation and activity accompany motility initiation and stimulation. J. Androl. 2004, 25, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Goswami, S.; Dey, S.; Gangoda, M.; Brothag, C.; Eisa, A.; Woodgett, J.; Phiel, C.; Kline, D.; Vijayaraghavan, S. Isoform-specific requirement for GSK3α in sperm for male fertility. Biol. Reprod. 2018, 99, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Kaidanovich-Beilin, O.; Lipina, T.V.; Takao, K.; Van Eede, M.; Hattori, S.; Laliberté, C.; Khan, M.; Okamoto, K.; Chambers, J.W.; Fletcher, P.J. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2009, 2, 35. [Google Scholar] [CrossRef] [Green Version]
- Kockeritz, L.; Doble, B.; Patel, S.; Woodgett, J.R. Glycogen synthase kinase-3-an overview of an over-achieving protein kinase. Curr. Drug Targets 2006, 7, 1377–1388. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef]
- Mukai, F.; Ishiguro, K.; Sano, Y.; Fujita, S.C. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3β. J. Neurochem. 2002, 81, 1073–1083. [Google Scholar] [CrossRef]
- Wood-Kaczmar, A.; Kraus, M.; Ishiguro, K.; Philpott, K.L.; Gordon-Weeks, P.R. An alternatively spliced form of glycogen synthase kinase-3β is targeted to growing neurites and growth cones. Mol. Cell. Neurosci. 2009, 42, 184–194. [Google Scholar] [CrossRef]
- Castaño, Z.; Gordon-Weeks, P.R.; Kypta, R.M. The neuron-specific isoform of glycogen synthase kinase-3β is required for axon growth. J. Neurochem. 2010, 113, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.G.; Gordon-Weeks, P.R. Microtubule-associated protein 1B phosphorylation by glycogen synthase kinase 3β is induced during PC12 cell differentiation. J. Cell Sci. 2001, 114, 4273–4284. [Google Scholar] [PubMed]
- Mao, Y.; Ge, X.; Frank, C.L.; Madison, J.M.; Koehler, A.N.; Doud, M.K.; Tassa, C.; Berry, E.M.; Soda, T.; Singh, K.K. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell 2009, 136, 1017–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.-Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultje, R.S.; Castaneda-Castellanos, D.R.; Jan, L.Y.; Jan, Y.-N.; Kriegstein, A.R.; Shi, S.-H. Mammalian Par3 regulates progenitor cell asymmetric division via notch signaling in the developing neocortex. Neuron 2009, 63, 189–202. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.-G.; Xu, C.; Gumbiner, B.M. Identification of targets of the Wnt pathway destruction complex in addition to β-catenin. Proc. Natl. Acad. Sci. USA 2009, 106, 5165–5170. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Kim, N.-G.; Gumbiner, B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J. Regulation of Hh/Gli signaling by dual ubiquitin pathways. Cell Cycle 2006, 5, 2457–2463. [Google Scholar] [CrossRef]
- Barth, A.I.; Caro-Gonzalez, H.Y.; Nelson, W.J. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 2008, 3, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Guo, W.; Liang, X.; Rao, Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: Critical roles of GSK-3β and its upstream regulators. Cell 2005, 120, 123–135. [Google Scholar] [PubMed] [Green Version]
- Etienne-Manneville, S.; Hall, A. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature 2003, 421, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Mimori-Kiyosue, Y.; Tsukita, S. Where is APC going? J. Cell Biol. 2001, 154, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.-Q.; Zhou, J.; Dedhar, S.; Wu, Y.-H.; Snider, W.D. NGF-induced axon growth is mediated by localized inactivation of GSK-3β and functions of the microtubule plus end binding protein APC. Neuron 2004, 42, 897–912. [Google Scholar] [CrossRef] [Green Version]
- Akhmanova, A.; Hoogenraad, C.C.; Drabek, K.; Stepanova, T.; Dortland, B.; Verkerk, T.; Vermeulen, W.; Burgering, B.M.; De Zeeuw, C.I.; Grosveld, F. Clasps are CLIP-115 and-170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 2001, 104, 923–935. [Google Scholar] [CrossRef]
- Trivedi, N.; Marsh, P.; Goold, R.G.; Wood-Kaczmar, A.; Gordon-Weeks, P.R. Glycogen synthase kinase-3β phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J. Cell Sci. 2005, 118, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2005, 1739, 280–297. [Google Scholar] [CrossRef] [Green Version]
- Zumbrunn, J.; Kinoshita, K.; Hyman, A.A.; Näthke, I.S. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3β phosphorylation. Curr. Biol. 2001, 11, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.; Gordon-Weeks, P.R. Inhibition of glycogen synthase kinase 3β in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol. Cell. Neurosci. 2003, 23, 626–637. [Google Scholar] [CrossRef]
- Garrido, J.J.; Simón, D.; Varea, O.; Wandosell, F. GSK3 alpha and GSK3 beta are necessary for axon formation. FEBS Lett. 2007, 581, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, A.; Huang, X.; Hall, A. Neuronal polarity is regulated by glycogen synthase kinase-3 (GSK-3β) independently of Akt/PKB serine phosphorylation. J. Cell Sci. 2006, 119, 3927–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.; Myers, K.R.; Yu, K.; Wise, A.; De Blas, A.L.; Hartzell, H.C.; Zheng, J.Q. Activity-dependent regulation of dendritic growth and maintenance by glycogen synthase kinase 3β. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T. LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Liu, W.; Yan, Z. Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 2010, 285, 26369–26376. [Google Scholar] [CrossRef] [Green Version]
- Tyagarajan, S.K.; Ghosh, H.; Yévenes, G.E.; Nikonenko, I.; Ebeling, C.; Schwerdel, C.; Sidler, C.; Zeilhofer, H.U.; Gerrits, B.; Muller, D. Regulation of GABAergic synapse formation and plasticity by GSK3β-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. USA 2011, 108, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Seo, A.Y.; Joseph, A.-M.; Dutta, D.; Hwang, J.C.; Aris, J.P.; Leeuwenburgh, C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 2010, 123, 2533–2542. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.-H.; Lin, C.-C.; Yang, M.-C.; Wei, C.-C.; Liao, H.-D.; Lin, R.-C.; Tu, W.-Y.; Kao, T.-C.; Hsu, C.-M.; Cheng, J.-T. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, e49112. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.L.; Hock, M.B.; Ekholm-Reed, S.; Wohlschlegel, J.A.; Dev, K.K.; Kralli, A.; Reed, S.I. SCFCdc4 acts antagonistically to the PGC-1α transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008, 22, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.A.; Souder, D.C.; Miller, K.N.; Clark, J.P.; Sagar, A.K.; Eliceiri, K.W.; Puglielli, L.; Beasley, T.M.; Anderson, R.M. GSK3β regulates brain energy metabolism. Cell Rep. 2018, 23, 1922.e1924–1931.e1924. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Yellowlees, D.; Kernohan, J.C.; Cohen, P. Purification of Glycogen Synthase Kinase 3 from Rabbit Skeletal Muscle. Copurification with the Activating Factor (FA) of the (Mg-ATP) Dependent Protein Phosphatase. Eur. J. Biochem. 1981, 119, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Llorens-Marítin, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Li, D.W.; Liu, Z.Q.; Chen, W.; Yao, M.; Li, G.R. Association of glycogen synthase kinase-3β with Parkinson’s disease. Mol. Med. Rep. 2014, 9, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in neurodegenerative diseases. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Leystra-Lantz, C.; Strong, M.J. Upregulation of GSK3β expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci). Brain Res. 2008, 1196, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, M.; Conte, A.; Zollino, M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin. Genet. 2013, 83, 408–416. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Philpott, K.L.; McCarthy, M.J.; Klippel, A.; Rubin, L.L. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J. Cell Biol. 1997, 139, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Crowder, R.J.; Freeman, R.S. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 1998, 18, 2933–2943. [Google Scholar] [CrossRef] [Green Version]
- Warita, H.; Manabe, Y.; Murakami, T.; Shiro, Y.; Nagano, I.; Abe, K. Early decrease of survival signal-related proteins in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Apoptosis 2001, 6, 345–352. [Google Scholar] [CrossRef]
- Yazdani, U.; Terman, J.R. The semaphorins. Genome Biol. 2006, 7, 211. [Google Scholar] [CrossRef] [Green Version]
- Dolma, K.; Iacobucci, G.J.; Hong Zheng, K.; Shandilya, J.; Toska, E.; White, J.A.; Spina, E.; Gunawardena, S. Presenilin influences glycogen synthase kinase-3 β (GSK-3β) for kinesin-1 and dynein function during axonal transport. Hum. Mol. Genet. 2014, 23, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.J.; Hebbar, S.; Gao, X.A.; Alexander, M.; Pandey, J.P.; Walla, M.D.; Cotham, W.E.; King, S.J.; Smith, D.S. GSK-3β phosphorylation of cytoplasmic dynein reduces Ndel1 binding to intermediate chains and alters dynein motility. Traffic 2015, 16, 941–961. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Nakamura, F.; Usui, H.; Takeuchi, K.; Yamashita, N.; Goshima, Y. Semaphorin3A-induced axonal transport mediated through phosphorylation of Axin-1 by GSK3β. Brain Res. 2015, 1598, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Venkova, K.; Christov, A.; Kamaluddin, Z.; Kobalka, P.; Siddiqui, S.; Hensley, K. Semaphorin 3A Signaling Through Neuropilin-1 Is an Early Trigger for Distal Axonopathy in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2014, 73, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allodi, I.; Comley, L.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Benitez, J.A.; Cao, M.; Corti, S.; Hedlund, E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 2016, 6, 25960. [Google Scholar] [CrossRef] [Green Version]
- Moszczynski, A.J.; Strong, W.; Xu, K.; McKee, A.; Brown, A.; Strong, M.J. Pathologic Thr175 tau phosphorylation in CTE and CTE with ALS. Neurology 2018, 90, e380–e387. [Google Scholar] [CrossRef] [Green Version]
- Cairns, N.J.; Neumann, M.; Bigio, E.H.; Holm, I.E.; Troost, D.; Hatanpaa, K.J.; Foong, C.; White III, C.L.; Schneider, J.A.; Kretzschmar, H.A. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 2007, 171, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.B.; Lee, V.M.-Y.; Trojanowski, J.Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ray, P.; Rao, E.J.; Shi, C.; Guo, W.; Chen, X.; Woodruff, E.A.; Fushimi, K.; Wu, J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2010, 107, 3169–3174. [Google Scholar] [CrossRef] [Green Version]
- Hanson, K.A.; Kim, S.H.; Wassarman, D.A.; Tibbetts, R.S. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J. Biol. Chem. 2010, 285, 11068–11072. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Neukomm, L.J.; Brown Jr, R.H.; Freeman, M.R. Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 2015, 25, 2130–2136. [Google Scholar] [CrossRef] [Green Version]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1669. [Google Scholar] [CrossRef] [Green Version]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 inhibitors: preclinical and clinical focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalecka-Franaszek, E.; Chuang, D.-M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 8745–8750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlenza, O.V.; De-Paula, V.d.J.R.; Diniz, B. Neuroprotective effects of lithium: implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Cho, S.I.; Lim, H.R.; Lee, J.K.; Lee, Y.A.; Noh, J.S.; Joo, I.S.; Kim, K.-W.; Gwag, B.J. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol. Pharmacol. 2007, 71, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, L.D.; Jiang, Y.M.; Manji, H.K. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J. Neurochem. 2000, 72, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Sugai, F.; Yamamoto, Y.; Miyaguchi, K.; Zhou, Z.; Sumi, H.; Hamasaki, T.; Goto, M.; Sakoda, S. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur. J. Neurosci. 2004, 20, 3179–3183. [Google Scholar] [CrossRef]
- Feng, H.-L.; Leng, Y.; Ma, C.-H.; Zhang, J.; Ren, M.; Chuang, D.-M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.-H.; Kim, Y.; Kim, H.Y.; Hwang, S.; Lee, C.H.; Kim, S.H. Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007, 205, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP 51 interaction and ER–mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef] [PubMed]
- Gohar, M.; Yang, W.; Strong, W.; Volkening, K.; Leystra-Lantz, C.; Strong, M.J. Tau phosphorylation at threonine-175 leads to fibril formation and enhanced cell death: implications for amyotrophic lateral sclerosis with cognitive impairment. J. Neurochem. 2009, 108, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Gupta, S.K.; Kim, K.J.; Powers, B.E.; Cerqueira, A.; Wainger, B.J.; Ngo, H.D.; Rosowski, K.A.; Schein, P.A.; Ackeifi, C.A. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 2013, 12, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, H.; Tamatani, M.; Yamaguchi, A.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J. 2001, 15, 1218–1220. [Google Scholar] [CrossRef]
- Jin, K.; Mao, X.; Batteur, S.; McEachron, E.; Leahy, A.; Greenberg, D. Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience 2001, 108, 351–358. [Google Scholar] [CrossRef]
- Lambrechts, D.; Carmeliet, P. VEGF at the neurovascular interface: Therapeutic implications for motor neuron disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2006, 1762, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, J.; Shah, S.; McCloskey, D.; Goodman, J.; Elkady, A.; Atassi, H.; Hylton, D.; Rudge, J.; Scharfman, H.; Croll, S. Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience 2008, 151, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Xu, W.; Luo, C.; Gozal, D.; Liu, R. VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Mol. Brain Res. 2003, 111, 155–164. [Google Scholar] [CrossRef]
- Zhang, B.; Rusciano, D.; Osborne, N.N. Orally administered epigallocatechin gallate attenuates retinal neuronal death in vivo and light-induced apoptosis in vitro. Brain Res. 2008, 1198, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-H.; Kim, S.H.; Kwon, H.; Park, Y.; Kim, K.S.; Song, C.W.; Kim, J.; Kim, M.-H.; Yu, H.-J.; Henkel, J.S. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/Akt and glycogen synthase kinase-3. Mol. Brain Res. 2003, 118, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-H.; Kwon, H.; Kim, K.S.; Kim, J.; Kim, M.-H.; Yu, H.-J.; Kim, M.; Lee, K.-W.; Do, B.R.; Jung, H.K. Epigallocatechin gallate prevents oxidative-stress-induced death of mutant Cu/Zn-superoxide dismutase (G93A) motoneuron cells by alteration of cell survival and death signals. Toxicology 2004, 202, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.H.; Lee, Y.B.; Kim, K.S.; Kim, H.J.; Kim, M.; Lee, Y.J.; Kim, J.; Lee, K.W.; Kim, S.H. Role of GSK-3β activity in motor neuronal cell death induced by G93A or A4V mutant hSOD1 gene. Eur. J. Neurosci. 2005, 22, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Ryves, W.J.; Harwood, A.J. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem. Biophys. Res. Commun. 2001, 280, 720–725. [Google Scholar] [CrossRef]
- Meijer, L.; Skaltsounis, A.-L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Gompel, M.; Leost, M.; Joffe, E.B.D.K.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First non-ATP competitive glycogen synthase kinase 3 β (GSK-3β) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef]
- Bebchuk, J.M.; Arfken, C.L.; Dolan-Manji, S.; Murphy, J.; Hasanat, K.; Manji, H.K. A preliminary investigation of a protein kinase C inhibitor in the treatment of acute mania. Arch. Gen. Psychiatry 2000, 57, 95–97. [Google Scholar] [CrossRef]
- Cechinel-Recco, K.; Valvassori, S.S.; Varela, R.B.; Resende, W.R.; Arent, C.O.; Vitto, M.F.; Luz, G.; de Souza, C.T.; Quevedo, J. Lithium and tamoxifen modulate cellular plasticity cascades in animal model of mania. J. Psychopharmacol. 2012, 26, 1594–1604. [Google Scholar] [CrossRef]
- Effect of Lithium and Divalproex in Alzheimer′s Disease. Available online: https://ClinicalTrials.gov/show/NCT00088387 (accessed on 14 September 2020).
- Forlenza, O.V.; Diniz, B.S.; Radanovic, M.; Santos, F.S.; Talib, L.L.; Gattaz, W.F. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br. J. Psychiatry 2011, 198, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Ewers, M.; Burger, K.; Annas, P.; Mortberg, A.; Bogstedt, A.; Frolich, L.; Schroder, J.; Schonknecht, P.; Riepe, M.W. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922. [Google Scholar] [CrossRef]
- Macdonald, A.; Briggs, K.; Poppe, M.; Higgins, A.; Velayudhan, L.; Lovestone, S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry A J. Psychiatry Late Life Allied Sci. 2008, 23, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Sereno, L.; Coma, M.; Rodriguez, M.; Sanchez-Ferrer, P.; Sanchez, M.; Gich, I.; Agullo, J.; Perez, M.; Avila, J.; Guardia-Laguarta, C. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andres, M.V.; Gomez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; Leon, T. Treatment of Alzheimer′s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer’s Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.-J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Type of Study | Model System | Key Findings | Reference |
|---|---|---|---|
| Ex Vivo | ALS patient sample (spinal cord, frontal and temporal cortices) | Increased GSK3β expression and cytosolic phospho-Y216 GSK3β in ALS patient sample | [61] |
| Ex Vivo | ALS patient sample (frontal and temporal cortices) | Increased GSK3β expression and cytosolic phospho-Y216 GSK3β in ALS patient sample | [62] |
| Ex Vivo | CTE-ALS patient sample (hippocampus and spinal cord) | Activated GSK3β, pThr175 tau, pThr231 tau, and oligomerized tau protein expression | [75] |
| In Vivo | SOD1G93A transgenic mouse | Increased GSK3β activity via decreasing the PI3-K/Akt expression in an age-dependent manner | [68] |
| In Vivo | SOD1G93A transgenic mouse | Inhibition of Sema3A/NRP1 signaling restored life span, motor function, and NMJ denervation | [73] |
| In Vivo | SOD1G93A transgenic mouse | IGF-2 prolonged survival by preserving motor neurons and inducing nerve regeneration | [74] |
| In Vivo | TDP-43Q331K transgenic fly | Loss of GSK3β/Shaggy suppressed TDP-43Q331K-mediated axon and NMJ degeneration | [80] |
| In Vivo | SOD1G93A transgenic mouse | Treatment with lithium (a GSK3β inhibitor) improved neuron survival, motor function, and mortality | [86] |
| In Vivo | SOD1G93A transgenic mouse | Treatment with VPA (a GSK3β inhibitor) rescued motor neuronal defects and delayed the disease progression | [88] |
| In Vivo | SOD1G93A transgenic mouse | Treatment with a combination of lithium and VPA strongly rescued the motor dysfunction and disease progression via upregulation of phospho-S9 GSK3β | [89] |
| In Vivo | SOD1G93A transgenic mouse | GSK3β inhibitor VIII treatment prolonged the life span via inhibition of GSK3β activity, preserved survival signals, and attenuated death and inflammatory signals | [91] |
| In Vitro | FUS/TDP43-transfected HEK293, NSC34 cells | ALS associated FUS, TDP-43 activated GSK3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations | [92,93] |
| In Vitro | Tau-transfected HEK293T and Neuro2A cells | ALSci is associated with tau phosphorylation at Thr175 and leads to the activation of GSK3β, which induces phosphorylation at tau Thr231. | [94] |
| In Vitro | ALS-patients iPSC-derived motor neuron | IGF-2 induced Akt phosphorylation, GSK3β phosphorylation, and β-catenin levels while protecting ALS patient motor neurons | [74] |
| In Vitro | SOD1G93A mESCs, ALS-patients iPSC-derived motor neuron | Kenpaullone treatment improved the survival of human motor neurons via inhibition of GSK3β | [95] |
| In Vitro | SOD1G93A-transfected NSC34 cells | GSK3β inhibitor: VEGF treatment activated PI3-K/Akt signaling and restored neuron cell death in motor neurons | [100] |
| In Vitro | PDC-induced mouse organotypic spinal cord | VPA (an inhibitor of GSK3β) treatment protected spinal motor neurons against glutamate toxicity | [88] |
| In Vitro | SOD1G93A-transfected VSC 4.1 cells | EGCG (an inhibitor of GSK3β) treatment restored viability in oxidative stress-induced cell death via activation of PI3-K/Akt signaling | [103] |
| In Vitro | SOD1G93A-transfected VSC 4.1 cells | 2-thio(3-iodobenzyl)-5-(1-pyridyl)-[1,3,4]-oxadiazole (an inhibitor of GSK3β) treatment restored viability via activation of HSF-1 and reduction of cytochrome c release, caspase-3 activation, and PARP cleavage | [104] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.-J.; Cha, S.J.; Lee, J.-W.; Kim, H.-J.; Kim, K. Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sci. 2020, 10, 675. https://doi.org/10.3390/brainsci10100675
Choi H-J, Cha SJ, Lee J-W, Kim H-J, Kim K. Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sciences. 2020; 10(10):675. https://doi.org/10.3390/brainsci10100675
Chicago/Turabian StyleChoi, Hyun-Jun, Sun Joo Cha, Jang-Won Lee, Hyung-Jun Kim, and Kiyoung Kim. 2020. "Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis" Brain Sciences 10, no. 10: 675. https://doi.org/10.3390/brainsci10100675
APA StyleChoi, H.-J., Cha, S. J., Lee, J.-W., Kim, H.-J., & Kim, K. (2020). Recent Advances on the Role of GSK3β in the Pathogenesis of Amyotrophic Lateral Sclerosis. Brain Sciences, 10(10), 675. https://doi.org/10.3390/brainsci10100675